
Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-α 334–352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Generation of TCR Transgenic (Tg) Mice
2.3. Genotyping
2.4. Peptide Synthesis
2.5. Immunization Procedures
2.6. IAb Binding Assay
2.7. Proliferation Assay
2.8. Determination of Antigen Specificity of Myhc-α 334–352-Reactive T Cells by IAb MHC Class II Dextramer Staining
2.9. Immunophenotyping by Flow Cytometry
2.10. Bulk TCR Sequencing
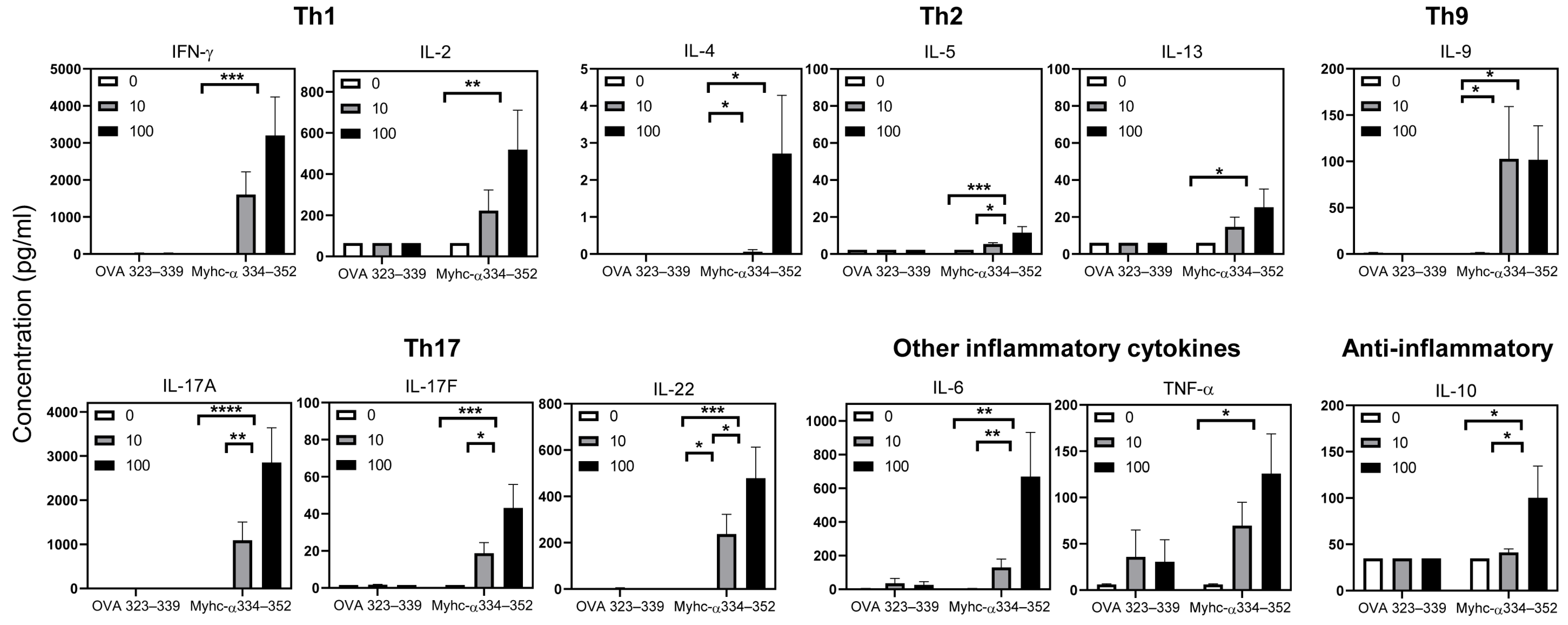
2.11. Cytokine Analysis
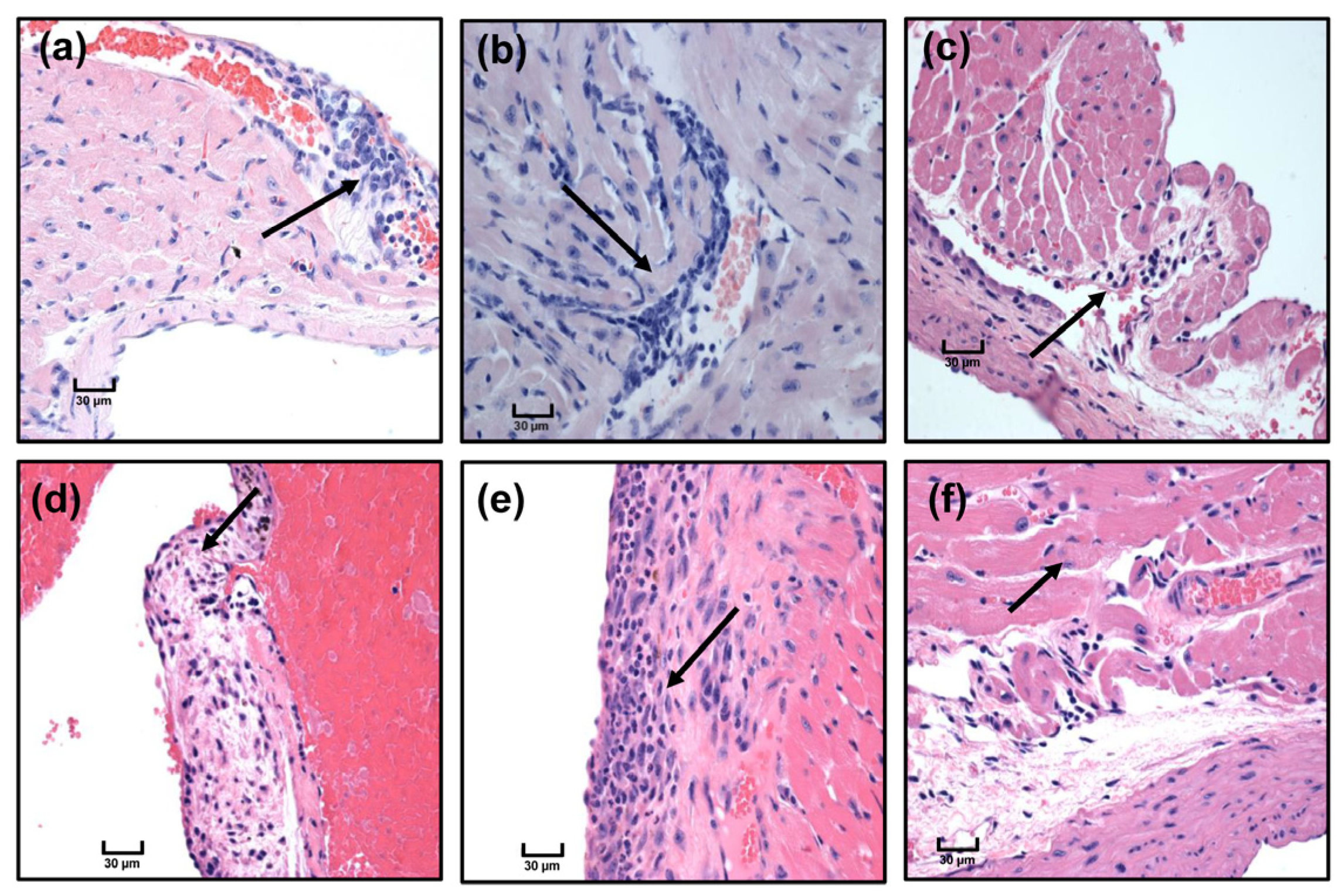
2.12. Histopathology
2.13. Statistical Analysis
3. Results
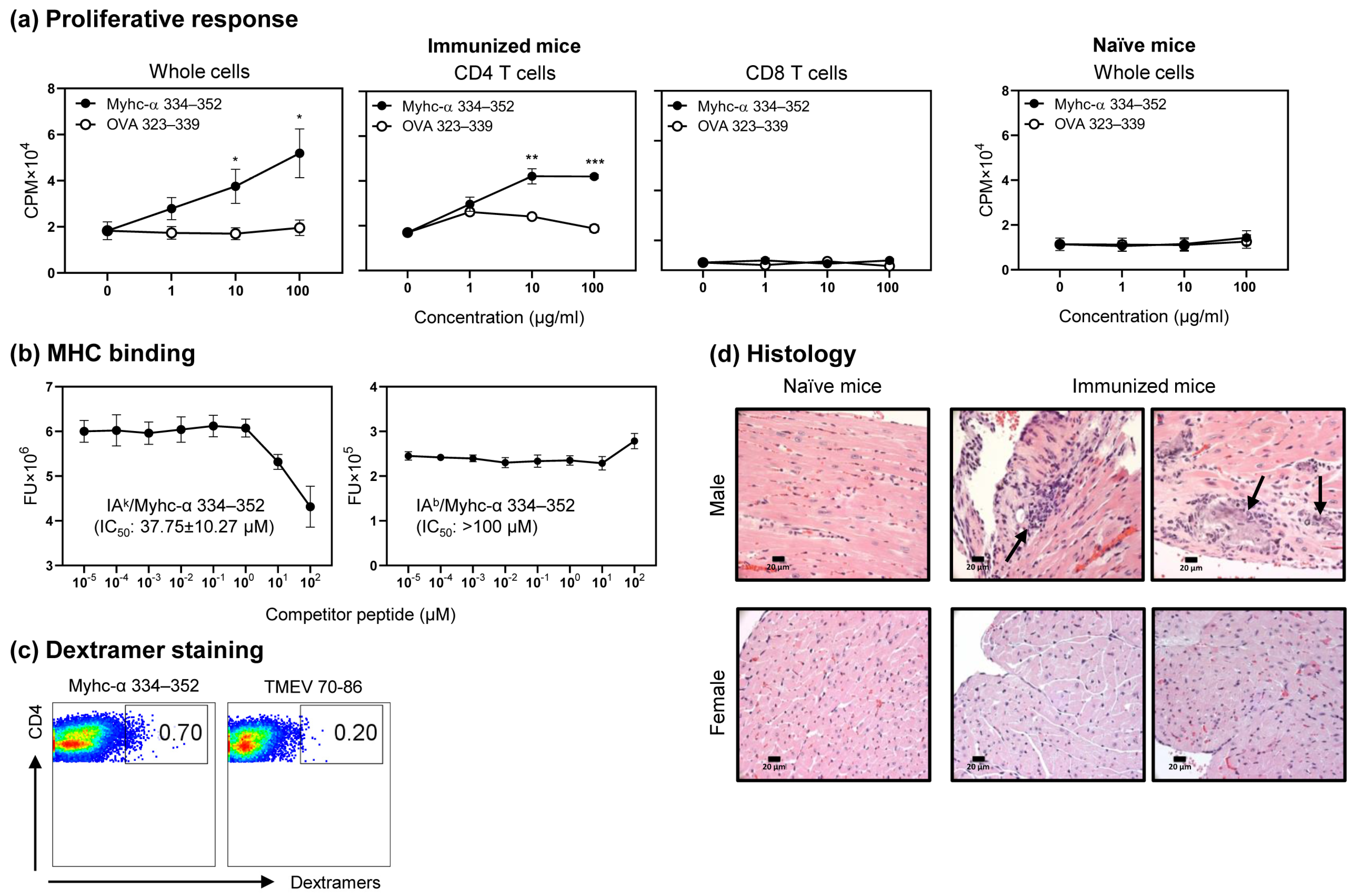
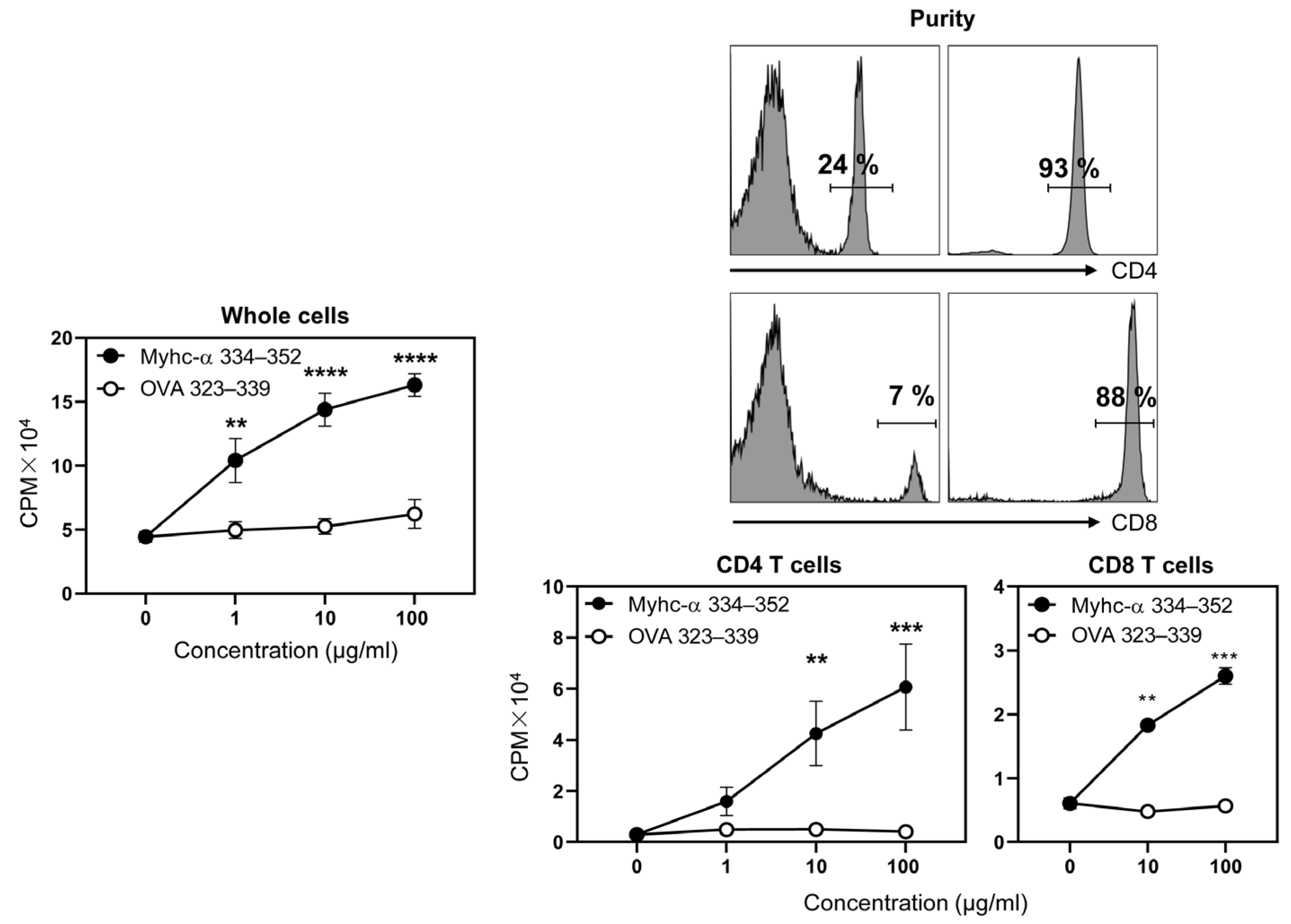
3.1. Myhc-α 334–352 Is a Promiscuous Immunogenic Epitope That Induces CD4 T Cell Response in Wt C57BL/6 Mice
3.2. Generation of Myhc-α 334–352 TCR Tg Mice
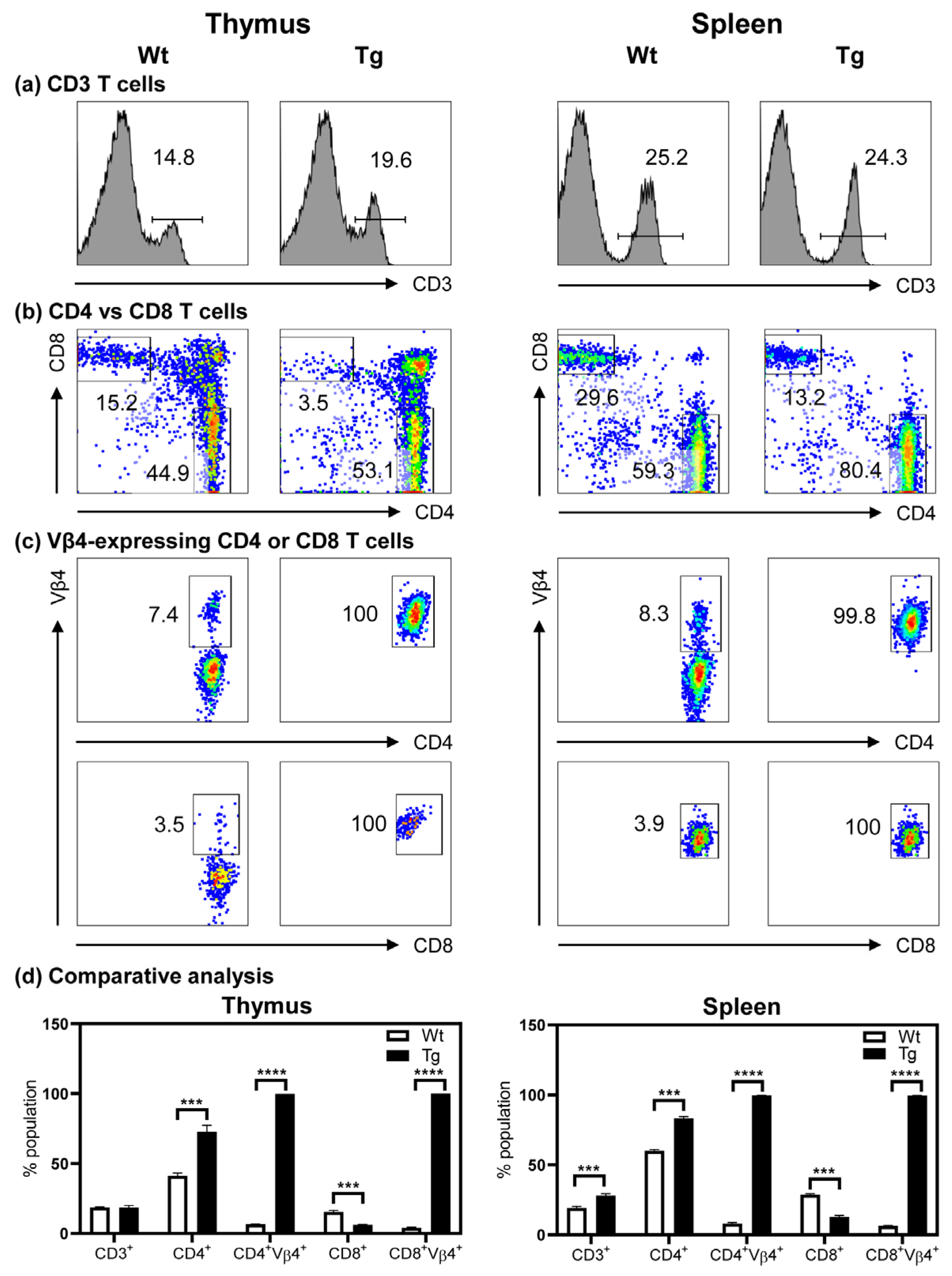
3.3. The Tg Mice Express Myhc-α 334–352-Specific TCR on Both CD4 and CD8 T Cells
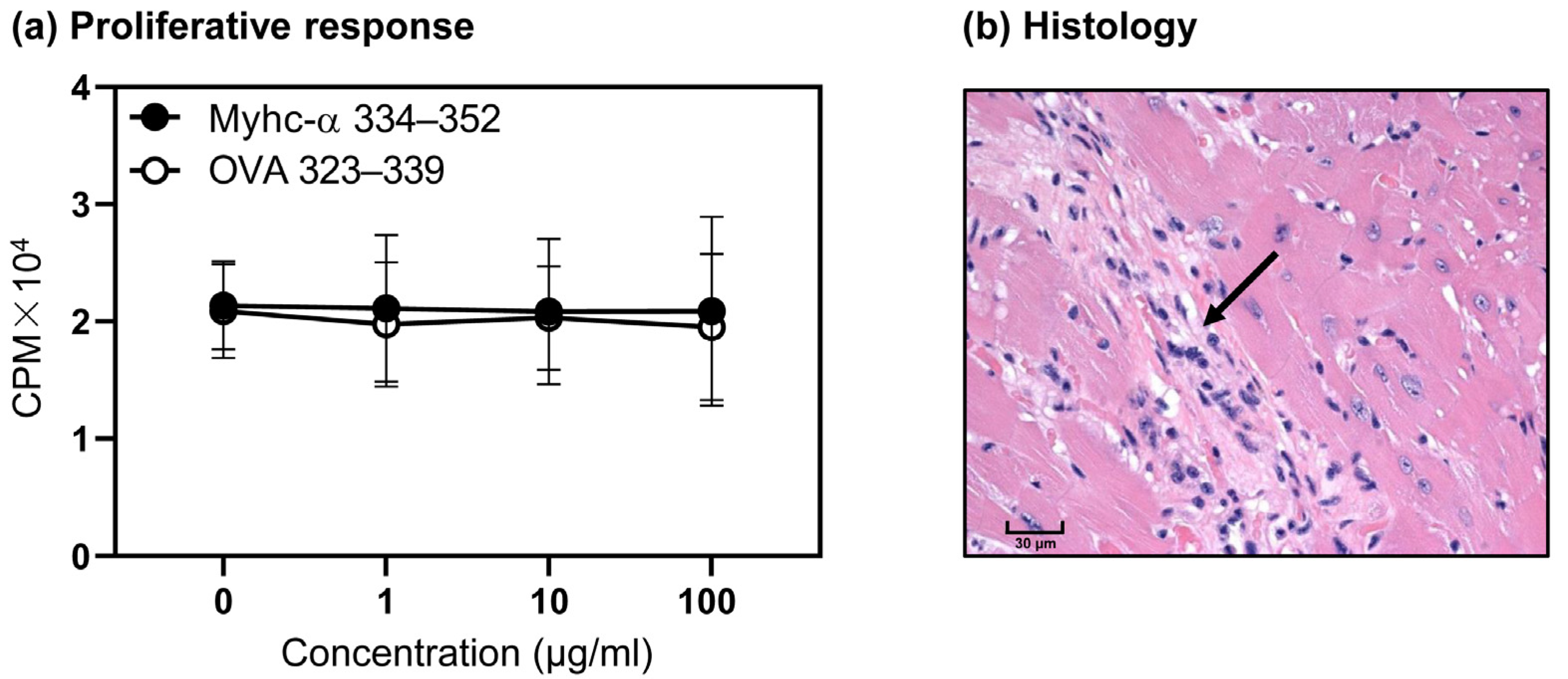
3.4. T Cells from Naïve Tg Mice Remain Tolerant, and They Do Not Develop Myocarditis Spontaneously
3.5. Immunization with Myhc-α 334–352 Breaks Tolerance in Tg Mice, and Immunized Animals Develop Myocarditis Albeit with Low Severity
3.6. Tg T Cells from Immunized Mice Preferentially Produce T Helper (Th)1 and Th17 Cytokines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J.; Doerer, J.J.; Haas, T.S.; Tierney, D.M.; Mueller, F.O. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980-2006. Circulation 2009, 119, 1085–1092. [Google Scholar] [CrossRef]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Thiene, G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol. Rev. 1999, 7, 127–135. [Google Scholar] [CrossRef]
- Koester, M.C. A Review of Sudden Cardiac Death in Young Athletes and Strategies for Preparticipation Cardiovascular Screening. J. Athl. Train 2001, 36, 197–204. [Google Scholar]
- Jensen-Urstad, M. Sudden death and physical activity in athletes and nonathletes. Scand. J. Med. Sci. Sports 1995, 5, 279–284. [Google Scholar] [CrossRef]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d. [Google Scholar] [CrossRef]
- Rroku, A.; Kottwitz, J.; Heidecker, B. Update on myocarditis—What we know so far and where we may be heading. Eur. Heart J. Acute Cardiovasc. Care 2020. [Google Scholar] [CrossRef]
- Kuhl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef]
- Krejci, J.; Mlejnek, D.; Sochorova, D.; Nemec, P. Inflammatory Cardiomyopathy: A Current View on the Pathophysiology, Diagnosis, and Treatment. Biomed. Res. Int. 2016, 2016, 4087632. [Google Scholar] [CrossRef]
- Mlejnek, D.; Krejci, J.; Hude, P.; Ozabalova, E.; Zampachova, V.; Stepanova, R.; Svobodova, I.; Freiberger, T.; Nemcova, E.; Spinarova, L. Viral genome changes and the impact of viral genome persistence in myocardium of patients with inflammatory cardiomyopathy. Arch. Med. Sci. 2018, 14, 1245–1253. [Google Scholar] [CrossRef]
