

Intermediate Molecular Phenotypes to Identify Genetic Markers of Anthracycline-Induced Cardiotoxicity Risk

 , , , ,
, , , ,  ,
,  , ,
, ,  , , ,
, , ,  , , , , , , , , ,
, , , , , , , , ,  , , ,
, , ,  , and add
Show full author list
, and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Generation and Chemotherapy
2.2. Heart-Tissue Processing and CDA Quantification
2.3. Quantification of Signaling Proteins via Multiplex Bead Arrays
2.4. miRNA Quantification via QPCR
2.5. Mouse QTL Genetic Analyses and Generation of Genetic Models
2.6. Patients
2.7. Cardiac Magnetic Resonance: Acquisition and Analysis
2.8. Human Genetic Analyses
2.9. General Statistical Analyses
3. Results
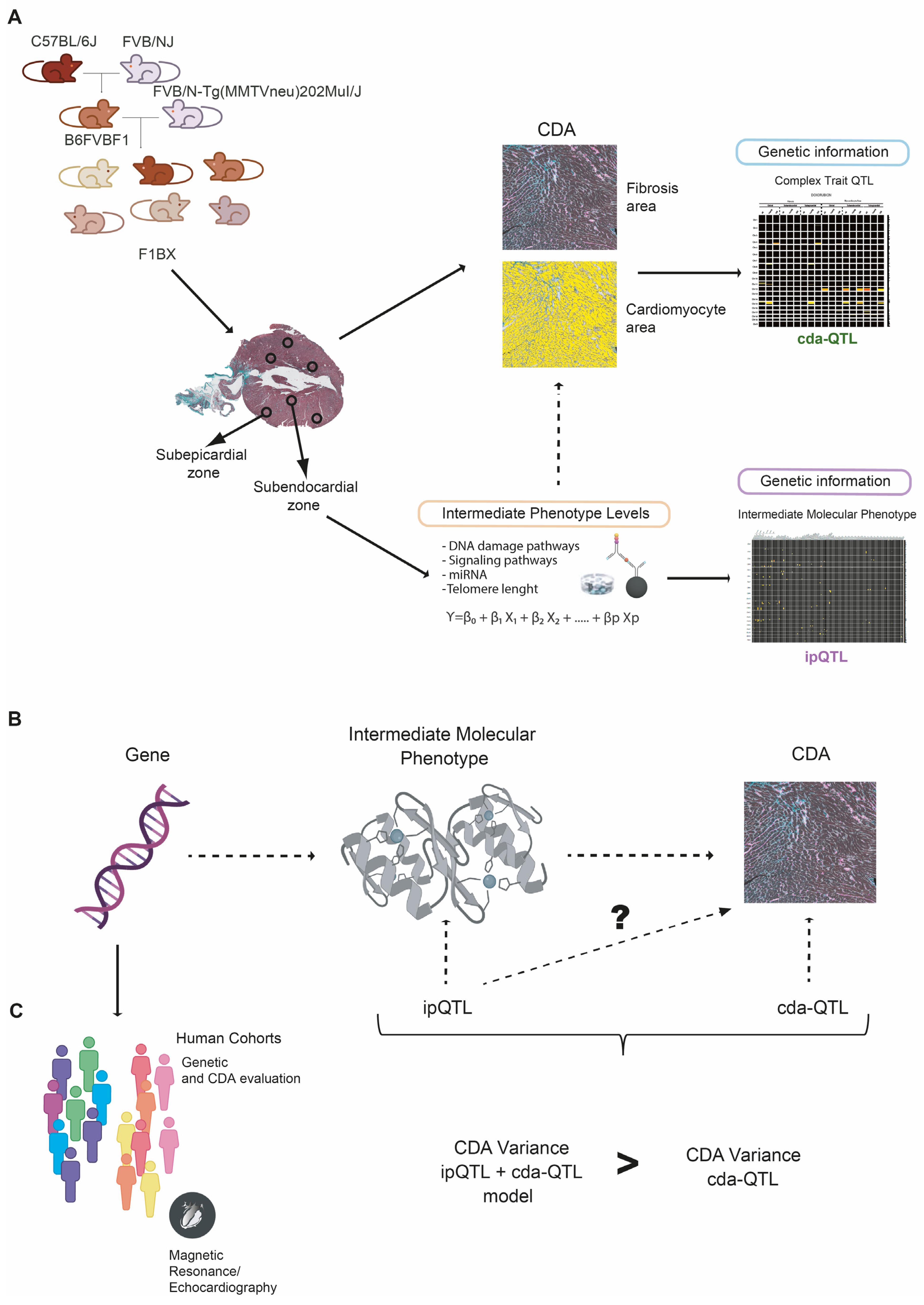
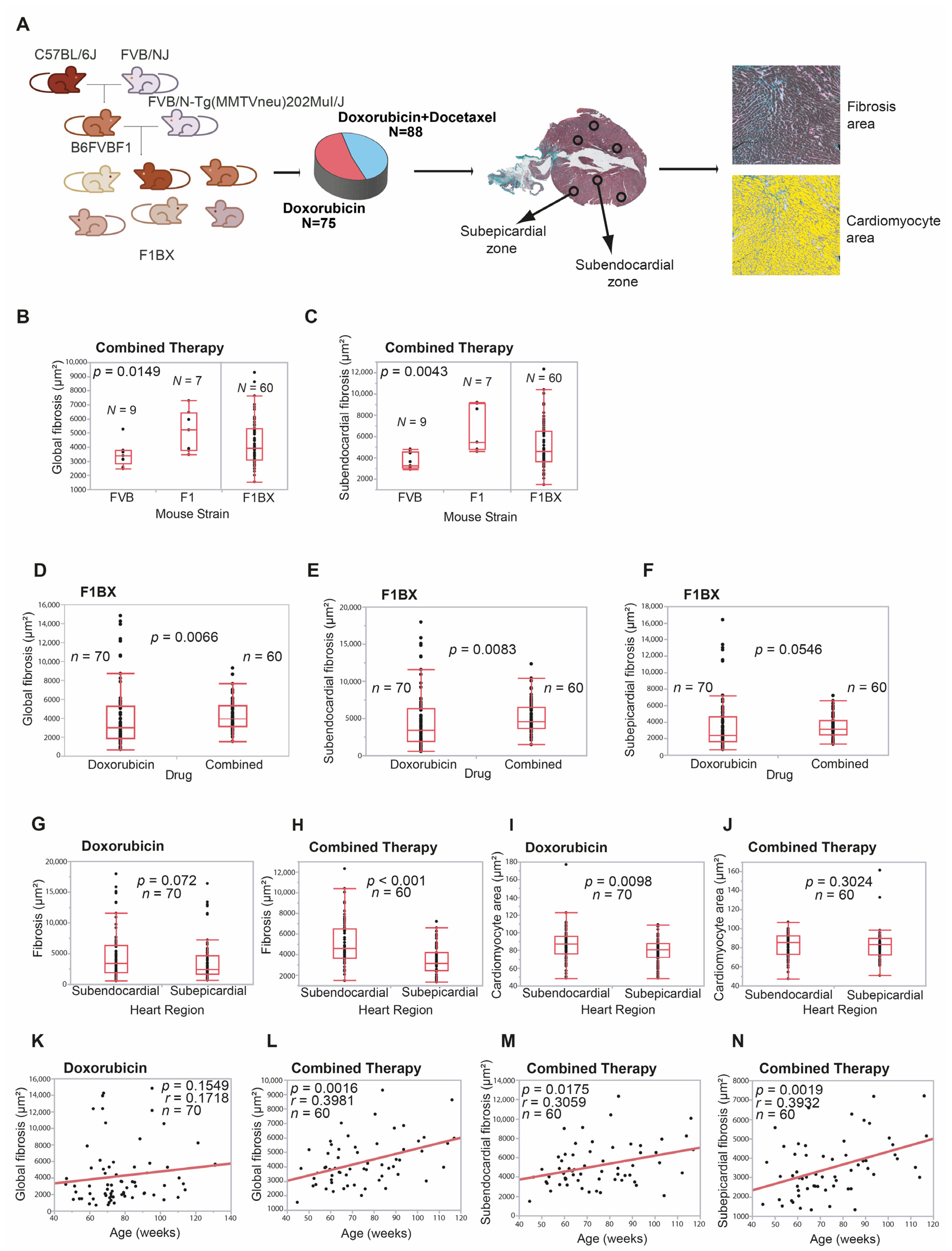
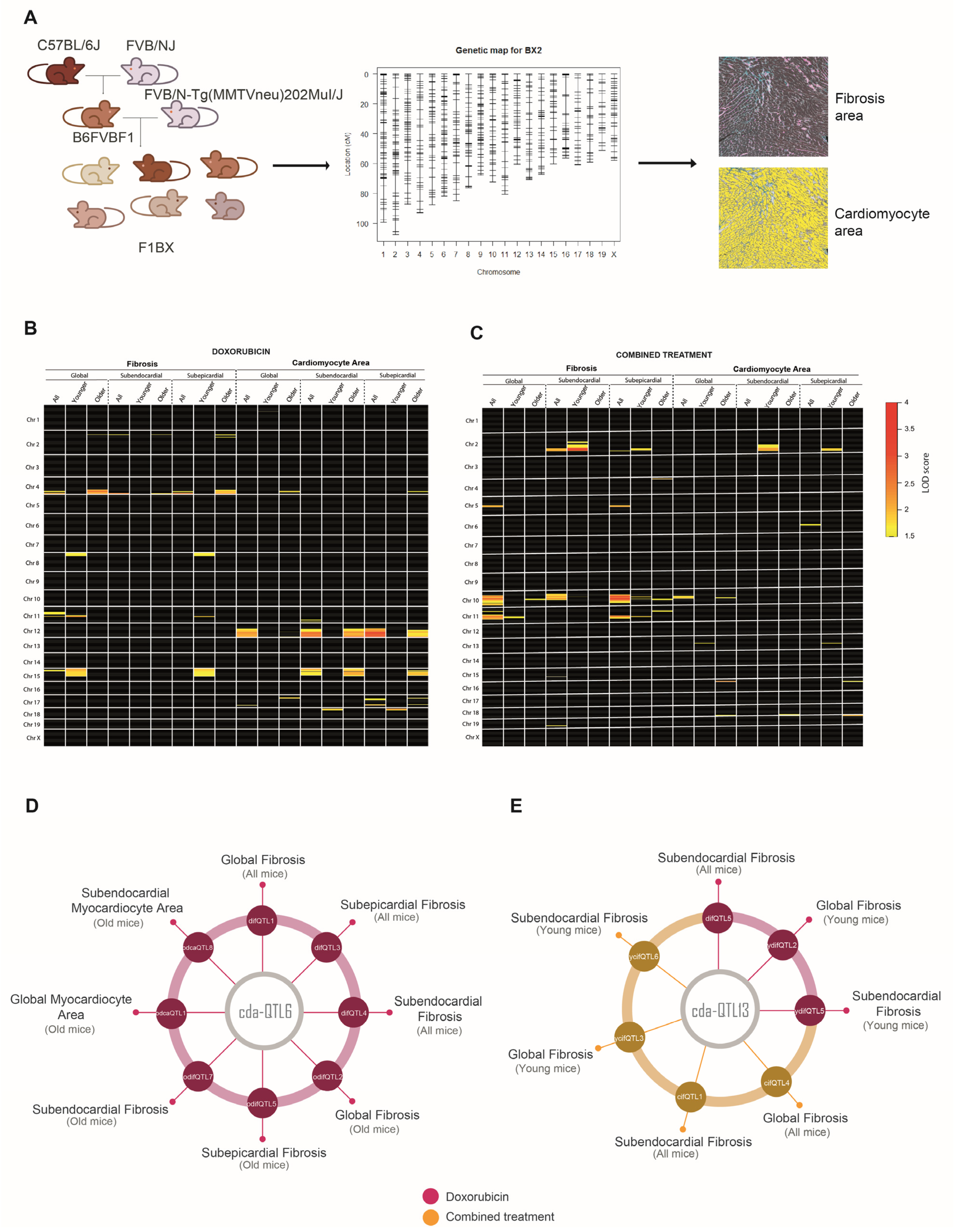
3.1. Cardiotoxicity Due to Anthracyclines Behaves as a Complex Trait in a Genetically Heterogeneous Mouse Cohort
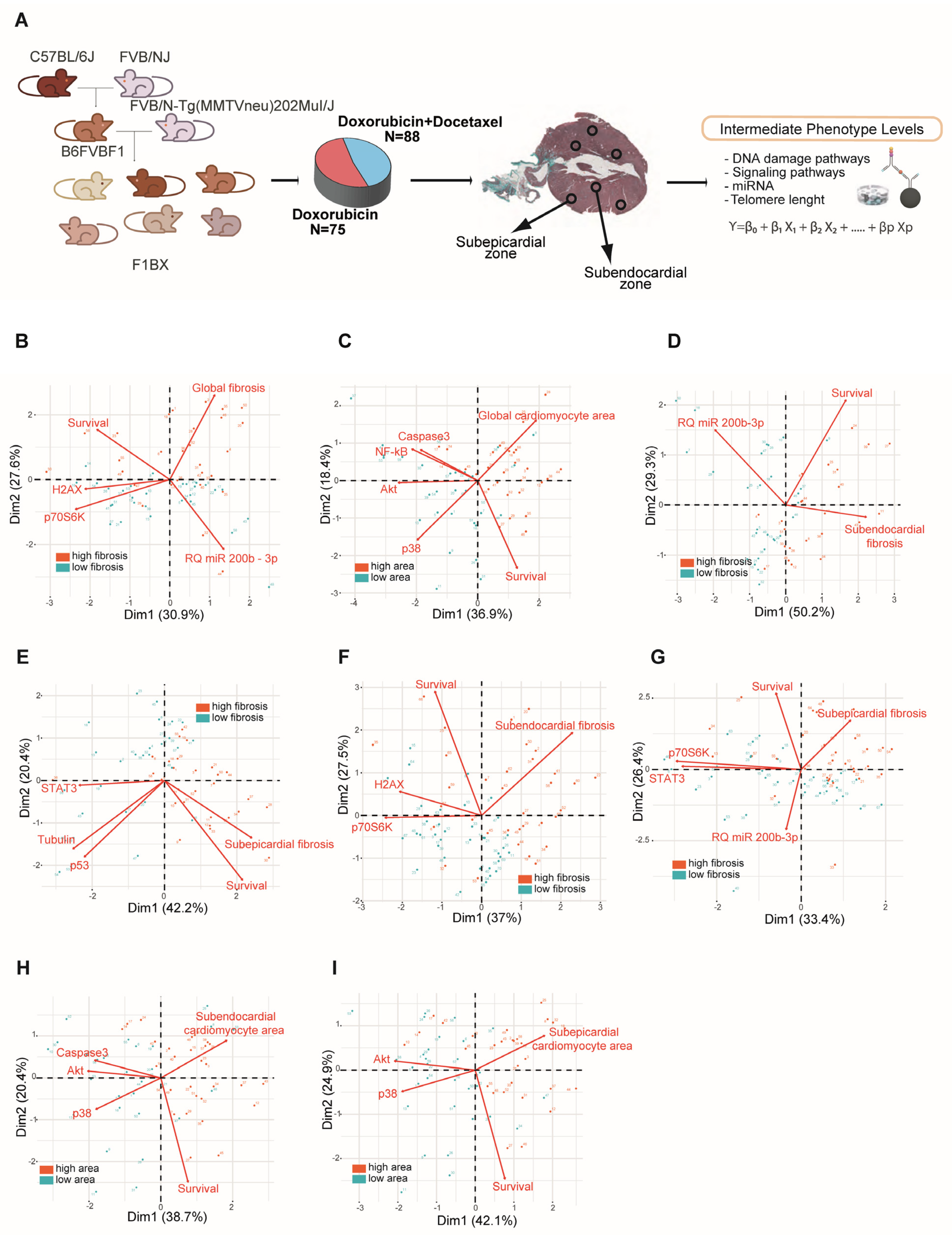
3.2. Intermediate Phenotype Levels of Molecular Origin in the Myocardium Are Associated with Chemotherapy-Induced Cardiotoxicity
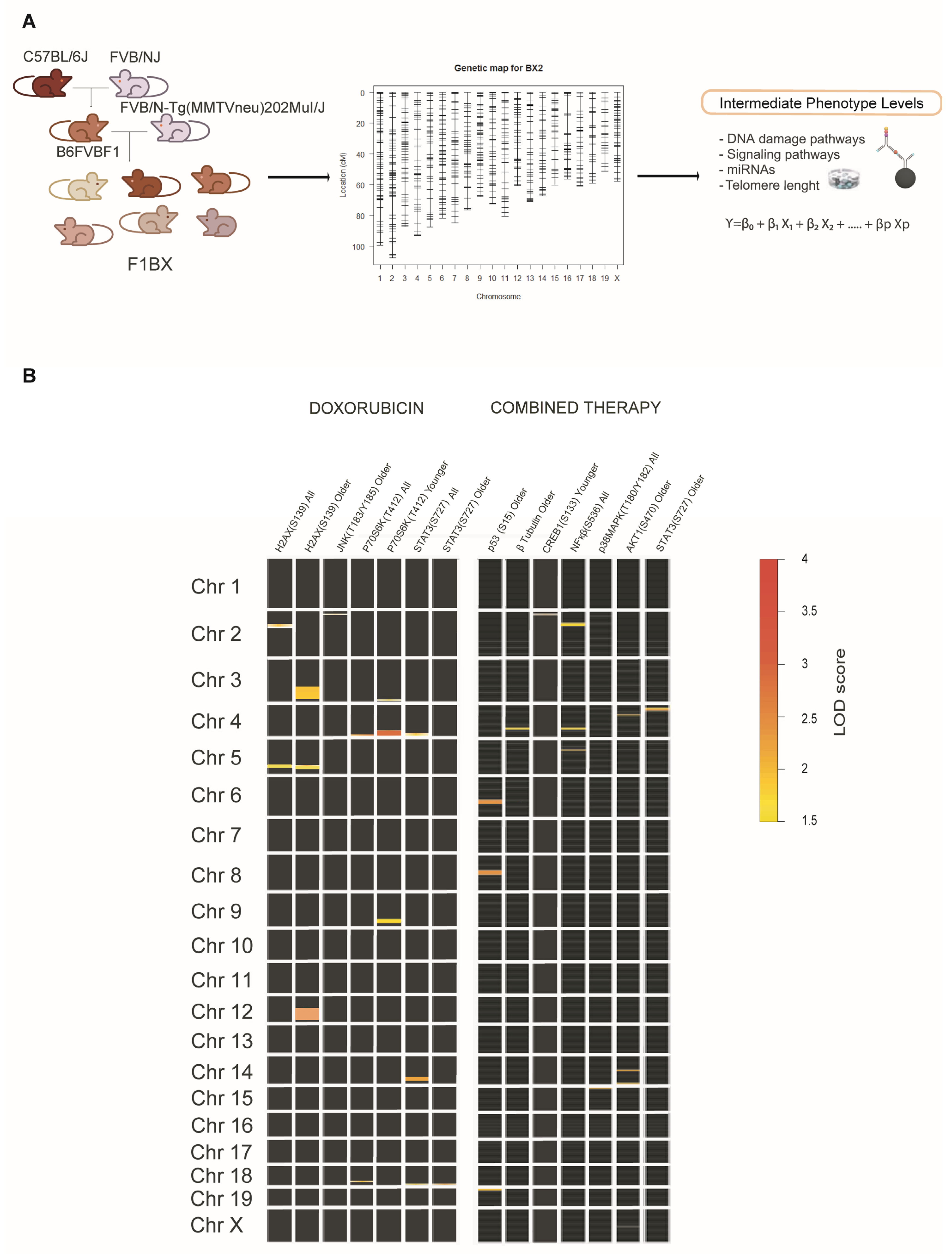
3.3. Identification of Genetic Determinants Linked to Intermediate Molecular Phenotypes of CDA
3.4. Identification of Genetic Determinants Directly Linked to CDA (cdaQTLs)
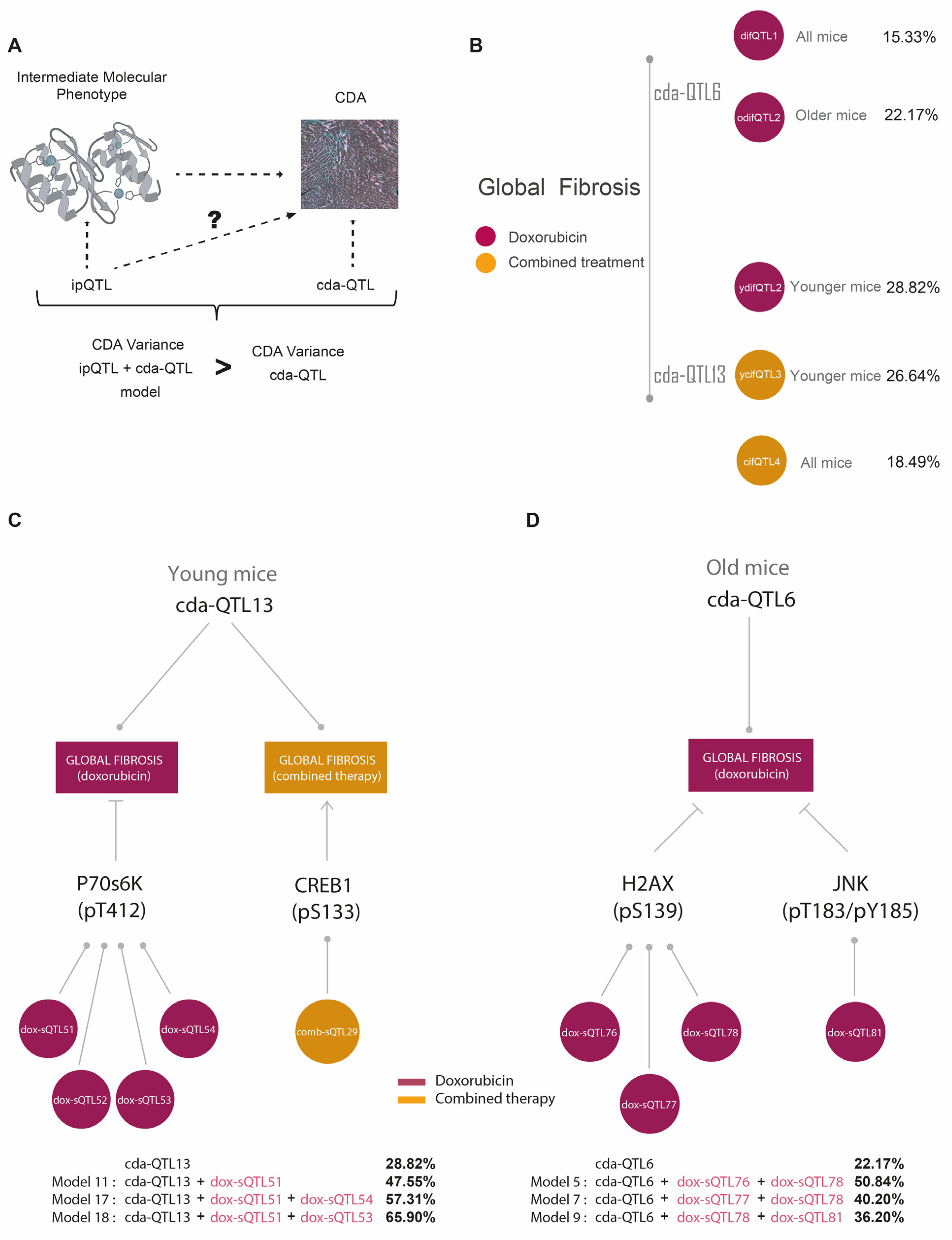
3.5. ipQTLs Integrated into Genetic Models with cdaQTLs Account for More Phenotypic Variation of CDA Than Explained by cda-QTLs Alone
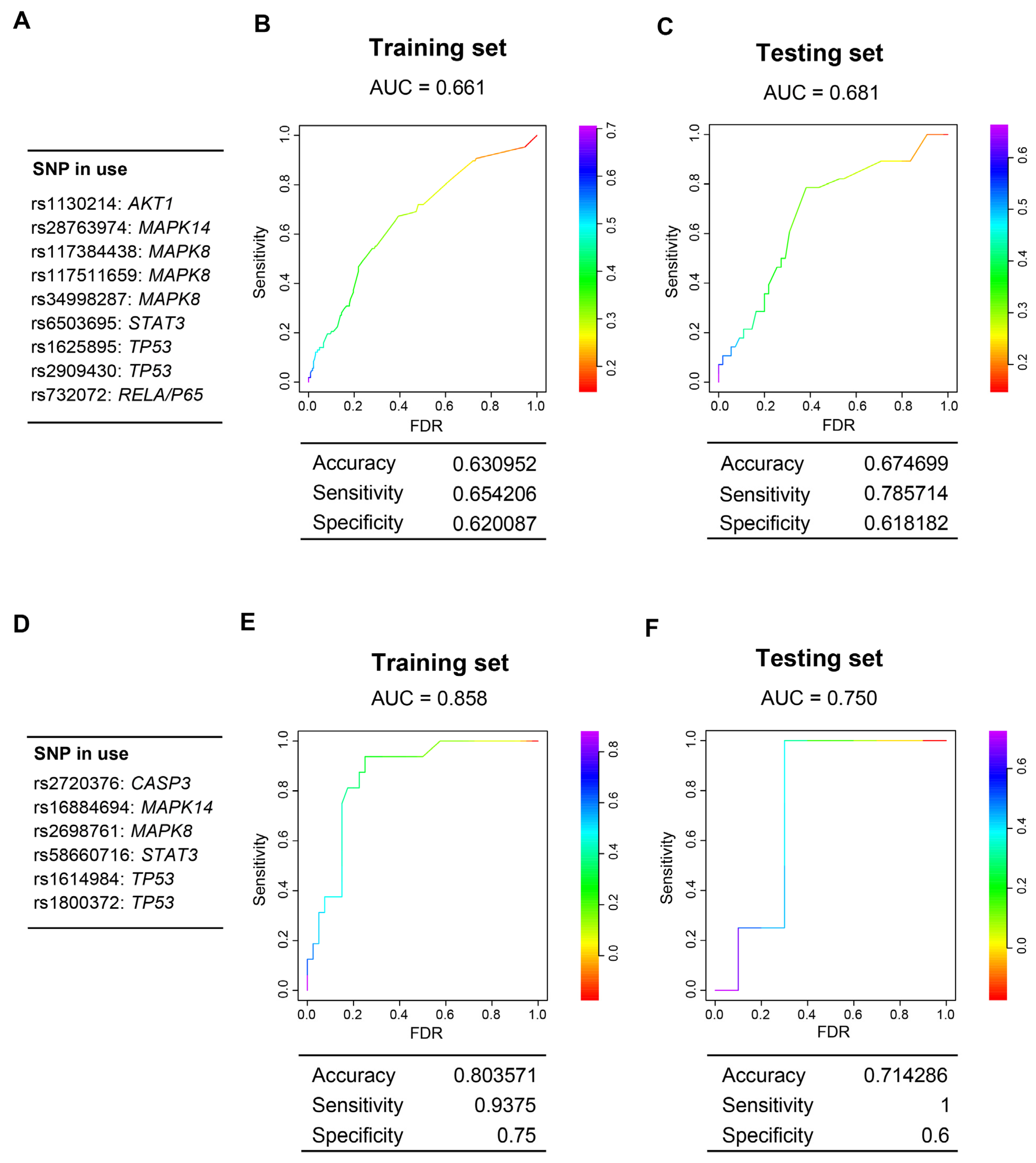
3.6. Genes Encoding Intermediate Molecular Phenotypes Associated with Myocardium Damage in Mice Can Be Genetic Determinants of CDA in Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caron, J.; Nohria, A. Cardiac Toxicity from Breast Cancer Treatment: Can We Avoid This? Curr. Oncol. Rep. 2018, 20, 61. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, J.L.; Byers, T.; DiGuiseppi, C.; Dabelea, D.; Denberg, T.D. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: A retrospective cohort study. Breast Cancer Res. 2011, 13, R64. [Google Scholar] [CrossRef] [PubMed]
