The Role of Mast Cells in Stroke


Abstract
1. Introduction
2. MCs Activation
3. Transcriptional and Epigenetic Regulation of MCs Response
4. MCs and Stroke
4.1. Ischemia in the Immature Brain
4.2. Ischemia in the Mature Brain
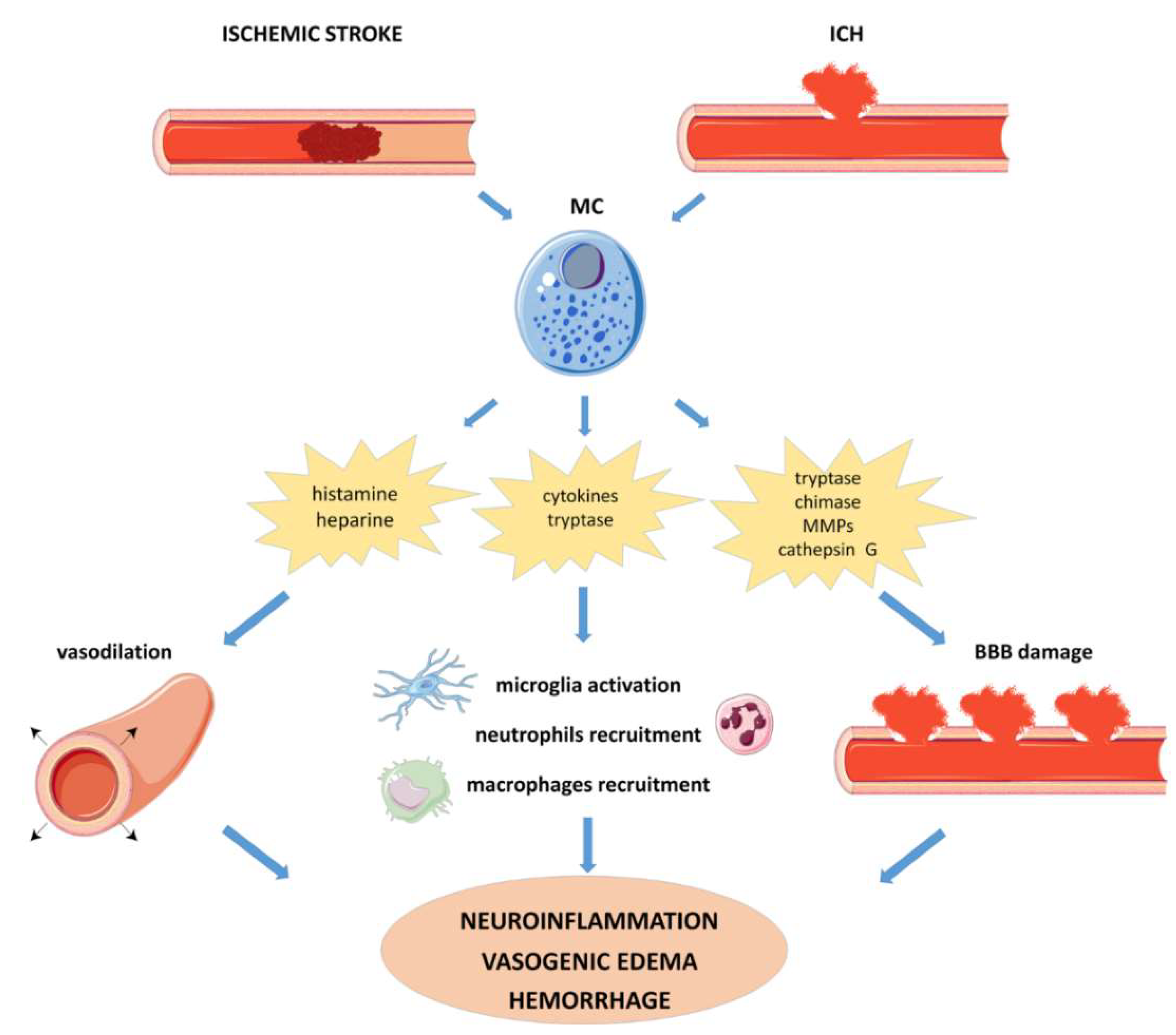
4.2.1. Ischemic Stroke
4.2.2. Intracerebral Hemorrhage (ICH)
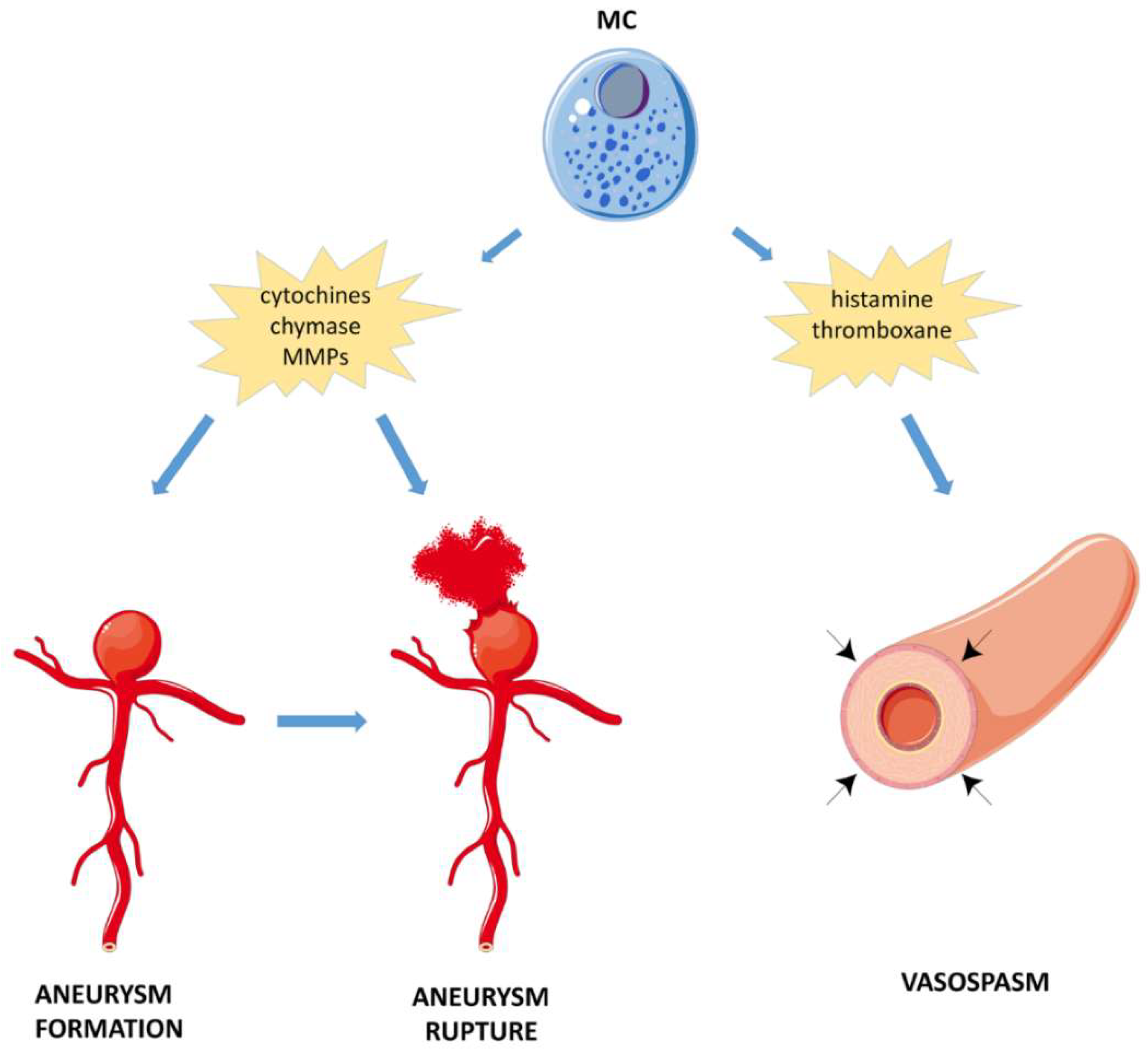
4.2.3. Subarachnoid Hemorrhage (SAH)
5. MCs Modulation: A Promising Strategy in Stroke Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Prussin, C.; Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2003, 111, S486–S494. [Google Scholar] [CrossRef]
- Metz, M.; Grimbaldeston, M.A.; Nakae, S.; Piliponsky, A.M.; Tsai, M.; Galli, S.J. Mast cells in the promotion and limitation of chronic inflammation. Immunol. Rev. 2007, 217, 304–328. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Urb, M.; Sheppard, D.C. The Role of Mast Cells in the Defence against Pathogens. PLoS Pathog. 2012, 8, e1002619. [Google Scholar] [CrossRef]
- Nelissen, S.; Lemmens, E.; Geurts, N.; Kramer, P.; Maurer, M.; Hendriks, J.; Hendrix, S. The role of mast cells in neuroinflammation. Acta Neuropathol. 2013, 125, 637–650. [Google Scholar] [CrossRef]
- Ehrlich, P. Über die spezifischen Granulationen des Blutes. Arch. Anat. Physiol. Abteil 1879, 571–579. [Google Scholar]
- Silver, R.; Curley, J.P. Mast cells on the mind: New insights and opportunities. Trends Neurosci. 2013, 36, 513–521. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, X.; Qian, Y. Mast cells and neuroinflammation. Med. Sci. Monit. 2014, 20, 200–206. [Google Scholar]
- Polyzoidis, S.; Koletsa, T.; Panagiotidou, S.; Ashkan, K.; Theoharides, T.C. Mast cells in meningiomas and brain inflammation. J. Neuroinflamm. 2015, 12, 170. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef]
- Silverman, A.J.; Sutherland, A.K.; Wilhelm, M.; Silver, R. Mast cells migrate from blood to brain. J. Neurosci. 2000, 20, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, K.M.; Liu, C.; Dong, X.; Silver, R. Blood-borne donor mast cell precursors migrate to mast cell-rich brain regions in the adult mouse. J. Neuroimmunol. 2011, 240, 142–146. [Google Scholar] [CrossRef]
- Da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.J.; Morrow, J.D. Thalamic mast cell activity is associated with sign-tracking behavior in rats. Brain Behav. Immun. 2017, 65, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, K.M.; Ribeiro, A.C.; Pfaff, D.W.; Silver, R. Brain mast cells link the immune system to anxiety-like behavior. Proc. Natl. Acad. Sci. USA 2008, 105, 18053–18057. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Pickett, L.A.; Wright, C.L.; Davis, K.T.; Joshi, A.; McCarthy, M.M. Mast Cells in the Developing Brain Determine Adult Sexual Behavior. J. Neurosci. 2018, 38, 8044–8059. [Google Scholar] [CrossRef]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Hendriksen, E.; van Bergeijk, D.; Oosting, R.S.; Redegeld, F.A. Mast cells in neuroinflammation and brain disorders. Neurosci. Biobehav Rev. 2017, 79, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; AhMed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef]
- Traina, G. Mast cells in the brain - Old cells, new target. J. Integr. Neurosci. 2017, 16, S69–S83. [Google Scholar] [CrossRef]
- Kempuraj, D.; Mentor, S.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Dubova, I.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Mast Cells in Stress, Pain, Blood-Brain Barrier, Neuroinflammation and Alzheimer’s Disease. Front. Cell Neurosci. 2019, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M. and Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Ocak, U.; Ocak, P.E.; Wang, A.; Zhang, J.H.; Boling, W.; Wu, P.; Mo, J.; Zhang, T.; Huang, L. Targeting mast cell as a neuroprotective strategy. Brain Inj. 2018, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P. Links between allergy and cardiovascular or hemostatic system. Int. J. Cardiol. 2014, 170, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Kounis, N.G. Kounis syndrome: An update on epidemiology, pathogenesis, diagnosis and therapeutic management. Clin. Chem. Lab. Med. 2016, 54, 1545–1559. [Google Scholar] [CrossRef]
- Abdelghany, M.; Subedi, R.; Shah, S.; Kozman, H. Kounis syndrome: A review article on epidemiology, diagnostic findings, management and complications of allergic acute coronary syndrome. Int. J. Cardiol. 2017, 232, 1–4. [Google Scholar] [CrossRef]
- AlKhater, S.A. CNS vasculitis and stroke as a complication of DOCK8 deficiency: A case report. BMC Neurol. 2016, 16, 54. [Google Scholar] [CrossRef]
- Yavuz, H.; Chee, R. A review on the vascular features of the hyperimmunoglobulin E syndrome. Clin. Exp. Immunol. 2010, 159, 238–244. [Google Scholar] [CrossRef]
- Yong, P.F.; Freeman, A.F.; Engelhardt, K.R.; Holland, S.; Puck, J.M.; Grimbacher, B. An update on the hyper-IgE syndromes. Arthritis Res. Ther. 2012, 14, 228. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Beaven, M.A. Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 2011, 31, 475–529. [Google Scholar] [CrossRef]
- Kalesnikoff, J.; Galli, S.J. New developments in mast cell biology. Nat. Immunol. Rev. 2008, 9, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Kalesnikoff, J.; Grimbaldeston, M.A.; Piliponsky, A.M.; Williams, C.M.; Tsai, M. Mast cells as “tunable” effector and immunoregulatory cells: Recent advances. Annu. Rev. Immunol. 2005, 23, 749–786. [Google Scholar] [CrossRef]
- Christy, A.L.; Brown, M.A. The multitasking mast cell: Positive and negative roles in the progression of autoimmunity. J. Immunol. 2007, 179, 2673–2679. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S. Mast-cell responses to pathogens. Nat. Rev. Immunol. 2004, 4, 787–799. [Google Scholar] [CrossRef]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, S.; Leoni, C. Epigenetic and transcriptional control of mast cell responses. F1000 Res. 2017, 6, 2064. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.L.; Walker, L.L. Dependence of mast cell IgE mediated cytokine production on nuclear factor-κB activity. J. Allergy Clin. Immunol. 2000, 105, 500–505. [Google Scholar] [CrossRef]