- de Leeuw, N.; Ruiter, D.J.; Balk, A.H.; de Jonge, N.; Melchers, W.J.; Galama, J.M. Histopathologic findings in explanted heart tissue from patients with end-stage idiopathic dilated cardiomyopathy. Transpl. Int. 2001, 14, 299–306. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Harhay, M.O.; Hayes, D., Jr.; Hsich, E.; Meiser, B.; Potena, L.; Robinson, A.; Rossano, J.W.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult heart transplantation report—2019; focus theme: Donor and recipient size match. J. Heart Lung Transplant. 2019, 38, 1056–1066. [Google Scholar] [CrossRef]
- Leone, O.; Pieroni, M.; Rapezzi, C.; Olivotto, I. The spectrum of myocarditis: From pathology to the clinics. Virchows Arch. 2019, 475, 279–301. [Google Scholar] [CrossRef]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef]
- Limas, C.J.; Goldenberg, I.F.; Limas, C. Autoantibodies against beta-adrenoceptors in human idiopathic dilated cardiomyopathy. Circ. Res. 1989, 64, 97–103. [Google Scholar] [CrossRef]
- Kaya, Z.; Leib, C.; Katus, H.A. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 2012, 110, 145–158. [Google Scholar] [CrossRef]
- Baba, A.; Yoshikawa, T.; Ogawa, S. Autoantibodies produced against sarcolemmal Na-K-ATPase: Possible upstream targets of arrhythmias and sudden death in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 1153–1159. [Google Scholar] [CrossRef]
- Shmilovich, H.; Danon, A.; Binah, O.; Roth, A.; Chen, G.; Wexler, D.; Keren, G.; George, J. Autoantibodies to cardiac troponin I in patients with idiopathic dilated and ischemic cardiomyopathy. Int. J. Cardiol. 2007, 117, 198–203. [Google Scholar] [CrossRef]
- Latif, N.; Smith, J.; Dunn, M.J.; Yacoub, M.H.; Rose, M.L. Complement-mediated cytotoxic activity of anti-heart antibodies present in the sera of patients with dilated cardiomyopathy. Autoimmunity 1994, 19, 99–104. [Google Scholar] [CrossRef]
- Klein, R.; Maisch, B.; Kochsiek, K.; Berg, P.A. Demonstration of organ specific antibodies against heart mitochondria (anti-M7) in sera from patients with some forms of heart diseases. Clin. Exp. Immunol. 1984, 58, 283–292. [Google Scholar]
- Matsui, S.; Fu, M.L.; Shimizu, M.; Fukuoka, T.; Teraoka, K.; Takekoshi, N.; Murakami, E.; Hjalmarson, A. Dilated cardiomyopathy defines serum autoantibodies against G-protein-coupled cardiovascular receptors. Autoimmunity 1995, 21, 85–88. [Google Scholar] [CrossRef]
- Ansari, A.A.; Neckelmann, N.; Villinger, F.; Leung, P.; Danner, D.J.; Brar, S.S.; Zhao, S.; Gravanis, M.B.; Mayne, A.; Gershwin, M.E.; et al. Epitope mapping of the branched chain alpha-ketoacid dehydrogenase dihydrolipoyl transacylase (BCKD-E2) protein that reacts with sera from patients with idiopathic dilated cardiomyopathy. J. Immunol. 1994, 153, 4754–4765. [Google Scholar] [CrossRef]