- Pein, F.; Sakiroglu, O.; Dahan, M.; Lebidois, J.; Merlet, P.; Shamsaldin, A.; Villain, E.; de Vathaire, F.; Sidi, D.; Hartmann, O. Cardiac abnormalities 15 years and more after adriamycin therapy in 229 childhood survivors of a solid tumour at the Institut Gustave Roussy. Br. J. Cancer 2004, 91, 37–44. [Google Scholar] [CrossRef]
- Mata Caballero, R.; Serrano Antolin, J.M.; Jimenez Hernandez, R.M.; Talavera Calle, P.; Curcio Ruigomez, A.; Del Castillo Arrojo, S.; Graupner Abad, C.; Cristobal Varela, C.; Alonso Martin, J.J. Incidence of long-term cardiotoxicity and evolution of the systolic function in patients with breast cancer treated with anthracyclines. Cardiol. J. 2022, 29, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Grenier, M.A.; Lipshultz, S.E. Epidemiology of anthracycline cardiotoxicity in children and adults. Semin. Oncol. 1998, 25, 72–85. [Google Scholar] [PubMed]
- Kabore, E.G.; Guenancia, C.; Vaz-Luis, I.; Di Meglio, A.; Pistilli, B.; Coutant, C.; Cottu, P.; Lesur, A.; Petit, T.; Dalenc, F.; et al. Association of body mass index and cardiotoxicity related to anthracyclines and trastuzumab in early breast cancer: French CANTO cohort study. PLoS Med. 2019, 16, e1002989. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Leong, S.L.; Chaiyakunapruk, N.; Lee, S.W. Candidate Gene Association Studies of Anthracycline-induced Cardiotoxicity: A Systematic Review and Meta-analysis. Sci. Rep. 2017, 7, 39. [Google Scholar] [CrossRef]
- Duan, S.; Bleibel, W.K.; Huang, R.S.; Shukla, S.J.; Wu, X.; Badner, J.A.; Dolan, M.E. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. 2007, 67, 5425–5433. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Gomez, A.; Castillo-Lluva, S.; Del Mar Saez-Freire, M.; Hontecillas-Prieto, L.; Mao, J.H.; Castellanos-Martin, A.; Perez-Losada, J. Missing heritability of complex diseases: Enlightenment by genetic variants from intermediate phenotypes. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 664–673. [Google Scholar] [CrossRef]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, I.I.; Gould, T.D. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406. [Google Scholar] [CrossRef]