- Lee, Y.N.; Tuckerman, J.; Nechushtan, H.; Schutz, G.; Razin, E.; Angel, P. c-Fos as a regulator of degranulation and cytokine production in FcεRI-activated mast cells. J. Immunol. 2004, 173, 2571–2577. [Google Scholar] [CrossRef]
- Klein, M.; Klein-Hessling, S.; Palmetshofer, A.; Serfling, E.; Tertilt, C.; Bopp, T.; Heib, V.; Becker, M.; Taube, C.H.; Schild, H.; et al. Specific and redundant roles for NFAT transcription factors in the expression of mast cellderived cytokines. J. Immunol. 2006, 177, 6667–6674. [Google Scholar] [CrossRef]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Invest. 2016, 126, 3981–3998. [Google Scholar] [CrossRef]
- Li, B.; Power, M.R.; Lin, T.J. De novo synthesis of early growth response factor-1 is required for the full responsiveness of mast cells to produce TNF and IL-13 by IgE and antigen stimulation. Blood 2006, 107, 2814–2820. [Google Scholar] [CrossRef]
- Li, B.; Berman, J.; Tang, J.T.; Lin, T.J. The early growth response factor-1 is involved in stem cell factor (SCF)-induced interleukin 13 production by mast cells, but is dispensable for SCF-dependent mast cell growth. J. Biol. Chem. 2007, 282, 22573–22581. [Google Scholar] [CrossRef]
- Barbu, E.A.; Zhang, J.; Berenstein, E.H.; Groves, J.R.; Parks, L.M.; Siraganian, R.P. The transcription factor Zeb2 regulates signaling in mast cells. J. Immunol. 2012, 188, 6278–6286. [Google Scholar] [CrossRef] [PubMed]
- Lorentz, A.; Baumann, A.; Vitte, J.; Blank, U. The SNARE machinery in mast cell secretion. Front. Immunol. 2012, 3, 143. [Google Scholar] [CrossRef] [PubMed]
- Montagner, S.; Leoni, C.; Emming, S.; Della Chiara, G.; Balestrieri, C.; Barozzi, I.; Piccolo, V.; Togher, S.; Ko, M.; Rao, A.; et al. TET2 regulates mast cell differentiation and proliferation through catalytic and non-catalytic activities. Cell Rep. 2016, 15, 1566–1579. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS ONE 2012, 7, e43090. [Google Scholar] [CrossRef]
- Melo, F.R.; Vita, F.; Berent-Maoz, B.; Levi-Schaffer, F.; Zabucchi, G.; Pejler, G. Proteolytic histone modification by mast cell tryptase, a serglycin proteoglycan-dependent secretory granule protease. J. Biol. Chem. 2014, 289, 7682–7690. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.R.; Wallerman, O.; Paivandy, A.; Calounova, G.; Gustafson, A.M.; Sabari, B.R.; Zabucchi, G.; Allis, C.D.; Pejler, G. Tryptase-catalyzed core histone truncation: A novel epigenetic regulatory mechanism in mast cells. J. Allergy Clin. Immunol. 2017, 140, 474–485. [Google Scholar] [CrossRef]
- Nelson, K.B.; Lynch, J.K. Stroke in newborn infants. Lancet Neurol. 2004, 3, 150–158. [Google Scholar] [CrossRef]
- Lehman, L.L.; Rivkin, M.J. Perinatal arterial ischemic stroke: Presentation, risk factors, evaluation, and outcome. Pediatr. Neurol. 2014, 51, 760–768. [Google Scholar] [CrossRef]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Zerna, C.; Thomalla, G.; Campbell, B.C.V.; Rha, J.H.; Hill, M.D. Current practice and future directions in the diagnosis and acute treatment of ischaemic stroke. Lancet 2018, 392, 1247–1256. [Google Scholar] [CrossRef]
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634A. [Google Scholar] [CrossRef]
- Hakim, A.M. Depression, strokes and dementia: New biological insights into an unfortunate pathway. Cardiovasc Psychiatry Neurol. 2011, 2011, 649629. [Google Scholar] [CrossRef][Green Version]
- Kalaria, R.N.; Akinyemi, R.; Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. ACTA 2016, 1862, 915–925. [Google Scholar] [CrossRef]
- Petrea, R.E.; Beiser, A.S.; Seshadri, S.; Kelly-Hayes, M.; Kase, C.S.; Wolf, P.A. Gender differences in stroke incidence and poststroke disability in the Framingham Heart Study. Stroke 2009, 40, 1032–1037. [Google Scholar] [CrossRef]
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Anderson, C.S. Stroke epidemiology: A review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003, 2, 43–53. [Google Scholar] [CrossRef]
- Krishnamurthi, R.V.; Barker-Collo, S.; Parag, V.; Parmar, P.; Witt, E.; Jones, A.; Mahon, S.; Anderson, C.S.; Barber, P.A.; Feigin, V.L. Stroke Incidence by Major Pathological Type and Ischemic Subtypes in the Auckland Regional Community Stroke Studies: Changes Between 2002 and 2011. Stroke. 2018, 49, 3–10. [Google Scholar] [CrossRef]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Kirton, A.; Deveber, G. Life after perinatal stroke. Stroke 2013, 44, 3265–3271. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.A.; Soul, J.S. Pharmacologic Prevention and Treatment of Neonatal Brain Injury. Clin. Perinatol. 2019, 46, 311–325. [Google Scholar] [CrossRef]
- Rainaldi, M.A.; Perlman, J.M. Pathophysiology of Birth Asphyxia. Clin. Perinatol. 2016, 43, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef]
- Ziemka-Nalecz, M.; Jaworska, J.; Zalewska, T. Insights Into the Neuroinflammatory Responses After Neonatal Hypoxia-Ischemia. J. Neuropathol. Exp. Neurol. 2017, 76, 644–654. [Google Scholar] [CrossRef]
- Lai, J.C.Y.; Rocha-Ferreira, E.; Ek, C.J.; Wang, X.; Hagberg, H.; Mallard, C. Immune responses in perinatal brain injury. Brain Behav. Immun. 2017, 63, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Hedtjarn, M.; Mallard, C.; Hagberg, H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2004, 24, 1333–1351. [Google Scholar] [CrossRef]
- Jin, Y.; Silverman, A.J.; Vannucci, S.J. Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev. Neurosci. 2007, 29, 373–384. [Google Scholar] [CrossRef]
