Abstract
Hearing loss (HL) is a common sensory disorder affecting over 5% of the global population. The etiology underlying HL includes congenital and acquired causes; genetic factors are the main cause in over 50% of congenital cases. Pathogenic variants in the GJB2 gene are a major cause of congenital non-syndromic hearing loss (NSHL), while their distribution is highly heterogeneous in different populations. To the best of our knowledge, there is no data regarding the genetic etiologies of HL in Peru. In this study, we screened 133 Peruvian families with NSHL living in Lima. We sequenced both exons of the GJB2 gene for all probands. Seven probands with familial NSHL that remained negative for GJB2 variants underwent whole genome sequencing (WGS). We identified biallelic pathogenic variants in GJB2 in 43 probands; seven were heterozygous for only one allele. The c.427C>T variant was the most common pathogenic variant followed by the c.35delG variant. WGS revealed three novel variants in MYO15A in two probands, one of them was predicted to affect splicing and the others produce a premature stop codon. The Peruvian population showed a complex profile for genetic variants in the GJB2 gene, this particular profile might be a consequence of the admixture history in Peru.
1. Introduction
Hearing loss (HL) is a common sensory disorder affecting over 5% of the global population [1]. The etiology underlying HL includes congenital and acquired causes; genetic factors are causative in over 50% of congenital cases [2]. The mode of inheritance can be autosomal recessive (~80%), autosomal dominant (~20%), mitochondrial, or X-linked (1%–1.5%) [3]. DNA variants in over 100 genes have been reported to cause non-syndromic hearing loss (NSHL) [4], and their distribution is highly heterogeneous in different populations [5].
Pathogenic variants in the GJB2 gene (NM_004004.6) are considered to be the major cause of congenital NSHL across populations [6]. The GJB2 gene has two exons; the second exon codes for the gap junction beta-2 (or Cx26) protein, involved in the maintenance of a high endocochlear potential [7]. The pathogenic variant c.35delG has been reported as the most common genetic cause of NSHL with European ancestry [8]. In populations from East Asia, such as Japan, China, and Korea, the c.235delC variant is the most common mutation [9,10,11,12]. In Latin American populations, such as Argentina and Mexico, c.35delG is the most common variant [13,14]. Around 1.8% of the Peruvian population have significant HL, of whom up to 11% of cases are congenital [15]. To the best of our knowledge, there is no data regarding genetic etiologies of HL in Peru.
Peru is a South American country hosting an admixed population with a significant Amerindian component. Due to many historical events, including migration before and during the Inca Empire and Spanish colonization, the global ethnic structure of the Peruvian population comprises ~80% of Amerindian, 7.4% of European, 4.4% of Oceanian, 3.5% of East Asian, and 1.7% of African ancestries. However, distribution varies across regions within Peru [16,17].
In this study, we screened 133 Peruvian families with NSHL living in Lima. We sequenced the two exons of the GJB2 gene for all probands. Seven probands from multiplex families, negative for GJB2 variants, underwent whole genome sequencing (WGS). In addition, we present ancestry analysis performed for the most common pathogenic variant (GJB2 NM_004004.6; c.427C>T) in order to determine the possible origin(s) of this variant in Peru.
2. Methods
2.1. Subjects
DNA samples from 133 unrelated probands (63 multiplex and 70 simplex cases) affected with NSHL were obtained from the biorepository of the Neurogenetics Research Center (NRC) at the Instituto Nacional de Ciencias Neurológicas (INCN), from Lima, Peru, collected for a previous study. Diagnosis of all probands with NSHL was based on clinical and audiology assessment performed by an otolaryngologist. All cases were screened for environmental factors through a survey given to the mothers of the cases. The survey queried prenatal, natal, and postnatal infections, aminoglycoside exposure, prematurity, perinatal hypoxia, need for neonatal intensive care, and head and sound trauma, for which all cases were negative. All DNA samples were negative for two GJB6 deletions by multiplex PCR from a previous study [18].