- Wu, B.B.; Leung, K.T.; Poon, E.N. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Martin, A.; Castillo-Lluva, S.; Saez-Freire Mdel, M.; Blanco-Gomez, A.; Hontecillas-Prieto, L.; Patino-Alonso, C.; Galindo-Villardon, P.; Perez Del Villar, L.; Martin-Seisdedos, C.; Isidoro-Garcia, M.; et al. Unraveling heterogeneous susceptibility and the evolution of breast cancer using a systems biology approach. Genome Biol. 2015, 16, 40. [Google Scholar] [CrossRef]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef]
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; Schizophrenia Working Group of Psychiatric Genomics, C.; de Candia, T.R.; Lee, S.H.; Wray, N.R.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kichaev, G.; Pasaniuc, B. Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am. J. Hum. Genet. 2016, 99, 139–153. [Google Scholar] [CrossRef]
- International Schizophrenia, C.; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef]
- Vosa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 2019, 177, 1022–1034.e1026. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W.; Crawford, N.P. The future of mouse QTL mapping to diagnose disease in mice in the age of whole-genome association studies. Annu. Rev. Genet. 2008, 42, 131–141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quigley, D.; Balmain, A. Systems genetics analysis of cancer susceptibility: From mouse models to humans. Nat. Rev. Genet. 2009, 10, 651–657. [Google Scholar] [CrossRef]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [PubMed]
- Mao, J.H.; Perez-Losada, J.; Wu, D.; Delrosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef]
- To, M.D.; Perez-Losada, J.; Mao, J.H.; Hsu, J.; Jacks, T.; Balmain, A. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nat. Genet. 2006, 38, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; To, M.D.; Perez-Losada, J.; Pelorosso, F.G.; Mao, J.H.; Nagase, H.; Ginzinger, D.G.; Balmain, A. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature 2009, 458, 505–508. [Google Scholar] [CrossRef]
- Rottenberg, S.; Nygren, A.O.; Pajic, M.; van Leeuwen, F.W.; van der Heijden, I.; van de Wetering, K.; Liu, X.; de Visser, K.E.; Gilhuijs, K.G.; van Tellingen, O.; et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12117–12122. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lander, E.; Kruglyak, L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995, 11, 241–247. [Google Scholar] [CrossRef]