- Jin, Y.; Silverman, A.J.; Vannucci, S.J. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke 2009, 40, 3107–3112. [Google Scholar] [CrossRef]
- Biran, V.; Cochois, V.; Karroubi, A.; Arrang, J.M.; Charriaut-Marlangue, C.; Héron, A. Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol. 2008, 18, 1–9. [Google Scholar] [CrossRef]
- Patkai, J.; Mesples, B.; Dommergues, M.A.; Fromont, G.; Thornton, E.M.; Renauld, J.C.; Evrard, P.; Gressens, P. Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr. Res. 2001, 50, 222–230. [Google Scholar] [CrossRef]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Kunz, A.; Dirnagl, U.; Mergenthaler, P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Ribatti, D. The crucial role of mast cells in blood–brain barrier alterations. Exp. Cell Res. 2015, 3, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lindsberg, P.J.; Strbian, D.; Karjalainen-Lindsberg, M.L. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J. Cereb. Blood Flow Metab. 2010, 30, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Porrini, V.; Iorio, R.; Benarese, M.; Lanzillotta, A.; Mota, M.; Fusco, M.; Tonin, P.; Spano, P.; Pizzi, M. PEA and luteolin synergistically reduce mast cell-mediated toxicity and elicit neuroprotection in cell-based models of brain ischemia. Brain Res. 2016, 1648, 409–417. [Google Scholar] [CrossRef]
- Hu, W.; Shen, Y.; Fu, Q.; Dai, H.; Tu, H.; Wei, E.; Luo, J.; Chen, Z. Effect of oxygen-glucose deprivation on degranulation and histamine release of mast cells. Cell Tissue Res. 2005, 322, 437–441. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Fu, L.; Hu, W.; Chen, Z. Carnosine attenuates mast cell degranulation and histamine release induced by oxygen-glucose deprivation. Cell Biochem. Funct. 2008, 26, 334–338. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Yaish-Ohad, S.; Sarau, H.M.; Barone, F.C.; Feuerstein, G.Z. Molecular cloning and expression of the rat monocyte chemotactic protein-3 gene: A possible role in stroke. Brain Res. Mol. Brain Res. 1999, 71, 304–312. [Google Scholar] [CrossRef]
- Shao, X.; Bao, W.; Hong, X.; Jiang, H.; Yu, Z. Identification and functional analysis of differentially expressed genes associated with cerebral ischemia/reperfusion injury through bioinformatics methods. Mol. Med. Rep. 2018, 18, 1513–1523. [Google Scholar] [CrossRef]
- Hu, W.; Xu, L.; Pan, J.; Zheng, X.; Chen, Z. Effect of cerebral ischemia on brain mast cells in rats. Brain Res. 2004, 1019, 275–280. [Google Scholar] [CrossRef]
- Strbian, D.; Karjalainen-Lindsberg, M.L.; Tatlisumak, T.; Lindsberg, P.J. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J. Cereb. Blood Flow Metab. 2006, 26, 605–612. [Google Scholar] [CrossRef] [PubMed]
- McKittrick, C.M.; Lawrence, C.E.; Carswell, H.V. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 638–647. [Google Scholar] [CrossRef]
- Mattila, O.S.; Strbian, D.; Saksi, J.; Pikkarainen, T.O.; Rantanen, V.; Tatlisumak, T.; Lindsberg, P.J. Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke 2011, 42, 3600–3605. [Google Scholar] [CrossRef]
- Arac, A.; Grimbaldeston, M.A.; Galli, S.J.; Bliss, T.M.; Steinberg, G.K. Meningeal Mast Cells as Key Effectors of Stroke Pathology. Front. Cell Neurosci. 2019, 13, 126. [Google Scholar] [CrossRef]
- Arac, A.; Grimbaldeston, M.A.; Nepomuceno, A.R.; Olayiwola, O.; Pereira, M.P.; Nishiyama, Y.; Tsykin, A.; Goodall, G.J.; Schlecht, U.; Vogel, H.; et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am. J. Pathol. 2014, 184, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Arsene, D.; Vasilescu, F.; Toader, C.; Bălan, A.; Popa, C.; Ardeleanu, C. Clinico-pathological correlations in fatal ischemic stroke. An immunohistochemical study of human brain penumbra. Rom. J. Morphol. Embryol. 2011, 52, 29–38. [Google Scholar] [PubMed]
- Grossman, A.W.; Broderick, J.P. Advances and challenges in treatment and prevention of ischemic stroke. Ann. Neurol. 2013, 74, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ferrari, F.; Villa, R.F. Neuroprotection for ischaemic stroke: Current status and challenges. Pharmacol. Ther. 2015, 146, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Saver, J.L. Future directions of acute ischaemic stroke therapy. Lancet Neurol. 2015, 14, 758–767. [Google Scholar] [CrossRef]
- Strbian, D.; Karjalainen-Lindsberg, M.L.; Kovanen, P.T.; Tatlisumak, T.; Lindsberg, P.J. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation 2007, 116, 411–418. [Google Scholar] [CrossRef]
- Cordonnier, C.; Demchuk, A.; Ziai, W.; Anderson, C.S. Intracerebral haemorrhage: Current approaches to acute management. Lancet Neurol. 2018, 392, 1257–1268. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet Neurol. 2009, 373, 1632–1644. [Google Scholar] [CrossRef]
- Sacco, S.; Marini, C.; Toni, D.; Olivieri, L.; Carolei, A. Incidence and 10-year survival of intracerebral hemorrhage in a population based registry. Stroke 2009, 40, 394–399. [Google Scholar] [CrossRef]
- Sansing, L.H. Intracerebral Hemorrhage. Semin. Neurol. 2016, 36, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Wang, J.; Anne Stetler, R.; Yang, Q.W. Inflammation in intracerebral hemorrhage: From mechanisms to clinical translation. Prog. Neurobiol. 2014, 115, 25–44. [Google Scholar] [CrossRef]
- Chen, S.; Yang, Q.; Chen, G.; Zhang, J.H. An update on inflammation in the acute phase of intracerebral hemorrhage. Transl. Stroke Res. 2015, 6, 4–8. [Google Scholar] [CrossRef]
- Yehya, M.; Torbey, M.T. The Role of Mast Cells in Intracerebral Hemorrhage. Neurocrit. Care. 2018, 28, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Strbian, D.; Tatlisumak, T.; Ramadan, U.A.; Lindsberg, P.J. Mast cell blocking reduces brain edema and hematoma volume and improves outcome after experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Manaenko, A.; Lekic, T.; Ma, Q.; Zhang, J.H.; Tang, J. Hydrogen inhalation ameliorated mast cell-mediated brain injury after intracerebral hemorrhage in mice. Crit. Care Med. 2013, 41, 1266–1275. [Google Scholar] [CrossRef]