- Caforio, A.L.; Grazzini, M.; Mann, J.M.; Keeling, P.J.; Bottazzo, G.F.; McKenna, W.J.; Schiaffino, S. Identification of alpha- and beta-cardiac myosin heavy chain isoforms as major autoantigens in dilated cardiomyopathy. Circulation 1992, 85, 1734–1742. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Bolte, H.D. Immunological analysis of auto-antibodies against the adenine nucleotide translocator in dilated cardiomyopathy. J. Mol. Cell. Cardiol. 1985, 17, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.; Savvatis, K.; Mohiddin, S.A.; Marelli-Berg, F.M. T-cell immunity in myocardial inflammation: Pathogenic role and therapeutic manipulation. Br. J. Pharmacol. 2017, 174, 3914–3925. [Google Scholar] [CrossRef]
- Bracamonte-Baran, W.; Cihakova, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar] [CrossRef]
- Blyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef]
- Krishnan, B.; Massilamany, C.; Basavalingappa, R.H.; Gangaplara, A.; Rajasekaran, R.A.; Afzal, M.Z.; Khalilzad-Sharghi, V.; Zhou, Y.; Riethoven, J.J.; Nandi, S.S.; et al. Epitope Mapping of SERCA2a Identifies an Antigenic Determinant That Induces Mainly Atrial Myocarditis in A/J Mice. J. Immunol. 2018, 200, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Basavalingappa, R.H.; Massilamany, C.; Krishnan, B.; Gangaplara, A.; Rajasekaran, R.A.; Afzal, M.Z.; Riethoven, J.J.; Strande, J.L.; Steffen, D.; Reddy, J. beta1-Adrenergic Receptor Contains Multiple IA(k) and IE(k) Binding Epitopes That Induce T Cell Responses with Varying Degrees of Autoimmune Myocarditis in A/J Mice. Front. Immunol. 2017, 8, 1567. [Google Scholar] [CrossRef]
- Krishnan, B.; Massilamany, C.; Basavalingappa, R.H.; Gangaplara, A.; Kang, G.; Li, Q.; Uzal, F.A.; Strande, J.L.; Delhon, G.A.; Riethoven, J.J.; et al. Branched chain alpha-ketoacid dehydrogenase kinase 111-130, a T cell epitope that induces both autoimmune myocarditis and hepatitis in A/J mice. Immun. Inflamm. Dis. 2017, 5, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Kaya, Z.; Katus, H.A.; Rose, N.R. Cardiac troponins and autoimmunity: Their role in the pathogenesis of myocarditis and of heart failure. Clin. Immunol. 2010, 134, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Massilamany, C.; Gangaplara, A.; Chapman, N.; Rose, N.; Reddy, J. Detection of cardiac myosin heavy chain-alpha-specific CD4 cells by using MHC class II/IA(k) tetramers in A/J mice. J. Immunol. Methods 2011, 372, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Basavalingappa, R.H.; Massilamany, C.; Krishnan, B.; Gangaplara, A.; Kang, G.; Khalilzad-Sharghi, V.; Han, Z.; Othman, S.; Li, Q.; Riethoven, J.J.; et al. Identification of an Epitope from Adenine Nucleotide Translocator 1 That Induces Inflammation in Heart in A/J Mice. Am. J. Pathol. 2016, 186, 3160–3175. [Google Scholar] [CrossRef] [PubMed]
- Pummerer, C.L.; Luze, K.; Grassl, G.; Bachmaier, K.; Offner, F.; Burrell, S.K.; Lenz, D.M.; Zamborelli, T.J.; Penninger, J.M.; Neu, N. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J. Clin. Investig. 1996, 97, 2057–2062. [Google Scholar] [CrossRef]