- Sen, S.; Satagopan, J.M.; Broman, K.W.; Churchill, G.A. R/qtlDesign: Inbred line cross experimental design. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2007, 18, 87–93. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [PubMed]
- Ruiz-Pinto, S.; Pita, G.; Patino-Garcia, A.; Alonso, J.; Perez-Martinez, A.; Carton, A.J.; Gutierrez-Larraya, F.; Alonso, M.R.; Barnes, D.R.; Dennis, J.; et al. Exome array analysis identifies GPR35 as a novel susceptibility gene for anthracycline-induced cardiotoxicity in childhood cancer. Pharmacogenet. Genom. 2017, 27, 445–453. [Google Scholar] [CrossRef]
- Vulsteke, C.; Pfeil, A.M.; Maggen, C.; Schwenkglenks, M.; Pettengell, R.; Szucs, T.D.; Lambrechts, D.; Dieudonne, A.S.; Hatse, S.; Neven, P.; et al. Clinical and genetic risk factors for epirubicin-induced cardiac toxicity in early breast cancer patients. Breast Cancer Res. Treat. 2015, 152, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Barreiro-Perez, M.; Tundidor-Sanz, E.; Martin-Garcia, A.; Diaz-Pelaez, E.; Iscar-Galan, A.; Merchan-Gomez, S.; Gallego-Delgado, M.; Jimenez-Candil, J.; Cruz-Gonzalez, I.; Sanchez, P.L. First Magnetic Resonance Managed by a Cardiology Department in the Spanish Public Healthcare System. Experience and Difficulties of an Innovative Model. Rev. Esp. De Cardiol. 2018, 71, 365–372. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- RC Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Gonzalez, J.R.; Armengol, L.; Sole, X.; Guino, E.; Mercader, J.M.; Estivill, X.; Moreno, V. SNPassoc: An R package to perform whole genome association studies. Bioinformatics 2007, 23, 644–645. [Google Scholar] [CrossRef] [PubMed]
- Balmain, A. Cancer as a complex genetic trait: Tumor susceptibility in humans and mouse models. Cell 2002, 108, 145–152. [Google Scholar] [CrossRef] [PubMed]
- De Laurentiis, M.; Cancello, G.; D’Agostino, D.; Giuliano, M.; Giordano, A.; Montagna, E.; Lauria, R.; Forestieri, V.; Esposito, A.; Silvestro, L.; et al. Taxane-based combinations as adjuvant chemotherapy of early breast cancer: A meta-analysis of randomized trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 44–53. [Google Scholar] [CrossRef]