- Akyol, G.Y.; Manaenko, A.; Akyol, O.; Solaroglu, I.; Ho, W.M.; Ding, Y.; Flores, J.; Zhang, J.H.; Tang, J. IVIG activates FcgammaRIIB-SHIP1-PIP3 Pathway to stabilize mast cells and suppress inflammation after ICH in mice. Sci. Rep. 2017, 7, 15583. [Google Scholar] [CrossRef]
- Marinkovic, I.; Mattila, O.S.; Strbian, D.; Meretoja, A.; Shekhar, S.; Saksi, J.; Abo-Ramadan, U.; Rantanen, V.; Lindsberg, P.J.; Tatlisumak, T. Evolution of intracerebral hemorrhage after intravenous tPA: Reversal of harmful effects with mast cell stabilization. J. Cereb. Blood Flow Metab. 2014, 34, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Nakano, F. To improve translational research in subarachnoid hemorrhage. Transl. Stroke Res. 2018, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Schweizer, T.A. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Penn, D.L.; Witte, S.R.; Komotar, R.J.; Sander Connolly, E., Jr. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J. Clin. Neurosci. 2014, 21, 28–32. [Google Scholar] [CrossRef]
- Chalouhi, N.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Koch, W.J.; Dumont, A.S. Biology of intracranial aneurysms: Role of inflammation. J. Cereb. Blood Flow Metab. 2012, 32, 1659–1676. [Google Scholar] [CrossRef]
- Hosaka, K.; Hoh, B.L. Inflammation and cerebral aneurysms. Transl. Stroke Res. 2014, 5, 190–198. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Wolters, P.J.; Yang, M.; Kitamoto, S.; Libby, P.; MacFarlane, L.A.; Mallen-St Clair, J.; Shi, G.P. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat. Med. 2007, 13, 719–724. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Yang, M.; Wolters, P.J.; MacFarlane, L.A.; Libby, P.; Sun, C.; Zhang, Y.; Liu, J.; Ennis, T.L.; et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J. Clin. Invest. 2007, 117, 3359–3368. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, J.; Lindholt, J.S.; Sukhova, G.K.; Liu, J.; He, A.; Abrink, M.; Pejler, G.; Stevens, R.L.; Thompson, R.W.; et al. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation 2009, 120, 973–982. [Google Scholar] [CrossRef]
- Johnson, J.L.; Jackson, C.L.; Angelini, G.D.; George, S.J. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1707–1715. [Google Scholar] [CrossRef]
- Ishibashi, R.; Aoki, T.; Nishimura, M.; Hashimoto, N.; Miyamoto, S. Contribution of mast cells to cerebral aneurysm formation. Curr. Neurovasc. Res. 2010, 7, 113–124. [Google Scholar] [CrossRef]
- Hasan, D.; Chalouhi, N.; Jabbour, P.; Hashimoto, T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: Preliminary results. J. Neuroinflamm. 2012, 9, 222. [Google Scholar] [CrossRef]
- Hasan, D.M.; Mahaney, K.B.; Magnotta, V.A.; Kung, D.K.; Lawton, M.T.; Hashimoto, T.; Winn, H.R.; Saloner, D.; Martin, A.; Gahramanov, S.; et al. Macrophage imaging within human cerebral aneurysms wall using ferumoxytol-enhanced MRI: A pilot study. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1032–1038. [Google Scholar] [CrossRef]
- Ollikainen, E.; Tulamo, R.; Frösen, J.; Lehti, S.; Honkanen, P.; Hernesniemi, J.; Niemelä, M.; Kovanen, P.T. Mast cells, neovascularization, and microhemorrhages are associated with saccular intracranial artery aneurysm wall remodeling. J. Neuropathol. Exp. Neurol. 2014, 73, 855–864. [Google Scholar] [CrossRef]
- Pluta, R.M.; Hansen-Schwartz, J.; Dreier, J.; Vajkoczy, P.; Macdonald, R.L.; Nishizawa, S.; Kasuya, H.; Wellman, G.; Keller, E.; Zauner, A.; et al. Cerebral vasospasm following subarachnoid hemorrhage: Time for a new world of thought. Neurol. Res. 2009, 31, 151–158. [Google Scholar] [CrossRef]
- Faleiro, L.C.; Machado, C.R.; Gripp, A., Jr.; Resende, R.A.; Rodrigues, P.A. Cerebral vasospasm: Presence of mast cells in human cerebral arteries after aneurysm rupture. J. Neurosurg. 1981, 54, 733–735. [Google Scholar] [CrossRef]
- Shepherd, R.K.; Linden, J.; Duling, B.R. Adenosine-induced vasoconstriction in vivo. Role of the mast cell and A3 adenosine receptor. Circ. Res. 1996, 78, 627–634. [Google Scholar] [CrossRef]
- Hansen, H.S. Palmitoylethanolamide and other anandamide congeners. Proposed role in the diseased brain. Exp. Neurol. 2010, 224, 48–55. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, D.; Luongo, L.; Cipriano, M.; Palazzo, E.; Cinelli, M.P.; de Novellis, V.; Maione, S.; Iuvone, T. Palmitoylethanolamide reduces granuloma-induced hyperalgesia by modulation of mast cell activation in rats. Mol. Pain 2011, 7, 3. [Google Scholar] [CrossRef]
- De Filippis, D.; Negro, L.; Vaia, M.; Cinelli, M.P.; Iuvone, T. New insights in mast cell modulation by palmitoylethanolamide. CNS Neurol. Disord. Drug Targets 2013, 12, 78–83. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol. Disord. Drug Targets 2013, 12, 55–61. [Google Scholar] [CrossRef]
- Facci, L.; Dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.D.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. U S A 1995, 92, 3376–3380. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Glia and Mast Cells as Targets for Palmitoylethanolamide, an Anti-inflammatory and Neuroprotective Lipid Mediator. Mol. Neurobiol. 2013, 48, 340–352. [Google Scholar] [CrossRef]
- Skaper, S.D.; Buriani, A.; Dal Toso, R.; Petrelli, L.; Romanello, S.; Facci, L.; Leon, A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 3984–3989. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Romanello, S.; Leon, A. Mast cell activation causes delayed neurodegeneration in mixed hippocampal cultuRes. via the nitric oxide pathway. J. Neurochem. 1996, 66, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef]