- Wegmann, K.W.; Zhao, W.; Griffin, A.C.; Hickey, W.F. Identification of myocarditogenic peptides derived from cardiac myosin capable of inducing experimental allergic myocarditis in the Lewis rat. The utility of a class II binding motif in selecting self-reactive peptides. J. Immunol. 1994, 153, 892–900. [Google Scholar] [CrossRef]
- Krishnan, B.; Massilamany, C.; Basavalingappa, R.H.; Rajasekaran, R.A.; Kuszynski, C.; Switzer, B.; Peterson, D.A.; Reddy, J. Versatility of using major histocompatibility complex class II dextramers for derivation and characterization of antigen-specific, autoreactive T cell hybridomas. J. Immunol. Methods 2015, 426, 86–94. [Google Scholar] [CrossRef]
- Kouskoff, V.; Signorelli, K.; Benoist, C.; Mathis, D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods 1995, 180, 273–280. [Google Scholar] [CrossRef]
- Massilamany, C.; Upadhyaya, B.; Gangaplara, A.; Kuszynski, C.; Reddy, J. Detection of autoreactive CD4 T cells using major histocompatibility complex class II dextramers. BMC Immunol. 2011, 12, 40. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Jia, T.; Elowsky, C.; Kang, G.; Riethoven, J.J.; Li, Q.; Zhou, Y.; Reddy, J. Direct staining with major histocompatibility complex class II dextramers permits detection of antigen-specific, autoreactive CD4 T cells in situ. PLoS ONE 2014, 9, e87519. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Reddy, J.; Gao, W.; Bettelli, E.; Awasthi, A.; Petersen, T.R.; Backstrom, B.T.; Sobel, R.A.; Wucherpfennig, K.W.; Strom, T.B.; et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007, 13, 423–431. [Google Scholar] [CrossRef]
- Reddy, J.; Bettelli, E.; Nicholson, L.; Waldner, H.; Jang, M.H.; Wucherpfennig, K.W.; Kuchroo, V.K. Detection of autoreactive myelin proteolipid protein 139-151-specific T cells by using MHC II (IAs) tetramers. J. Immunol. 2003, 170, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Seth, N.P.; Wucherpfennig, K.W. Ex vivo analysis of thymic CD4 T cells in nonobese diabetic mice with tetramers generated from I-A(g7)/class II-associated invariant chain peptide precursors. J. Immunol. 2003, 171, 4175–4186. [Google Scholar] [CrossRef]
- Gangaplara, A.; Massilamany, C.; Steffen, D.; Reddy, J. Mimicry epitope from Ehrlichia canis for interphotoreceptor retinoid-binding protein 201-216 prevents autoimmune uveoretinitis by acting as altered peptide ligand. J. Neuroimmunol. 2013, 263, 98–107. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Donermeyer, D.L.; Beisel, K.W.; Allen, P.M.; Smith, S.C. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J. Exp. Med. 1995, 182, 1291–1300. [Google Scholar] [CrossRef]
- Afanasyeva, M.; Wang, Y.; Kaya, Z.; Park, S.; Zilliox, M.J.; Schofield, B.H.; Hill, S.L.; Rose, N.R. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am. J. Pathol. 2001, 159, 193–203. [Google Scholar] [CrossRef]