- Zito, C.; Manganaro, R.; Cusma Piccione, M.; Madonna, R.; Monte, I.; Novo, G.; Mercurio, V.; Longobardo, L.; Cadeddu Dessalvi, C.; Deidda, M.; et al. Anthracyclines and regional myocardial damage in breast cancer patients. A multicentre study from the Working Group on Drug Cardiotoxicity and Cardioprotection, Italian Society of Cardiology (SIC). Eur. Heart J. Cardiovasc. Imaging 2021, 22, 406–415. [Google Scholar] [CrossRef]
- Friedman, M.A.; Bozdech, M.J.; Billingham, M.E.; Rider, A.K. Doxorubicin cardiotoxicity. Serial endomyocardial biopsies and systolic time intervals. JAMA 1978, 240, 1603–1606. [Google Scholar] [CrossRef]
- Billingham, M.E.; Mason, J.W.; Bristow, M.R.; Daniels, J.R. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat. Rep. 1978, 62, 865–872. [Google Scholar] [PubMed]
- Ferreira de Souza, T.; Quinaglia, A.C.S.T.; Osorio Costa, F.; Shah, R.; Neilan, T.G.; Velloso, L.; Nadruz, W.; Brenelli, F.; Sposito, A.C.; Matos-Souza, J.R.; et al. Anthracycline Therapy Is Associated With Cardiomyocyte Atrophy and Preclinical Manifestations of Heart Disease. JACC. Cardiovasc. Imaging 2018, 11, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Goorin, A.M.; Chauvenet, A.R.; Perez-Atayde, A.R.; Cruz, J.; McKone, R.; Lipshultz, S.E. Initial congestive heart failure, six to ten years after doxorubicin chemotherapy for childhood cancer. J. Pediatr. 1990, 116, 144–147. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Colan, S.D.; Gelber, R.D.; Perez-Atayde, A.R.; Sallan, S.E.; Sanders, S.P. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N. Engl. J. Med. 1991, 324, 808–815. [Google Scholar] [CrossRef]
- Piek, A.; de Boer, R.A.; Sillje, H.H. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Segura, A.M.; Radovancevic, R.; Demirozu, Z.T.; Frazier, O.H.; Buja, L.M. Anthracycline treatment and ventricular remodeling in left ventricular assist device patients. Tex. Heart Inst. J. 2015, 42, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F. Q&A: Genetic analysis of quantitative traits. J. Biol. 2009, 8, 23. [Google Scholar] [CrossRef]