- Portavella, M.; Rodriguez-Espinosa, N.; Galeano, P.; Blanco, E.; Romero, J.I.; Holubiec, M.I.; Rodriguez de Fonseca, F.; Fernández-Espejo, E. Oleoylethanolamide and Palmitoylethanolamide Protect Cultured Cortical Neurons Against Hypoxia. Cannabis. Cannabinoid. Res. 2018, 3, 171–178. [Google Scholar] [CrossRef]
- Holubiec, M.I.; Romero, J.I.; Suárez, J.; Portavella, M.; Fernández-Espejo, E.; Blanco, E.; Galeano, P.; de Fonseca, F.R. Palmitoylethanolamide prevents neuroinflammation, reduces astrogliosis and preserves recognition and spatial memory following induction of neonatal anoxia-ischemia. Psychopharmacology 2018, 235, 2929–2945. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Genovese, T.; Impellizzeri, D.; Crupi, R.; Velardi, E.; Marino, A.; Esposito, E.; Cuzzocrea, S. Reduction of ischemic brain injury by administration of palmitoylethanolamide after transient middle cerebral artery occlusion in rats. Brain Res. 2012, 1477, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Dajas, F.; Rivera-Megret, F.; Blasina, F.; Arredondo, F.; Abin-Carriquiry, J.A.; Costa, G.; Echeverry, C.; Lafon, L.; Heizen, H.; Ferreira, M.; et al. Neuroprotection by flavonoids. Braz. J. Med. Biol. Res. 2003, 36, 1613–1620. [Google Scholar] [CrossRef]
- Guerra-Araiza, C.; Alvarez-Mejia, A.L.; Sanchez-Torres, S.; Farfan-Garcia, E.; Mondragon-Lozano, R.; Pinto-Almazan, R.; Salgado-Ceballos, H. Effect of natural exogenous antioxidants on aging and on neurodegenerative diseases. Free Radic. Res. 47, 451–462. [CrossRef]
- Qiao, H.; Dong, L.; Zhang, X.; Zhu, C.; Wang, L.; Liu, Z.; Chen, L.; Xing, Y.; Wang, C.; Li, Y. Protective effect of luteolin in experimental ischemic stroke: Upregulated SOD1, CA.T.; Bcl-2 and claudin-5, down-regulated MDA and Bax expression. Neurochem. Res. 2012, 37, 2014–2024. [Google Scholar] [CrossRef]
- Qiao, H.; Zhang, X.; Zhu, C.; Dong, L.; Wang, L.; Xing, Y.; Wang, C.; Ji, Y.; Cao, X. Luteolin downregulates TLR4, TLR5, NF-kappaB and p-p38MAPK expression, upregulates the p-ERK expression, and protects rat brains against focal ischemia. Brain Res. 2012, 1448, 71–81. [Google Scholar] [CrossRef]
- Zhao, G.; Zang, S.Y.; Jiang, Z.H.; Chen, Y.Y.; Ji, X.H.; Lu, B.F.; Wu, J.H.; Qin, G.W.; Guo, L.H. Postischemic administration of liposome-encapsulated luteolin prevents against ischemia-reperfusion injury in a rat middle cerebral artery occlusion model. J. Nutr. Biochem. 2011, 22, 929–936. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, P.; Yu, H.; Shi, L. Luteolin protects PC-12 cells from H2O2-induced injury by up-regulation of microRNA-21. BioMed. Pharmacother. 2019, 112, 108698. [Google Scholar] [CrossRef]
- Tian, T.; Zeng, J.; Zhao, G.; Zhao, W.; Gao, S.; Liu, L. Neuroprotective effects of orientin on oxygen-glucose deprivation/reperfusion-induced cell injury in primary culture of rat cortical neurons. Exp. Biol. Med. 2018, 243, 78–86. [Google Scholar] [CrossRef]
- Jeon, I.H.; Kim, H.S.; Kang, H.J.; Lee, H.S.; Jeong, S.I.; Kim, S.J.; Jang, S.I. Anti-inflammatory and antipruritic effects of luteolin from Perilla (P. frutescens L.) leaves. Molecules 2014, 19, 6941–6951. [Google Scholar] [CrossRef]
- Jin, M.; Son, K.H.; Chang, H.W. Luteolin-7-O-glucoside suppresses leukotriene C(4) production and degranulation by inhibiting the phosphorylation of mitogen activated protein kinases and phospholipase Cgamma1 in activated mouse bone marrow-derived mast cells. Biol. Pharm. Bull. 2011, 34, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Kritas, S.K.; Saggini, A.; Varvara, G.; Murmura, G.; Caraffa, A.; Antinolfi, P.; Toniato, E.; Pantalone, A.; Neri, G.; Frydas, S.; et al. Luteolin inhibits mast cell-mediated allergic inflammation. J. Biol. Regul. Homeost Agents 2013, 27, 955–959. [Google Scholar] [PubMed]
- Caltagirone, C.; Cisari, C.; Schievano, C.; Di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S.; Stroke Study Group. Coultramicronized palmitoylethanolamide/luteolin in the treatment of cerebral ischemia: From rodent to man. Transl. Stroke Res. 2016, 7, 54–69. [Google Scholar] [CrossRef]
- Zhang, T.; Finn, D.F.; Barlow, J.W.; Walsh, J.J. Mast cell stabilisers. Eur. J. Pharmacol. 2016, 778, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, E.W. Intravenous immune globulin in autoimmune and inflammatory diseases. N. Engl. J. Med. 2012, 367, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Selvaraj, P.K.; Woodruff, T.M.; Mattson, M.P. Targeting ischemic brain injury with intravenous immunoglobulin. Expert Opin. Ther. Targets 2008, 12, 19–29. [Google Scholar] [CrossRef]
- Widiapradja, A.; Vegh, V.; Lok, K.Z.; Manzanero, S.; Thundyil, J.; Gelderblom, M.; Cheng, Y.L.; Pavlovski, D.; Tang, S.C.; Jo, D.G.; et al. Intravenous immunoglobulin protects neurons against amyloid beta-peptide toxicity and ischemic stroke by attenuating multiple cell death pathways. J. Neurochem. 2012, 122, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef]
- Chen, X.; Arumugam, T.V.; Cheng, Y.L.; Lee, J.H.; Chigurupati, S.; Mattson, M.P.; Basta, M. Combination Therapy with Low-Dose IVIG and a C1-esterase Inhibitor Ameliorates Brain Damage and Functional Deficits in Experimental Ischemic Stroke. Neuromol. Med. 2018, 20, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.; Towers, T.L.; Ravetch, J.V. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 2001, 291, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S. Molecular hydrogen as a preventive and therapeutic medical gas: Initiation, development and potential of hydrogen medicine. Pharmacol. Ther. 2014, 144, 1–11. [Google Scholar] [CrossRef]