- Smith, S.C.; Allen, P.M. Myosin-induced acute myocarditis is a T cell-mediated disease. J. Immunol. 1991, 147, 2141–2147. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Basavalingappa, R.H.; Rajasekaran, R.A.; Khalilzad-Sharghi, V.; Han, Z.; Othman, S.; Steffen, D.; Reddy, J. Localization of CD8 T cell epitope within cardiac myosin heavy chain-alpha334-352 that induces autoimmune myocarditis in A/J mice. Int. J. Cardiol 2016, 202, 311–321. [Google Scholar] [CrossRef]
- McFarland, B.J.; Sant, A.J.; Lybrand, T.P.; Beeson, C. Ovalbumin(323-339) peptide binds to the major histocompatibility complex class II I-A(d) protein using two functionally distinct registers. Biochemistry 1999, 38, 16663–16670. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Fitch, P.M.; Leech, M.D.; Ilchmann, A.; Wilson, C.; McFarlane, A.J.; Howie, S.E.; Anderton, S.M.; Schwarze, J. Combination peptide immunotherapy based on T-cell epitope mapping reduces allergen-specific IgE and eosinophilia in allergic airway inflammation. Immunology 2013, 138, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, O.; Shimizu, J.; Unanue, E.R. The role of I-Ag7 beta chain in peptide binding and antigen recognition by T cells. Int. Immunol. 1997, 9, 1523–1526. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massilamany, C.; Steffen, D.; Reddy, J. An epitope from Acanthamoeba castellanii that cross-react with proteolipid protein 139-151-reactive T cells induces autoimmune encephalomyelitis in SJL mice. J. Neuroimmunol. 2010, 219, 17–24. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massilamany, C.; Gangaplara, A.; Steffen, D.; Reddy, J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-alpha that induce autoimmune myocarditis in A/J mice. Cell Immunol. 2011, 271, 438–449. [Google Scholar] [CrossRef]
- Crenshaw, E.B., 3rd; Ryan, A.; Dillon, S.R.; Kalla, K.; Rosenfeld, M.G. Wocko, a neurological mutant generated in a transgenic mouse pedigree. J. Neurosci. 1991, 11, 1524–1530. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ting, C.N.; Kohrman, D.; Burgess, D.L.; Boyle, A.; Altschuler, R.A.; Gholizadeh, G.; Samuelson, L.C.; Jang, W.; Meisler, M.H. Insertional mutation on mouse chromosome 18 with vestibular and craniofacial abnormalities. Genetics 1994, 136, 247–254. [Google Scholar] [CrossRef]
- Silaeva, Y.Y.; Kalinina, A.A.; Khromykh, L.M.; Kazansky, D.B. Functional properties of CD8+ T cells under lymphopenic conditions. bioRxiv 2018, 353011. [Google Scholar] [CrossRef]
- Sakai, K.; Zamvil, S.S.; Mitchell, D.J.; Lim, M.; Rothbard, J.B.; Steinman, L. Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J. Neuroimmunol. 1988, 19, 21–32. [Google Scholar] [CrossRef]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar] [CrossRef]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef]