- Aapro, M.; Bernard-Marty, C.; Brain, E.G.; Batist, G.; Erdkamp, F.; Krzemieniecki, K.; Leonard, R.; Lluch, A.; Monfardini, S.; Ryberg, M.; et al. Anthracycline cardiotoxicity in the elderly cancer patient: A SIOG expert position paper. Ann. Oncol. 2011, 22, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Salvatorelli, E.; Menna, P.; Cascegna, S.; Liberi, G.; Calafiore, A.M.; Gianni, L.; Minotti, G. Paclitaxel and docetaxel stimulation of doxorubicinol formation in the human heart: Implications for cardiotoxicity of doxorubicin-taxane chemotherapies. J. Pharmacol. Exp. Ther. 2006, 318, 424–433. [Google Scholar] [CrossRef]
- Salvatorelli, E.; Menna, P.; Gianni, L.; Minotti, G. Defective taxane stimulation of epirubicinol formation in the human heart: Insight into the cardiac tolerability of epirubicin-taxane chemotherapies. J. Pharmacol. Exp. Ther. 2007, 320, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar] [CrossRef]
- Ichihara, S.; Yamada, Y.; Kawai, Y.; Osawa, T.; Furuhashi, K.; Duan, Z.; Ichihara, G. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochem. Biophys. Res. Commun. 2007, 359, 27–33. [Google Scholar] [CrossRef]
- Kang, Y.J.; Zhou, Z.X.; Wang, G.W.; Buridi, A.; Klein, J.B. Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J. Biol. Chem. 2000, 275, 13690–13698. [Google Scholar]
- Ghigo, A.; Li, M.; Hirsch, E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta 2016, 1863, 1916–1925. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffre, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart Fail. Rev. 2018, 23, 109–122. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Marino, L.; Filippelli, A.; Berrino, L.; Ferreira-Martins, J.; Zheng, H.; Hosoda, T.; Rota, M.; et al. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation 2010, 121, 276–292. [Google Scholar] [CrossRef]
- Xu, X.; Chen, K.; Kobayashi, S.; Timm, D.; Liang, Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J. Pharmacol. Exp. Ther. 2012, 341, 183–195. [Google Scholar] [CrossRef]
- Yu, S.Y.; Liu, L.; Li, P.; Li, J. Rapamycin inhibits the mTOR/p70S6K pathway and attenuates cardiac fibrosis in adriamycin-induced dilated cardiomyopathy. Thorac. Cardiovasc. Surg. 2013, 61, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Oh, J.; Kang, S.K.; Park, S.; Lee, S.H.; Choi, D.; Chung, J.H.; Chung, Y.W.; Kang, S.M. Insulin Protects Cardiac Myocytes from Doxorubicin Toxicity by Sp1-Mediated Transactivation of Survivin. PLoS ONE 2015, 10, e0135438. [Google Scholar] [CrossRef]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; Del Alamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef]