This study was approved by the Ethics Committee of the Instituto Nacional de Ciencias Neurológicas (INCN) and the IRB of the University of Miami.
2.2. Sanger Sequencing of GJB2 Exon 1 and Exon 2
All samples were analyzed at the Hussman Institute for Human Genomics at the University of Miami. We performed Sanger sequencing of both exons of the GJB2 gene using primers 5’-CCGGGAAGCTCTGAGGAC-3’, 5’-GCAACCGCTCTGGGTCTC-3’ for exon 1; and 5’-TTGGTGTTTGCTCAGGAAGA-3’, 5’-GGCCTACAGGGGTTTCAAAT-3’ for exon 2. PCR conditions were the same as previously described [19]. Visualization of PCR products was on agarose gels followed by cleaning over Sephadex columns according to the manufacturer’s recommendations. We performed sequence analysis using the Big Dye Terminator v3.1 Cycle Sequencing Kit and the ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Foster City, USA). Sequence traces were analyzed using the Sequencher® 5.0 program (Gene Codes Corporation, Ann Arbor, USA). Classification of genetic variants was performed following the American College of Medical Genetics (ACMG) guidelines [20].
2.3. Whole Genome Sequencing Analysis
Seven unrelated probands from multiplex families, negative for any pathogenic or likely pathogenic variant in GJB2, were subjected to WGS using a BGISEQ-500 instrument with paired-end 100 bp protocol [21]. Reads were mapped to the human reference genome (NCBI build37/hg 19 version). Burrows-Wheeler Aligner (BWA) software was used for alignment and the Genome Analysis Toolkit (GATK) was used for variant calling [22,23]. Copy number variants (CNVs) were called using the CNVnator software [24]. Structural variations (SV) were screened with Breakdancer [25].
Analysis of WGS data was limited to NSHL genes listed on the Hereditary Hearing Loss Homepage [4]. Only exons and up to 20 bp flanking intronic regions were analyzed in these genes. Minor allele frequency thresholds of 0.005 for recessive and 0.0005 for dominant variants were considered by using the gnomAD (http://gnomad.broadinstitute.org/) and dbSNP (https://www.ncbi.nlm.nih.gov/projects /SNP/) databases, as well as the HIHG internal WES/WGS database that includes >4000 exomes/genomes from different ethnicities. We also filtered variants by using the combination criteria of damaging for SIFT (http://sift.jcvi.org/), disease-causing for MutationTaster (http://www.mutationtaster.org/), CADD score >20 (https://cadd.gs.washington.edu/) and GERP score >2 (http://mendel.stanford.edu/SidowLab/downloads/gerp/). Sanger sequencing was used to confirm detected variants.
2.4. Genome-Wide Genotyping and Quality Control
Genome-wide genotyping was performed by using the Global Screening Array (GSA) v1 (Illumina, San Diego, CA, USA). We assessed the quality control using the PLINK software, v.2 [26]. Firstly, we excluded samples with a low call rate (>90%), and markers with a genotype missingness of 3%, and Hardy–Weinberg equilibrium departure (p < 10−6) and minor allele frequency less than 0.05. Then, we applied the linkage disequilibrium pruning method at pairwise r2 < 0.4 to select the tagging SNPs from the array dataset.
2.5. Admixture Analysis
The Peruvian population is an admixture of four parental ancestries: Native Americans (AI), European (EU), West African (AF), and East Asian (EA). To estimate the proportion of the parental ancestries within Peruvians, we used a model-based clustering algorithm implemented in the ADMIXTURE software [27]. We performed supervised ADMIXTURE analysis at K = 4 by including the reference panels (AI, EU, AF, and EA populations) from the Human Genome Diversity Project [28].