- Waldner, H.; Whitters, M.J.; Sobel, R.A.; Collins, M.; Kuchroo, V.K. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 3412–3417. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Chandwaskar, R.; Lee, D.H.; Sullivan, J.M.; Solomon, A.; Rodriguez-Manzanet, R.; Greve, B.; Sobel, R.A.; Kuchroo, V.K. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J. Immunol. 2012, 188, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Allen, P.M. Expression of myosin-class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc. Natl. Acad. Sci. USA 1992, 89, 9131–9135. [Google Scholar] [CrossRef] [PubMed]
- Flatz, L.; Hegazy, A.N.; Bergthaler, A.; Verschoor, A.; Claus, C.; Fernandez, M.; Gattinoni, L.; Johnson, S.; Kreppel, F.; Kochanek, S.; et al. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 2010, 16, 339–345. [Google Scholar] [CrossRef]
- Goverman, J.; Woods, A.; Larson, L.; Weiner, L.P.; Hood, L.; Zaller, D.M. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell 1993, 72, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Brabb, T.; Goldrath, A.W.; von Dassow, P.; Paez, A.; Liggitt, H.D.; Goverman, J. Triggers of autoimmune disease in a murine TCR-transgenic model for multiple sclerosis. J. Immunol. 1997, 159, 497–507. [Google Scholar] [CrossRef]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef]
- Barin, J.G.; Baldeviano, G.C.; Talor, M.V.; Wu, L.; Ong, S.; Fairweather, D.; Bedja, D.; Stickel, N.R.; Fontes, J.A.; Cardamone, A.B.; et al. Fatal eosinophilic myocarditis develops in the absence of IFN-gamma and IL-17A. J. Immunol. 2013, 191, 4038–4047. [Google Scholar] [CrossRef]
- Guenova, E.; Skabytska, Y.; Hoetzenecker, W.; Weindl, G.; Sauer, K.; Tham, M.; Kim, K.W.; Park, J.H.; Seo, J.H.; Ignatova, D.; et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2015, 112, 2163–2168. [Google Scholar] [CrossRef]
- Ye, D.; Wang, Z.; Ye, J.; Wang, M.; Liu, J.; Xu, Y.; Jiang, H.; Chen, J.; Wan, J. Interleukin-5 levels are decreased in the plasma of coronary artery disease patients and inhibit Th1 and Th17 differentiation in vitro. Rev. Esp. Cardiol. Engl. Ed. 2020, 73, 393–402. [Google Scholar] [CrossRef]
- Leyva-Castillo, J.M.; Das, M.; Artru, E.; Yoon, J.; Galand, C.; Geha, R.S. Mast cell-derived IL-13 downregulates IL-12 production by skin dendritic cells to inhibit the T(H)1 cell response to cutaneous antigen exposure. J. Allergy Clin. Immunol. 2021, 147, 2305–2315 e2303. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Yang, J.; Ouyang, X.; Liu, W.; Li, H.; Yang, J.; Bromberg, J.; Chen, S.H.; Mayer, L.; Unkeless, J.C.; et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur. J. Immunol. 2008, 38, 1807–1813. [Google Scholar] [CrossRef]