- Buchner, D.A.; Nadeau, J.H. Contrasting genetic architectures in different mouse reference populations used for studying complex traits. Genome Res. 2015, 25, 775–791. [Google Scholar] [CrossRef]
- McOwan, T.N.; Craig, L.A.; Tripdayonis, A.; Karavendzas, K.; Cheung, M.M.; Porrello, E.R.; Conyers, R.; Elliott, D.A. Evaluating anthracycline cardiotoxicity associated single nucleotide polymorphisms in a paediatric cohort with early onset cardiomyopathy. Cardio-Oncology 2020, 6, 5. [Google Scholar] [CrossRef]
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 2013, 14, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Gatti, D.M.; Toro-Salazar, O.; Davis, C.; Lutz, C.M.; Spinale, F.; Stearns, T.; Furtado, M.B.; Churchill, G.A. Doxorubicin-Induced Cardiotoxicity in Collaborative Cross (CC) Mice Recapitulates Individual Cardiotoxicity in Humans. G3 2019, 9, 2637–2646. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) QTLs and CDA Conditions Used in the Genetic Models | |||||
|---|---|---|---|---|---|
| cda-QTL | Therapy Type | Mouse Age Group | Pathophenotype of CDA | (a) Molecular Intermediate Phenotypes Associated with Global Fibrosis | (b) ipQTLs |
| cda-QTL6 | Doxorubicin | Old Mice | Global Fibrosis | γH2AX(S139) | dox-ipQTL76 |
| dox-ipQTL77 | |||||
| dox-ipQTL78 | |||||
| JNK(T183/Y185) | dox-ipQTL81 | ||||
| miR200b-3p | N.I. | ||||
| cda-QTL13 | Doxorubicin | Young Mice | Global Fibrosis | p70S6K(T412) | dox-ipQTL51 |
| dox-ipQTL52 | |||||
| dox-ipQTL53 | |||||
| dox-ipQTL54 | |||||
| Combined Therapy | Young Mice | Global Fibrosis | CREB(S133) | comb-ipQTL29 | |
| (B) Improvement of the Global Fibrosis Variation Explained by cda-QTL6 and cda-QTL13 with ipQTLs in Genetics Models | |||||
| Basal Effect/Model Effect | Model Components | LOD Score | Fibrosis Variation (%) | p-Value | |
| cda-QTL13 | Basal effect | n.a. | 2.38 | 28.82 | n.a. |