2.6. Local Ancestry Analysis
We used the same reference panel (selected for global ancestry analysis) to calculate the local ancestral components of chromosome 13 in 20 unrelated probands (1 homozygous and 19 heterozygous) positive for the c.427C>T variant. We assessed local ancestry using a reference panel from three continental populations (AI, EU, and AF). We first phased the reference panel with the Peruvian dataset by using the Segmented Haplotype Estimation and Imputation tool ver. 2 (ShapeIT) [29]. Then, we used RFMix [30], a discriminative modeling approach, to infer the local ancestry across chromosome 13. To improve the accuracy of haplotypes, we ran the RFMix with the PopPhased option in a minimum node size 5. Then, we defined a 2 Mb region (chr13:19,000,000–21,000,000) that stretched the local ancestral block around the c.427C>T variant [31]. The region included 25 variants common in both the reference panel and the Peruvian genotyped dataset (Table S1).
3. Results
Hearing loss in all cases had a prelingual or congenital onset (average age of onset was 18.2 months), with severity ranging from moderate to profound. Around 52% of probands described here were males and 48% were females (Table S2). Lima was the city of birth in 81.2% of cases.
3.1. GJB2 Sequencing
Among the 133 analyzed cases, 43 (32%) had biallelic pathogenic variants in GJB2 and seven were heterozygous for only one allele (Tables S3 and S4). The overall allele frequency of pathogenic variants in GJB2 was 35% (93/266). We found eight pathogenic and six likely pathogenic GJB2 variants. The c.427C>T variant, causing a change in codon 143 from an arginine to a tryptophan p.(Arg143Trp), was identified in 20 probands (19 heterozygous and 1 homozygous), representing 22.6% (21 out of 93) of alleles. The c.35delG variant was the second most common variant, identified in 17.2% of mutated alleles. Remarkably, we identified six heterozygous probands for the rarely reported pathogenic variant c.645delT (Table 1). In addition, we identified three benign variants and four variants of uncertain significance (VUS) (Table S3). Benign variants were found with a pathogenic variant or alone. Only the VUS c.154G>T p.(Val52Phe) was found together with a likely pathogenic variant, c.94C>A (p.Arg32Ser).

Table 1.
Pathogenic variants identified in GJB2.
3.2. Ancestry Analysis
We examined the population structure of Peruvians using the supervised ADMIXTURE analysis at K = 4. Figure 1a illustrates the results from the ADMIXTURE analysis in a bar-plot figure. Each vertical bar represents an individual, which shows an estimate of the fraction of continental ancestries (AI, AF, EU, and EA). On average, the fraction of AI ancestry in Peruvians was the highest with the mean 76.9% (SD = 16.9). The EU, AF, and EA ancestry proportions were split, with the mean values 18.7% (SD = 13.8), 2.9% (SD = 4), and 1.5% (SD = 7.8), respectively (Figure 1b). Results of the admixture analysis agree with the recent genetic studies showing a four-way admixture (AI, EU, AF, and EA) structure in Peruvians.
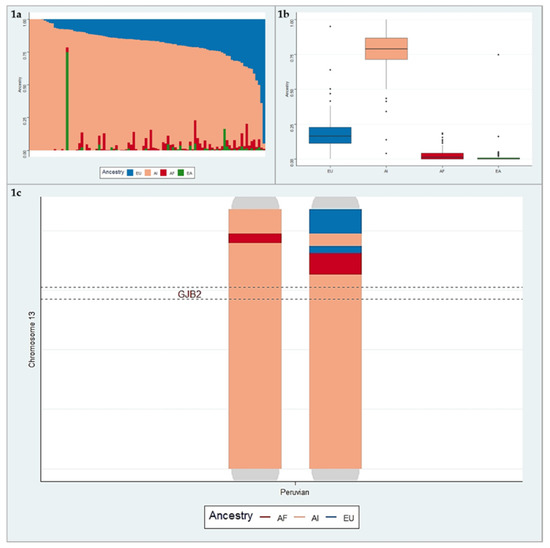
Figure 1.
a. Bar-plot of four-way admixed Peruvian individuals estimated using ADMIXTURE software at K = 4. b. The box-plot of the average ancestries in Peruvian individuals. c. Illustration of local ancestry on chromosome 13. This figure represents admixture blocks “local ancestry”, with each ancestry coded by a different color (red: African (AF), blue: European (EU), pink: American Indian (AI)), from a Peruvian individual.