- Nindl, V.; Maier, R.; Ratering, D.; De Giuli, R.; Zust, R.; Thiel, V.; Scandella, E.; Di Padova, F.; Kopf, M.; Rudin, M.; et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur. J. Immunol. 2012, 42, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.C.; Hansen, B.E.; Jacobsen, H.; Madsen, L.S.; Andersen, C.B.; Engberg, J.; Rothbard, J.B.; McDevitt, G.S.; Malmstrom, V.; Holmdahl, R.; et al. Definition of MHC and T cell receptor contacts in the HLA-DR4restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7574–7579. [Google Scholar] [CrossRef] [PubMed]
- Fugger, L.; Rothbard, J.B.; Sonderstrup-McDevitt, G. Specificity of an HLA-DRB1*0401-restricted T cell response to type II collagen. Eur. J. Immunol. 1996, 26, 928–933. [Google Scholar] [CrossRef]
- Rosloniec, E.F.; Brand, D.D.; Myers, L.K.; Whittington, K.B.; Gumanovskaya, M.; Zaller, D.M.; Woods, A.; Altmann, D.M.; Stuart, J.M.; Kang, A.H. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 1997, 185, 1113–1122. [Google Scholar] [CrossRef]
- Cope, A.P.; Patel, S.D.; Hall, F.; Congia, M.; Hubers, H.A.; Verheijden, G.F.; Boots, A.M.; Menon, R.; Trucco, M.; Rijnders, A.W.; et al. T cell responses to a human cartilage autoantigen in the context of rheumatoid arthritis-associated and nonassociated HLA-DR4 alleles. Arthritis Rheum. 1999, 42, 1497–1507. [Google Scholar] [CrossRef]
- Wicker, L.S.; Chen, S.L.; Nepom, G.T.; Elliott, J.F.; Freed, D.C.; Bansal, A.; Zheng, S.; Herman, A.; Lernmark, A.; Zaller, D.M.; et al. Naturally processed T cell epitopes from human glutamic acid decarboxylase identified using mice transgenic for the type 1 diabetes-associated human MHC class II allele, DRB1*0401. J. Clin. Investig. 1996, 98, 2597–2603. [Google Scholar] [CrossRef]
- Patel, S.D.; Cope, A.P.; Congia, M.; Chen, T.T.; Kim, E.; Fugger, L.; Wherrett, D.; Sonderstrup-McDevitt, G. Identification of immunodominant T cell epitopes of human glutamic acid decarboxylase 65 by using HLA-DR(alpha1*0101,beta1*0401) transgenic mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8082–8087. [Google Scholar] [CrossRef]
- Altmann, D.M.; Douek, D.C.; Frater, A.J.; Hetherington, C.M.; Inoko, H.; Elliott, J.I. The T cell response of HLA-DR transgenic mice to human myelin basic protein and other antigens in the presence and absence of human CD4. J. Exp. Med. 1995, 181, 867–875. [Google Scholar] [CrossRef]
- Hofmann, U.; Frantz, S. Role of T-cells in myocardial infarction. Eur. Heart J. 2016, 37, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Tae Yu, H.; Youn, J.C.; Lee, J.; Park, S.; Chi, H.S.; Lee, J.; Choi, C.; Park, S.; Choi, D.; Ha, J.W.; et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell Mol. Immunol. 2015, 12, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Basavalingappa, R.H.; Arumugam, R.; Lasrado, N.; Yalaka, B.; Massilamany, C.; Gangaplara, A.; Riethoven, J.J.; Xiang, S.H.; Steffen, D.; Reddy, J. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol. Immunol. 2020, 124, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wei, G.; Pan, W.; Tabel, H. Experimental African trypanosomiasis: A subset of pathogenic, IFN-gamma-producing, MHC class II-restricted CD4+ T cells mediates early mortality in highly susceptible mice. J. Immunol. 2006, 176, 1724–1732. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCR Genes | Primers | Sequence (5′ to 3′) |