- Chen, C.H.; Manaenko, A.; Zhan, Y.; Liu, W.W.; Ostrowki, R.P.; Tang, J.; Zhang, J.H. Hydrogen gas reduced acute hyperglycemia-enhanced hemorrhagic transformation in a focal ischemia rat model. Neuroscience 2010, 169, 402–414. [Google Scholar] [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Nishijima, Y.; Adachi, N.; Sakamoto, M.; Kudo, Y.; Kaneko, K.; Nakao, A.; Imaoka, T. A basic study on molecular hydrogen (H2) inhalation in acute cerebral ischemia patients for safety check with physiological parameters and measurement of blood H2 level. Med. Gas Res. 2012, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Nishijima, Y.; Ohta, S.; Sakamoto, M.; Kinone, K.; Horikosi, T.; Tamaki, M.; Takeshita, H.; Futatuki, T.; Ohishi, W.; et al. Hydrogen Gas Inhalation Treatment in Acute Cerebral Infarction: A Randomized Controlled Clinical Study on Safety and Neuroprotection. J. Stroke Cerebrovasc. Dis. 2017, 26, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Nagatani, K.; Nawashiro, H.; Takeuchi, S.; Tomura, S.; Otani, N.; Osada, H.; Wada, K.; Katoh, H.; Tsuzuki, N.; Mori, K. Safety of intravenous administration of hydrogen-enriched fluid in patients with acute cerebral ischemia: Initial clinical studies. Med. Gas Res. 2013, 3, 13. [Google Scholar] [CrossRef]
- Itoh, T.; Fujita, Y.; Ito, M.; Masuda, A.; Ohno, K.; Ichihara, M.; Kojima, T.; Nozawa, Y.; Ito, M. Molecular hydrogen suppresses FcepsilonRI-mediated signal transduction and prevents degranulation of mast cells. Biochem. Biophys. Res. Commun. 2009, 389, 651–656. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef]
- Shen, Y.; He, P.; Fan, Y.Y.; Zhang, J.X.; Yan, H.J.; Hu, W.W.; Ohtsu, H.; Chen, Z. Carnosine protects against permanent cerebral ischemia in histidine decarboxylase knockout mice by reducing glutamate excitotoxicity. Free Radic. Biol. Med. 2010, 48, 727–735. [Google Scholar] [CrossRef]
- Rajanikant, G.K.; Zemke, D.; Senut, M.C.; Frenkel, M.B.; Chen, A.F.; Gupta, R.; Majid, A. Carnosine Is Neuroprotective Against Permanent Focal Cerebral Ischemia in Mice. Stroke 2007, 38, 3023–3031. [Google Scholar] [CrossRef]
- Min, J.; Senut, M.C.; Rajanikant, K.; Greenberg, E.; Bandagi, R.; Zemke, D.; Mousa, A.; Kassab, M.; Farooq, M.U.; Gupta, R.; et al. Differential Neuroprotective Effects of Carnosine, Anserine, and N-Acetyl Carnosine against Permanent Focal Ischemia. J. Neurosci. Res. 2008, 86, 2984–2991. [Google Scholar] [CrossRef]
- Ou-Yang, L.; Liu, Y.; Wang, B.Y.; Cao, P.; Zhang, J.J.; Huang, Y.Y.; Shen, Y.; Lyu, J.X. Carnosine suppresses oxygen-glucose deprivation/recovery-induced proliferation and migration of reactive astrocytes of rats in vitro. ACTA Pharmacol. Sin. 2018, 39, 24–34. [Google Scholar] [CrossRef]
- Noguchi, K.; Ali, T.F.; Miyoshi, J.; Orito, K.; Negoto, T.; Biswas, T.; Taira, N.; Koga, R.; Okamoto, Y.; Fujita, M.; et al. Neuroprotective effects of a novel carnosine-hydrazide derivative on hippocampal CA1 damage after transient cerebral ischemia. Eur. J. Med. Chem. 2019, 1, 207–214. [Google Scholar] [CrossRef]
- Davis, C.K.; Laud, P.J.; Bahor, Z.; Rajanikant, G.K.; Majid, A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 1686–1694. [Google Scholar] [CrossRef]
- Akhalaya, M.Y.; Baizhumanov, A.A.; Graevskaya, E.E. Effects of Taurine, Carnosine, and Casomorphine on Functional Activity of Rat Peritoneal Mast Cells. Bull. Exp. Biol. Med. 2006, 141, 302–305. [Google Scholar] [CrossRef]
- Nishigaki, T. Mast cell degranulation and its inhibition by an anti-allergic agent tranilast. An electron microscopic study. Virchows Arch. B 1988, 55, 311–322. [Google Scholar] [PubMed]
- Saito, T.; Hagihara, A.; Igarashi, N.; Matsuda, N.; Yamashita, A.; Ito, K.; Mio, M.; Tasaka, K. Inhibitory effects of emedastine difumarate on histamine release. JPN J. Pharmacol. 1993, 62, 137–143. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Cao, W. Mesenchymal stem cell-mediated immunomodulation in cell therapy of neurodegenerative diseases. Cell Immunol. 2018, 326, 8–14. [Google Scholar] [CrossRef]
- Kuwabara, A.; Liu, J.; Kamio, Y.; Liu, A.; Lawton, M.T.; Lee, J.W.; Hashimoto, T. Protective Effect of Mesenchymal Stem Cells Against the Development of Intracranial Aneurysm Rupture in Mice. Neurosurgery 2017, 81, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Marech, I.; Patruno, R.; Zizzo, N.; Gadaleta, C.; Introna, M.; Zito, A.F.; Gadaleta, C.D.; Ranieri, G. Masitinib (AB1010), from canine tumor model to human clinical development: Where we are? Crit. Rev. Oncol. Hematol. 2014, 91, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Kocic, I.; Kowianski, P.; Rusiecka, I.; Lietzau, G.; Mansfield, C.; Moussy, A.; Hermine, O.; Dubreuil, P. Neuroprotective effect of masitinib in rats with postischemic stroke. Naunyn. Schmiedebergs Arch. Pharmacol. 2015, 388, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Pae, H.O.; Jeong, S.J.; Yoo, J.C.; Choi, B.M.; Jun, C.D.; Chung, H.T.; Miyamoto, T.; Higuchi, R.; Kim, Y.C. Scopoletin: An inducible nitric oxide synthesis inhibitory active constituent from Artemisia feddei. Planta Med. 1999, 65, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Mogana, R.; Teng-Jin, K.; Wiart, C. Anti-inflammatory, anticholinesterase, and antioxidant potential of scopoletin isolated from canarium patentinervium miq. (burseaceae kunth). Evid. Based Complement. Alter. Nat. Med. 2013, 2013, 734824. [Google Scholar]
- Connell, B.J.; Saleh, M.C.; Rajagopal, D.; Saleh, T.M. UPEI-400, a conjugate of lipoic acid and scopoletin, mediates neuroprotection in a rat model of ischemia/reperfusion. Food Chem. Toxicol. 2017, 100, 175–182. [Google Scholar] [CrossRef]
- Moon, P.D.; Lee, B.H.; Jeong, H.J.; An, H.J.; Park, S.J.; Kim, H.R.; Ko, S.G.; Um, J.Y.; Hong, S.H.; Kim, H.M. Use of scopoletin to inhibit the production of inflammatory cytokines through inhibition of the IkappaB/NF-kappaB signal cascade in the human mast cell line HMC-1. Eur. J. Pharmacol. 2007, 555, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Suleria, H.A.R.; Ahmad, B.; Peters, D.G.; Mubarak, M.S. A comprehensive review of the health perspectives of resveratrol. Food Funct. 2017, 8, 4284–4305. [Google Scholar] [CrossRef]