3.3. Haplotype Analysis for Variant c.427C>T
We found that Peruvian cases have two haplotypes for the c.427C>T variant, a European and an Amerindian haplotype. The Amerindian haplotype is present in 79% of cases carrying the c.427C>T (Figure 2). The African haplotype is absent in the studied sample.
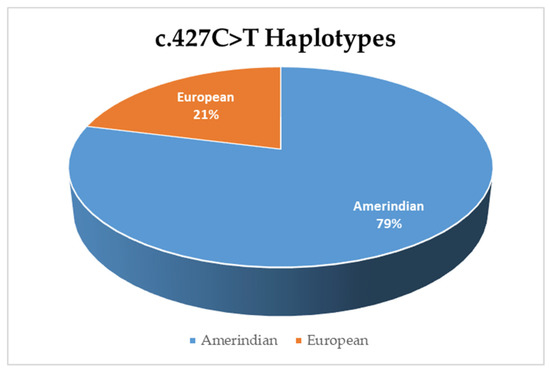
Figure 2.
Frequency of the haplotypes in cases positive for the c.427C>T variant.
3.4. Whole Genome Sequencing
We did not detect pathogenic or likely pathogenic variants (single nucleotide, indel, or copy number variants) in known NSHL genes in five probands. We found three causative variants within the MYO15A gene (NM_016239.3) in the remaining two probands. One proband was homozygous for the c.3757-2A>G variant, located in a canonical splice site two base pairs upstream of exon 5. The second proband was heterozygous for two variants: c.843C>A, which is a nonsense variant in exon 2, and c.8798delT, which produces a frameshift in the open reading frame of exon 50 (Table S5). These genetic variants have not been reported in the public databases gnomAD or 1000 genomes project.
4. Discussion
This is the first study where a Peruvian population with NSHL was screened for genetic variants in the GJB2 gene. We found 14 causative variants in 50 cases among 133 probands in a Peruvian sample affected with NSHL. The most frequent variant was c.427C>T; haplotype analysis suggests both Amerindian and European origin for this variant. Among the seven probands that completed WGS, we found three novel causative variants in MYO15A.
Our Peruvian cohort presents a complex profile for GJB2 variants, with two pathogenic variants being the most frequent ones. The c.427C>T variant was the most common of the pathogenic variants (22.6%). It has been reported as the main genetic cause of HL in Ghana [32]. In Japanese and Korean populations, the c.427C>T variant was reported as the second most common pathogenic variant with a frequency of 21.4% and 26.8% among 70 and 41 mutated alleles, respectively [12,33].
In contrast, studies in Latin-American populations reported that c.427C>T is an infrequent cause of HL. In an Argentinian study, the c.427C>T variant was reported as the second most common pathogenic variant after c.35delG, with only 4 of 68 mutated alleles [14]. In Ecuador, this variant was present in three alleles from a total of 100 mutated alleles [34]. In Mexican and Brazilian populations, this variant is almost absent, reported only once in 78 Mexican subjects [13,35,36] and in 1 in 74 Brazilian cases [37]. On the other hand, this variant was not reported in Colombian and Venezuelan populations where GJB2 was sequenced [38,39]. In this regard, the high frequency of c.427C>T found in Peruvian subjects contrasts with other Latin-American populations, where c.35delG is reported as the most common variant and c.427C>T has a low frequency. Although none of these studies included admixture analysis, the difference in allelic frequencies might be explained by a different ancestral composition of populations in each study. We cannot discard biases due to sample size and lack of representability of the population.