| Models with cda-QTL13 in young mice | Model 11 | cda-QTL13; dox-ipQTL51 | 3.78 | 47.55 | 0.0006 |
| Model 17 | cda-QTL13; dox-ipQTL51; dox-ipQTL54 | 4.99 | 57.31 | 0.002 | |
| Model 18 | cda-QTL13; dox-ipQTL51; dox-ipQTL53 | 6.3 | 65.90 | 0.0001 | |
| cda-QTL13 | Basal effect | n.a. | 2.29 | 22.17 | n.a. |
| Models with cda-QTL6 in old mice | Model 5 | cda-QTL6; dox-ipQTL76; dox-ipQTL78 | 6.63 | 50.84 | 0.00007 |
| Model 7 | cda-QTL6; dox-ipQTL77; dox-ipQTL78 | 4.8 | 40.20 | 0.0024 | |
| Model 9 | cda-QTL6; dox-ipQTL78; dox-ipQTL81 | 4.2 | 36.20 | 0.0075 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Vecino, A.; Corchado-Cobos, R.; Blanco-Gómez, A.; García-Sancha, N.; Castillo-Lluva, S.; Martín-García, A.; Mendiburu-Eliçabe, M.; Prieto, C.; Ruiz-Pinto, S.; Pita, G.; et al. Intermediate Molecular Phenotypes to Identify Genetic Markers of Anthracycline-Induced Cardiotoxicity Risk. Cells 2023, 12, 1956. https://doi.org/10.3390/cells12151956
Gómez-Vecino A, Corchado-Cobos R, Blanco-Gómez A, García-Sancha N, Castillo-Lluva S, Martín-García A, Mendiburu-Eliçabe M, Prieto C, Ruiz-Pinto S, Pita G, et al. Intermediate Molecular Phenotypes to Identify Genetic Markers of Anthracycline-Induced Cardiotoxicity Risk. Cells. 2023; 12(15):1956. https://doi.org/10.3390/cells12151956
Chicago/Turabian StyleGómez-Vecino, Aurora, Roberto Corchado-Cobos, Adrián Blanco-Gómez, Natalia García-Sancha, Sonia Castillo-Lluva, Ana Martín-García, Marina Mendiburu-Eliçabe, Carlos Prieto, Sara Ruiz-Pinto, Guillermo Pita, and et al. 2023. "Intermediate Molecular Phenotypes to Identify Genetic Markers of Anthracycline-Induced Cardiotoxicity Risk" Cells 12, no. 15: 1956. https://doi.org/10.3390/cells12151956
APA StyleGómez-Vecino, A., Corchado-Cobos, R., Blanco-Gómez, A., García-Sancha, N., Castillo-Lluva, S., Martín-García, A., Mendiburu-Eliçabe, M., Prieto, C., Ruiz-Pinto, S., Pita, G., Velasco-Ruiz, A., Patino-Alonso, C., Galindo-Villardón, P., Vera-Pedrosa, M. L., Jalife, J., Mao, J.-H., Macías de Plasencia, G., Castellanos-Martín, A., Sáez-Freire, M. d. M., ... Pérez Losada, J. (2023). Intermediate Molecular Phenotypes to Identify Genetic Markers of Anthracycline-Induced Cardiotoxicity Risk. Cells, 12(15), 1956. https://doi.org/10.3390/cells12151956