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189. [Google Scholar] [CrossRef]
- Faggi, L.; Pignataro, G.; Parrella, E.; Porrini, V.; Vinciguerra, A.; Cepparulo, P.; Cuomo, O.; Lanzillotta, A.; Mota, M.; Benarese, M.; et al. Synergistic Association of Valproate and Resveratrol. Reduces Brain Injury in Ischemic Stroke. Int. J. Mol. Sci. 2018, 19, 172. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in PC12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11. [Google Scholar] [CrossRef]
- Yu, P.; Wang, L.; Tang, F.; Zeng, L.; Zhou, L.; Song, X.; Jia, W.; Chen, J.; Yang, Q. Resveratrol Pretreatment Decreases Ischemic Injury and Improves Neurological Function Via Sonic Hedgehog Signaling After Stroke in Rats. Mol. Neurobiol. 2017, 54, 212–226. [Google Scholar] [CrossRef]
- Wan, D.; Zhou, Y.; Wang, K.; Hou, Y.; Hou, R.; Ye, X. Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res. Bull. 2016, 121, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Dempsey, R.J.; Vemuganti, R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem. Int. 2015, 89, 75–82. [Google Scholar] [CrossRef]
- Ruan, W.; Li, J.; Xu, Y.; Wang, Y.; Zhao, F.; Yang, X.; Jiang, H.; Zhang, L.; Saavedra, J.M.; Shi, L.; et al. MALAT1 Up-Regulator Polydatin Protects Brain Microvascular Integrity and Ameliorates Stroke Through C/EBPβ/MALAT1/CREB/PGC-1α/PPARγ Pathway. Cell Mol. Neurobiol. 2019, 39, 265–286. [Google Scholar] [CrossRef]
- Tang, K.S.; Tan, J.S. The protective mechanisms of polydatin in cerebral ischemia. Eur. J. Pharmacol. 2019, 842, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Li, S.; Hu, Y.; Zhang, H.; Liu, Y.; Jiang, H.; Fang, M.; Li, Z.; Xu, K.; Zhang, H.; et al. Resveratrol. post-treatment protects against neonatal brain injury after hypoxia-ischemia. Oncotarget 2016, 7, 79247–79261. [Google Scholar] [CrossRef] [PubMed]
- Faggi, L.; Porrini, V.; Lanzillotta, A.; Benarese, M.; Mota, M.; Tsoukalas, D.; Parrella, E.; Pizzi, M. A Polyphenol-Enriched Supplement Exerts Potent Epigenetic-Protective Activity in a Cell-Based Model of Brain Ischemia. Nutrients 2019, 11, 345. [Google Scholar] [CrossRef]
- Chen, J.; Bai, Q.; Zhao, Z.; Sui, H.; Xie, X. Resveratrol improves delayed r-tPA treatment outcome by reducing MMPs. Acta Neurol. Scand. 2016, 134, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Koo, N.; Cho, D.; Kim, Y.; Choi, H.J.; Kim, K.M. Effects of resveratrol on mast cell degranulation and tyrosine phosphorylation of the signaling components of the IgE receptor. Planta Med. 2006, 72, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Li, J.; Lv, J.; Mo, X.; Yang, C.; Chen, X.; Liu, Z.; Liu, J. Polydatin (PD) inhibits IgE-mediated passive cutaneous anaphylaxis in mice by stabilizing mast cells through modulating Ca²⁺ mobilization. Toxicol. Appl Pharmacol. 2012, 264, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Bae, J.Y.; Park, S.H.; Kim, Y.H.; Park, J.H.; Kang, Y.H. Resveratrol inhibits IgE-mediated basophilic mast cell degranulation and passive cutaneous anaphylaxis in mice. J. Nutr. 2013, 145, 632–639. [Google Scholar] [CrossRef]
- Han, S.Y.; Choi, Y.J.; Kang, M.K.; Park, J.H.; Kang, Y.H. Resveratrol Suppresses Cytokine Production Linked to FcεRI-MAPK Activation in IgE-Antigen Complex-Exposed Basophilic Mast Cells and Mice. Am. J. Chin. Med. 2015, 43, 1605–1623. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.; McHale, C.; Gomez, G. Resveratrol preferentially inhibits IgE-dependent PGD2 biosynthesis but enhances TNF production from human skin mast cells. Biochim. Biophys. ACTA 2016, 1860, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, W.; Hu, D.; Han, X.; Wang, H.; Yang, J.; Xu, Y.; Li, Y.; Yao, W.; Chen, C. Resveratrol efficiently improves pulmonary function via stabilizing mast cells in a rat intestinal injury model. Life Sci. 2017, 185, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Huang, X.; Han, X.; Hu, D.; Hu, X.; Li, Y.; Huang, P.; Yao, W. Resveratrol Suppresses Gut-Derived NLRP3 Inflammasome Partly through Stabilizing Mast Cells in a Rat Model. Mediators Inflamm. 2018, 2018, 6158671. [Google Scholar] [CrossRef]
- Baba, A.; Tachi, M.; Ejima, Y.; Endo, Y.; Toyama, H.; Matsubara, M.; Saito, K.; Yamauchi, M.; Miura, C.; Kazama, I. Anti-allergic drugs tranilast and ketotifen dose-dependently exert mast cell-stabilizing properties. Cell Physiol. Biochem. 2016, 38, 15–27. [Google Scholar] [CrossRef]
- Grant, S.M.; Goa, K.L.; Fitton, A.; Sorkin, E.M. Ketotifen. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in asthma and allergic disorders. Drugs 1990, 40, 412–448. [Google Scholar] [CrossRef] [PubMed]
- Craps, L.P. Immunologic and therapeutic aspects of ketotifen. J. Allergy Clin. Immunol. 1985, 76, 389–393. [Google Scholar] [CrossRef]
- Schoch, C. In vitro inhibition of human conjunctival mast-cell degranulation by ketotifen. J. Ocul. Pharmacol. Ther. 2003, 19, 75–81. [Google Scholar] [CrossRef]
- Lambiase, A.; Micera, A.; Bonini, S. Multiple action agents and the eye: Do they really stabilize mast cells? Curr. Opin. Allergy Clin. Immunol. 2009, 9, 454–465. [Google Scholar] [CrossRef]
- Hei, Z.Q.; Gan, X.L.; Huang, P.J.; Wei, J.; Shen, N.; Gao, W.L. Influence of ketotifen, cromolyn sodium, and compound 48/80 on the survival rates after intestinal ischemia reperfusion injury in rats. BMC Gastroenterol. 2008, 8, 42. [Google Scholar]
- Reber, L.L.; Frossard, N. Targeting mast cells in inflammatory diseases. Pharmacol. Ther. 2014, 142, 416–435. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Stroke | Experimental Model | Findings | References |
|---|---|---|---|
| NHIBI | Carotid ligation mouse model | MCs associated genes upregulated | [70] |
| Carotid ligation rat model | Rapid increase of activated MCs in the brain | [71,72] | |
| MCs pharmacological inhibition reduced MCs migration, brain damage and glial activation | |||