The cohort analyzed in this study was mainly born in Lima and showed an Amerindian composition of 76.9%, which is higher than the average for a Peruvian population from Lima. The high percent of Amerindians might explain the high frequency of an Amerindian haplotype in Peruvian cases carrying the c.427C>T variant. These findings are remarkable as the c.427C>T variant is the most frequent causative GJB2 variant in the African population of Ghana and relatively common in East Asians. Our haplotype analysis suggests that the most common deafness variant in Peru has multiple origins, perhaps one in America and another that has migrated from Europe. Alternatively, this variant may have a single ancient origin and was brought to the Americas twice: first during the migration from the Behring strait thousands of years ago and subsequently by Europeans five centuries ago.
Interestingly, six probands (five multiplex and one simplex cases) were heterozygous for the rare variant c.645delT. Clinical data available for three of these cases showed profound HL. This variant was previously reported in two studies from the U.S., and was identified in only one case among 119 and 384 probands with severe to profound HL, respectively. The authors did not specify the ethnic origin of these subjects [40,41]. While more studies are needed to assess the origin of c.645delT, its presence in six probands suggests a founder effect in this population.
We identified three novel variants in MYO15A in two of seven GJB2 negative cases (Table S5). There are more than 150 reported pathogenic and likely pathogenic variants in MYO15A, which is considered to be a relatively common gene for recessive NSHL [42,43]. The c.3757-2A>G variant is predicted to most likely affect splicing (www.umd.be/HSF/) within the splice acceptor region located 2 bp upstream of exon 5. The c.843C>A variant is located in exon 2 and causes a premature stop codon. The c.8798delT variant causes a frameshift in exon 50, leading to a premature stop codon. In silico analysis shows that both variants likely trigger nonsense mediated decay with a loss of 3250 and 498 amino acids, respectively (http://www.mutationtaster.org/). Our findings suggest that MYO15A variants are relatively common, following those in GJB2, in the Peruvian population, which should be considered in further mutation screenings. In addition, the absence of causative variants in known deafness genes in five multiplex cases suggests the existence of novel genes to be discovered in the Peruvian population.
This is the first report of the genetics of HL in a Peruvian cohort; the results show new and remarkable findings in comparison to other Latin-American populations. However, some limitations should be noted. First, the samples included in this study are not truly representative of Peru as all samples were collected in Lima, and ~80% of subjects were born in Lima. In addition, we did not have access to parental samples to confirm compound heterozygosity.
5. Conclusions
The Peruvian population shows a complex profile for genetic variants in the GJB2 gene, which is likely caused by the admixed composition of the Peruvian cohort. The dual origin, Amerindian and European, of the c.427C>T in the Peruvian sample suggests that HL mutations in Peru have heterogeneous origins throughout history.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/10/8/581/s1, Table S1: List of SNPs used for haplotype construction, Table S2: Summary of clinical features of cases, Table S3: Characteristics of identified GJB2 variants, Table S4: List of GJB2 genotypes, Table S5: Causative genetic variants identified via WGS.
Author Contributions
Conceptualization, E.F.-I., M.C.-O., R.D.C.V., and M.T.; data curation, E.F.-I., G.B., F.R., R.D.C.V., M.I.-M. and K.M.N.; Methodology, E.F.-I., G.B., M.C.-O., M.I.-M. and K.M.N.; Software, F.R. and G.B.; formal analysis, E.F.-I., G.B., F.R, M.C.-O., and M.T.; investigation, E.F.-I., G.B., R.D.C.V., R.B.-C. and C.S.; resources, E.F.-I., G.B., R.D.C.V., R.B.-C. and M.T.; writing—original draft preparation, E.F.-I., G.B., F.R., M.C.-O. and M.T.; writing—review and editing, E.F.-I., G.B., F.R., M.C.-O., R.D.C.V., R.B.-C., M.I.-M., K.M.N., C.S., M.T.; visualization, E.F.-I. and F.R.; supervision, E.F.-I., M.C.-O. and M.T
Funding
This study was supported by R01DC09645 and R01DC012836 from the National Institutes of Health/National Institute on Deafness and Other Communication Disorders to MT.