|---|---|---|
| Qualitative PCR | ||
| α chain | P2 set: | |
| Forward | GCAGCAGGAGAAACGTGAC | |
| Reverse | GCTTTGCATTGCTTCCTTCC | |
| P3 set: | ||
| Forward | CCTTGCACATCACAGACTC | |
| Reverse | TGAAGTGAGSTGGGGAATA | |
| β chain | P2 set: | |
| Forward | CGACCCAAAATTATCCAG | |
| Reverse | CAGTGCCTGGCCCAAAGTAC | |
| P3 set: | ||
| Forward | CCATTTAGACCTTCAGATCA | |
| Reverse | CCCAATCCCGCTGAGAAC | |
| qPCR | ||
| α chain | Forward | GTGCCTTTCCCGAAGGTTA |
| Reverse | TCTCCTTGCACATCACAGAC | |
| Probe | 56-FAM/TGCAGCAAG/ZEN/TGCGGAAGGAAG/3IABkFQ | |
| β chain | Forward | TAACACGAGGAGCCGAGT |
| Reverse | AGACCTTCAGATCACAGCTCTA | |
| Probe | 5SUN/AGCAGCCAA/ZEN/AGTTTGGGCCAAG/3IABkFQ | |
| Internal positive control | Forward | TCACCAGTCATTTCTGCTTTG |
| Reverse | CACGTGGGCTCCAGATTT | |
| Probe | 5Cy5/CCAATGGTC/TAO/GGGCACTGCTCAA/3lAbRQSp | |
| Cell Type | TCR Chains | Variable (V) Segment | Diversity (D) Segment | Joining (J) Segment | Constant Region | CDRs | ||
|---|---|---|---|---|---|---|---|---|
| CDR1 | CDR2 | CDR3 | ||||||
| CD4 or CD8 | α | TRAV14-1*02 | - | TRAJ42*01 | TRAC*01 | DSTFNY | ISSVSDK | CAASAEGSNAKLTF |
| β | TRBV2*01 | D | TRBJ2-5*01 | TRBC2*01 | LGHNA | YSYQKL | CASSQSLGQDTQYF | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sur, M.; Rasquinha, M.T.; Arumugam, R.; Massilamany, C.; Gangaplara, A.; Mone, K.; Lasrado, N.; Yalaka, B.; Doiphode, A.; Gurumurthy, C.; et al. Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-α 334–352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background. Cells 2023, 12, 2346. https://doi.org/10.3390/cells12192346
Sur M, Rasquinha MT, Arumugam R, Massilamany C, Gangaplara A, Mone K, Lasrado N, Yalaka B, Doiphode A, Gurumurthy C, et al. Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-α 334–352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background. Cells. 2023; 12(19):2346. https://doi.org/10.3390/cells12192346
Chicago/Turabian StyleSur, Meghna, Mahima T. Rasquinha, Rajkumar Arumugam, Chandirasegaran Massilamany, Arunkumar Gangaplara, Kiruthiga Mone, Ninaad Lasrado, Bharathi Yalaka, Aakash Doiphode, Channabasavaiah Gurumurthy, and et al. 2023. "Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-α 334–352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background" Cells 12, no. 19: 2346. https://doi.org/10.3390/cells12192346
APA StyleSur, M., Rasquinha, M. T., Arumugam, R., Massilamany, C., Gangaplara, A., Mone, K., Lasrado, N., Yalaka, B., Doiphode, A., Gurumurthy, C., Steffen, D., & Reddy, J. (2023). Transgenic Mice Expressing Functional TCRs Specific to Cardiac Myhc-α 334–352 on Both CD4 and CD8 T Cells Are Resistant to the Development of Myocarditis on C57BL/6 Genetic Background. Cells, 12(19), 2346. https://doi.org/10.3390/cells12192346