| Transient focal ischemia rat model | Rapid increase of activated MCs and histamine in the brain | [73] | |
| Ibotenate mouse model | IL-9 exacerbated brain damage by activating MCs | [74] | |
| MCs pharmacological inhibition reduced brain damage | |||
| Ischemic Stroke | OGD mouse MCs | OGD promoted MCs activation | [83,84,85] |
| OGD mouse MCs and neurons | OGD-activated MCs induced neurotoxicity | [83] | |
| MCs pharmacological inhibition reduced MCs-induced neurotoxicity | |||
| MCAO mouse model | MCs associated gene upregulated | [87] | |
| MC-deficient mice showed decreased BBB leakage, brain edema and neutrophils infiltration | [90] | ||
| MCs pharmacological inhibition decreased BBB leakage, brain edema and neutrophils infiltration | |||
| Meningeal MCs worsen infiltration of granulocytes and macrophages, brain swelling, and infarct size | [93] | ||
| Four-vessel occlusion rat model | Modulation of MCs number and histamine levels | [88] | |
| MCAO rat model | MCs pharmacological activation increased edema formation | [89] | |
| MCs pharmacological inhibition decreased brain swelling, BBB leakage and neutrophils infiltration | |||
| MC-deficient rats showed decreased brain swelling, BBB leakage, and neutrophils infiltration | |||
| MCAO rat model | Increased MCs gelatinase activity | [91] | |
| MCs pharmacological activation increased gelatinase activity | |||
| MCs pharmacological inhibition decreased gelatinase activity | |||
| MC-deficient rats displayed decreased gelatinase activity | |||
| MCAO rat model treated with rtPA | MCs pharmacological inhibition reduced rtPA-induced hemorrhagic conversion, brain swelling, and neutrophil infiltration. | [98] | |
| MC-deficient rats displayed decreased rtPA-induced hemorrhagic conversion, brain swelling, and neutrophil infiltration. | |||
| Patients | Lack of MCs in penumbra brain region | [94] | |
| ICH | Blood infusion rat model | MCs pharmacological activation increased brain damage. | [106] |
| MCs pharmacological inhibition decreased brain damage, improved neurologic outcome | |||
| MC-deficient rats displayed decreased brain damage, improved neurologic outcome | |||
| Collagenase infusion mouse model | MCs activation | [107,108] | |
| MCs pharmacological inhibition decreased brain damage, improved neurologic outcome | |||
| Collagenase infusion rat model treated with rtPA | MCs pharmacological inhibition reduced rtPA-induced hematoma growth, hemispheric expansion, mortality, and neurologic deficits. | [109] | |
| SAH | CA rat model | MCs in aneurysm wall | [119] |
| MCs pharmacological inhibition reduced inflammation and CA size and thinning | |||
| Co-culture rat MCs and smooth muscle cells | Histamine and thromboxane inhibitors decreased MCs-mediated vasoconstriction | [119] | |
| Patients | MCs in aneurysm wall | [120,121,122] | |
| MCs in the muscular layer of cerebral arteries | [123] |
| Drugs | Experimental Model | Findings | References |
|---|---|---|---|
| PEA | MCAO rat model | PEA reduced MCs derived chymase and tryptase | [137] |
| Luteolin | OGD mouse MCs and neurons | Luteolin reduced OGD-activated MCs degranulation and induced neurotoxicity | [83] |
| PEA/Luteolin | OGD mouse MCs and neurons | PEA/Luteolin reduced OGD-activated MCs degranulation and MCs-induced neurotoxicity | [83] |
| MCAO rat model | PEA/Luteolin reduced ischemia-induced MCs infiltration and expression of chymase and tryptase | [148] | |
| Cromoglycate | Carotid ligation rat model | Cromoglycate reduced MCs migration, glial activation and brain atrophy | [71,72] |
| Ibotenate mouse model | Cromoglycate reduced MCs migration, glial activation and brain atrophy | [74] | |
| MCAO rat model | Cromoglycate reduced brain swelling, perivascular gelatinase activity, BBB leakage and neutrophil accumulation | [53,98] | |
| MCAO mouse model | Cromoglycate decreased BBB leakage, brain edema and neutrophils infiltration | [90] | |
| MCAO rat model treated with rtPA | Cromoglycate reduced rtPA-induced hemorrhagic conversion, brain swelling and neutrophil infiltration. | [98] | |
| Blood infusion rat model | Cromoglycate inhibited hematoma growth, decreased neurological deficits and mortality | [106] | |
| Collagenase infusion rat model treated with rtPA | Cromoglycate reduced rtPA-induced hematoma growth, hemispheric expansion, mortality and neurologic deficits. | [109] | |
| IVIG | Collagenase infusion mouse model | IVIG attenuated BBB damage, brain edema, infarct area and pro-inflammatory cytokines levels | [108] |
| H2 | Collagenase infusion mouse model | H2 decreased MCs accumulation and degranulation, BBB damage and improved neurobehavioral function | [107] |
| Carnosine | OGD rat MCs | Carnosine reduced degranulation and histamine release in OGD-activated MCs | [85] |
| Emedastine | CA rat model | Emedastine decreased MCs activation, inflammation and CA size and thinning. | [119] |
| Tranilast | CA rat model | Tranilast decreased MCs activation, inflammation and CA size and thinning. | [119] |
| MSCs | CA mouse model | Intravenous injection of MSCs reduced aneurysm rupture rate and CA MCs infiltration | [175] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrella, E.; Porrini, V.; Benarese, M.; Pizzi, M. The Role of Mast Cells in Stroke. Cells 2019, 8, 437. https://doi.org/10.3390/cells8050437
Parrella E, Porrini V, Benarese M, Pizzi M. The Role of Mast Cells in Stroke. Cells. 2019; 8(5):437. https://doi.org/10.3390/cells8050437
Chicago/Turabian StyleParrella, Edoardo, Vanessa Porrini, Marina Benarese, and Marina Pizzi. 2019. "The Role of Mast Cells in Stroke" Cells 8, no. 5: 437. https://doi.org/10.3390/cells8050437
APA StyleParrella, E., Porrini, V., Benarese, M., & Pizzi, M. (2019). The Role of Mast Cells in Stroke. Cells, 8(5), 437. https://doi.org/10.3390/cells8050437