Acknowledgments
We are grateful to the families participating in this study. Special thanks to Diego Véliz-Otani for his revision and suggestions to this work and the members of the laboratory of neurogenetics Maria Victoria-Marca and Olimpio Ortega-Dávila for helping in the recruitment and sample processing. The authors thanks to Jorge La Serna for his help writing the project and drawing pedigrees.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Deafness and hearing loss. Available online: http://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 30 October 2018).
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Egilmez, O.K.; Kalcioglu, M.T. Genetics of Nonsyndromic Congenital Hearing Loss. Available online: https://www.hindawi.com/journals/scientifica/2016/7576064/ (accessed on 30 October2018).
- Welcome to the Hereditary Hearing Loss Homepage | Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 7 April 2019).
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J.; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 2016, 135, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Jones, M.-K.N. Nonsyndromic Hearing Loss and Deafness, DFNB1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, R.L.; Huygen, P.L.M.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S.; Van Laer, L.; Van Camp, G.; Usami, S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 2003, 112, 329–333. [Google Scholar] [PubMed]
- Taniguchi, M.; Matsuo, H.; Shimizu, S.; Nakayama, A.; Suzuki, K.; Hamajima, N.; Shinomiya, N.; Nishio, S.; Kosugi, S.; Usami, S.-I.; et al. Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J. Hum. Genet. 2015, 60, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.-X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han Chinese Subjects with Non-Syndromic Hearing Loss. PloS One 2015, 10, e0128691. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, A.R.; Han, K.H.; Kim, M.Y.; Jeon, E.-H.; Koo, J.-W.; Oh, S.H.; Choi, B.Y. Residual Hearing in DFNB1 Deafness and Its Clinical Implication in a Korean Population. PloS One 2015, 10, e0125416. [Google Scholar] [CrossRef]
- de la Luz Arenas-Sordo, M.; Menendez, I.; Hernández-Zamora, E.; Sirmaci, A.; Gutiérrez-Tinajero, D.; McGetrick, M.; Murphy-Ruiz, P.; Leyva-Juárez, X.; Huesca-Hernández, F.; Dominguez-Aburto, J.; et al. Unique spectrum of GJB2 mutations in Mexico. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1678–1680. [Google Scholar] [CrossRef]
- Gravina, L.P.; Foncuberta, M.E.; Prieto, M.E.; Garrido, J.; Barreiro, C.; Chertkoff, L. Prevalence of DFNB1 mutations in Argentinean children with non-syndromic deafness. Report of a novel mutation in GJB2. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 250–254. [Google Scholar] [CrossRef]
- Instituto Nacional de Estadística e Informática. Primera Encuesta Nacional Especializada sobre DISCAPACIDAD 2012; Instituto Nacional de Estadística e Informática: Lima, Peru, 2014. [Google Scholar]
- Harris, D.N.; Song, W.; Shetty, A.C.; Levano, K.S.; Cáceres, O.; Padilla, C.; Borda, V.; Tarazona, D.; Trujillo, O.; Sanchez, C.; et al. Evolutionary genomic dynamics of Peruvians before, during, and after the Inca Empire. Proc. Natl. Acad. Sci. USA 2018, 115, E6526–E6535. [Google Scholar] [CrossRef]
- Sandoval, J.R.; Salazar-Granara, A.; Acosta, O.; Castillo-Herrera, W.; Fujita, R.; Pena, S.D.J.; Santos, F.R. Tracing the genomic ancestry of Peruvians reveals a major legacy of pre-Columbian ancestors. J. Hum. Genet. 2013, 58, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Chacón Villanueva, R.; Badillo, R.; Cornejo-Olivas, M.; Ortega, O.; Marca, V.; Descailleaux, R.; Rubio, J.; Mazzetti-Soler, P. Mutations of Connexin Genes 26(GJB2) and 30(GJB6) in a Peruvian Deaf Population. In Proceedings of the 12th International Congress of Human Genetics and the American Society of Human Genetics 61st Annual Meeting, Montreal, QC, Canada, 11–15 October 2011. [Google Scholar]
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 mutations in Mongolia: complex alleles, low frequency, and reduced fitness of the deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Reich, D.; Patterson, N.; Campbell, D.; Tandon, A.; Mazieres, S.; Ray, N.; Parra, M.V.; Rojas, W.; Duque, C.; Mesa, N.; et al. Reconstructing Native American population history. Nature 2012, 488, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Marchini, J. 1000 Genomes Project Consortium; 1000 Genomes Project Consortium Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun. 2014, 5, 3934. [Google Scholar] [CrossRef] [PubMed]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Rajabli, F.; Feliciano, B.E.; Celis, K.; Hamilton-Nelson, K.L.; Whitehead, P.L.; Adams, L.D.; Bussies, P.L.; Manrique, C.P.; Rodriguez, A.; Rodriguez, V.; et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 2018, 14, e1007791. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Funayama, M.; Li, Y.; Kamiya, K.; Kawano, A.; Suzuki, M.; Hattori, N.; Ikeda, K. Prevalence of GJB2 causing recessive profound non-syndromic deafness in Japanese children. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Paz-y-Miño, C.; Beaty, D.; López-Cortés, A.; Proaño, I. Frequency of GJB2 and del(GJB6-D13S1830) mutations among an Ecuadorian mestizo population. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1648–1654. [Google Scholar] [CrossRef]
- Hernández-Juárez, A.A.; Lugo-Trampe, J.J.; Campos-Acevedo, L.D.; Lugo-Trampe, A.; Treviño-González, J.L.; de-la-Cruz-Ávila, I.; Martínez-de-Villarreal, L.E. GJB2 and GJB6 mutations are an infrequent cause of autosomal-recessive nonsyndromic hearing loss in residents of Mexico. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 2107–2112. [Google Scholar]
- Loeza-Becerra, F.; Rivera-Vega, M.R.; Martínez-Saucedo, M.; Gonzalez-Huerta, L.M.; Urueta-Cuellar, H.; Berrruecos-Villalobos, P.; Cuevas-Covarrubias, S. Particular distribution of the GJB2/GJB6 gene mutations in Mexican population with hearing impairment. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1057–1060. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Alexandrino, F.; Christiani, T.V.; Steiner, C.E.; Cunha, J.L.R.; Guerra, A.T.M.; Sartorato, E.L. Molecular genetics study of deafness in Brazil: 8-year experience. Am. J. Med. Genet. A. 2007, 143A, 1574–1579. [Google Scholar] [CrossRef]
- Utrera, R.; Ridaura, V.; Rodríguez, Y.; Rojas, M.J.; Mago, L.; Angeli, S.; Henríquez, O. Detection of the 35delG/GJB2 and del(GJB6-D13S1830) mutations in Venezuelan patients with autosomal recessive nonsyndromic hearing loss. Genet. Test. 2007, 11, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, M.L.; Olarte, M.; Gelvez, N.; Gómez, M.; Frías, J.L.; Bernal, J.E.; Florez, S.; Medina, D. Molecular studies in the GJB2 gene (Cx26) among a deaf population from Bogotá, Colombia: results of a screening program. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Angeli, S.I. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope 2008, 118, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Ouyang, X.; Du, L.; Yan, D.; Angeli, S.I.; Liu, X.Z. Etiologic Diagnosis of Nonsyndromic Genetic Hearing Loss in Adult vs Pediatric Populations. Otolaryngol.--Head Neck Surg. Off. J. Am. Acad. Otolaryngol.-Head Neck Surg. 2012, 147, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Bird, J.E.; Faridi, R.; Shahzad, M.; Shah, S.; Lee, K.; Khan, S.N.; Imtiaz, A.; Ahmed, Z.M.; Riazuddin, S.; et al. Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum. Mutat. 2016, 37, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, J.; Wang, H.; Yin, L.; Wang, D.; Zhao, L.; Zhou, H.; Wang, Q. Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med. Genet. 2019, 20, 60. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).