



Stimulator of Interferon Genes (STING) Triggers Adipocyte Autophagy


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals and Cells
2.2. Human Samples
2.3. Cell Treatment
2.4. Imaging of Mitochondria
2.5. Autophagy Assays
2.6. mRNA Analysis
2.7. Histology, Immunofluorescence and Flow Cytometry
2.8. Western Blotting
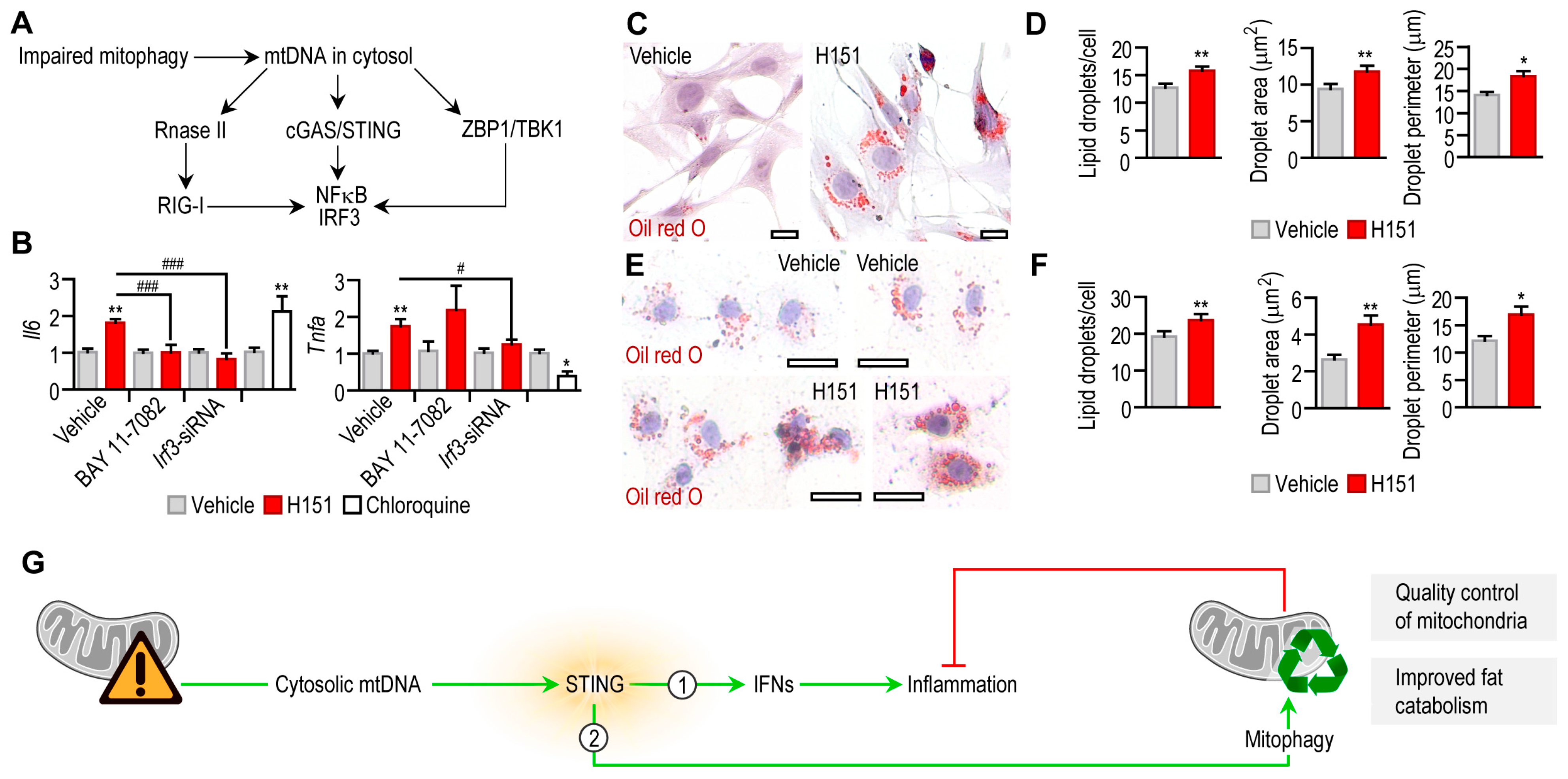
3. Results
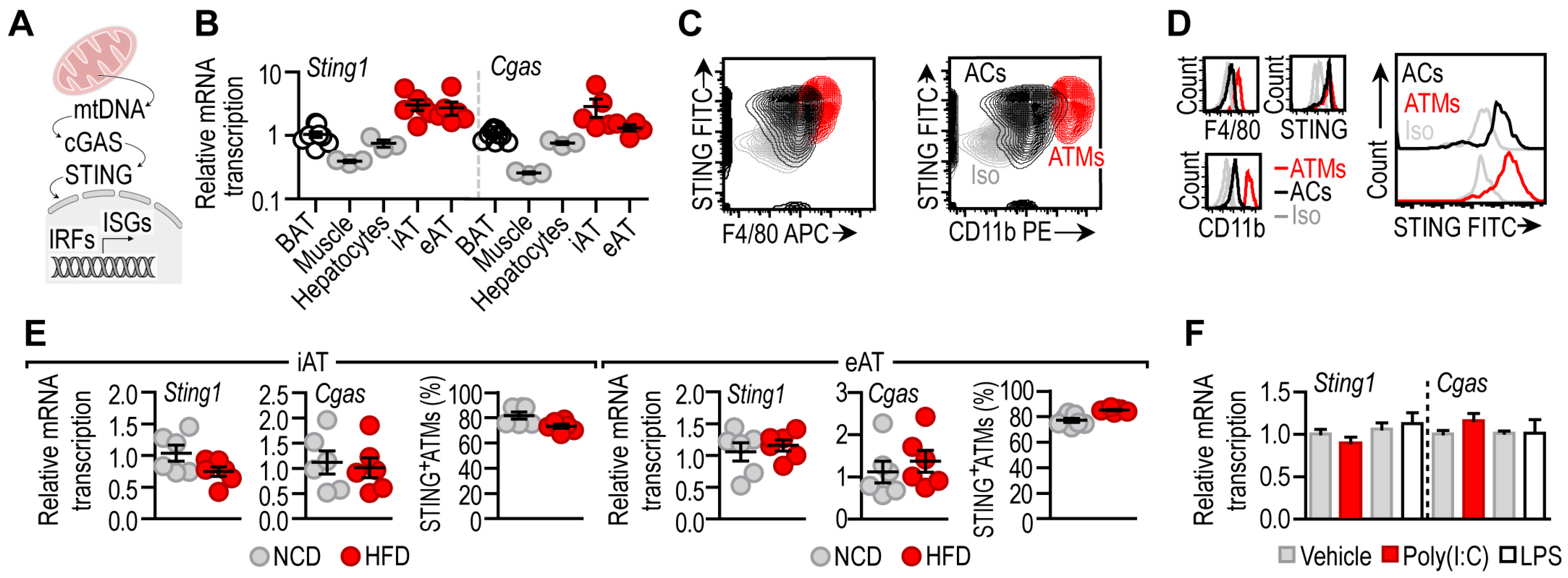
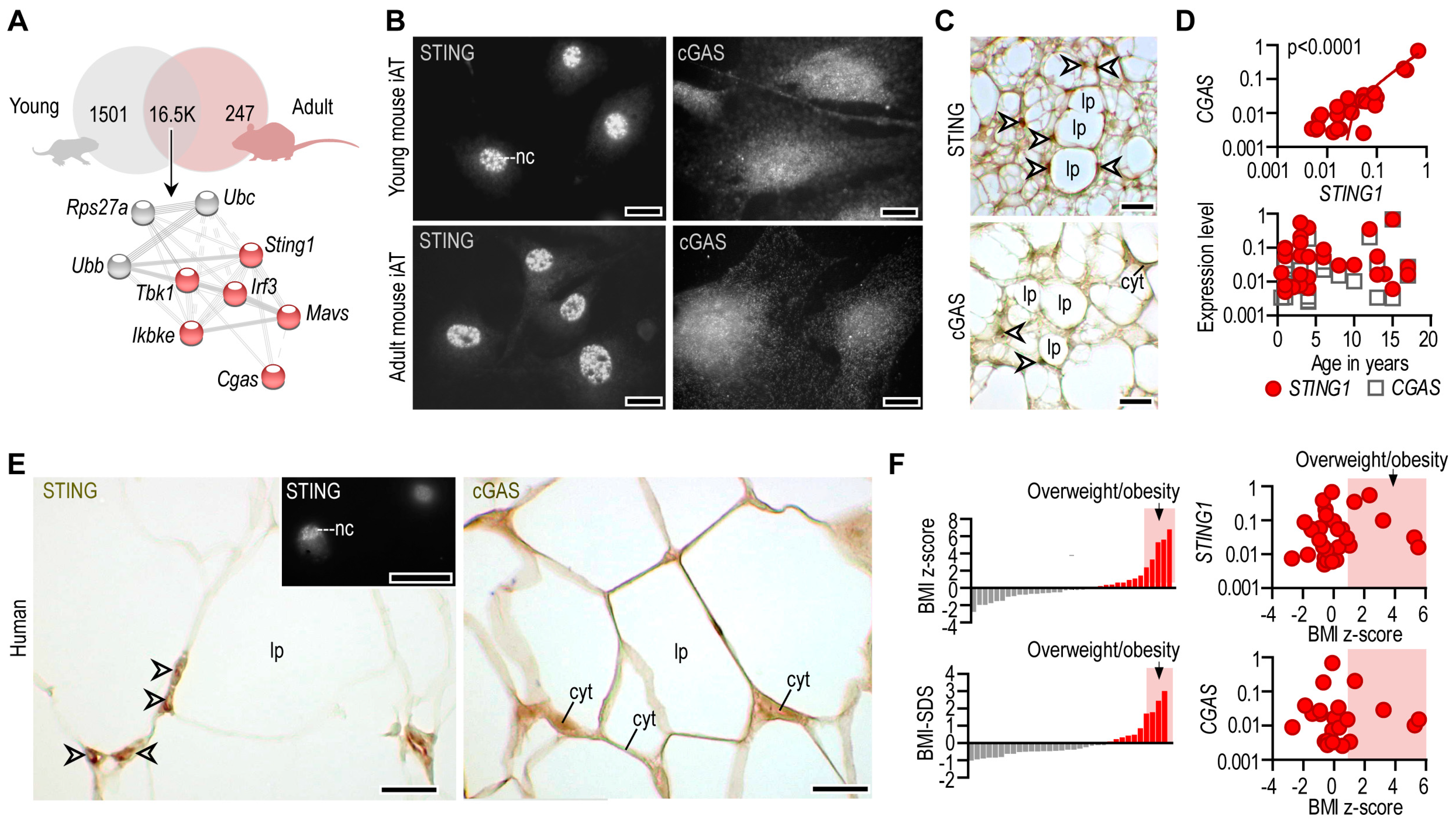
3.1. STING Is Constitutively Expressed in Adipocytes
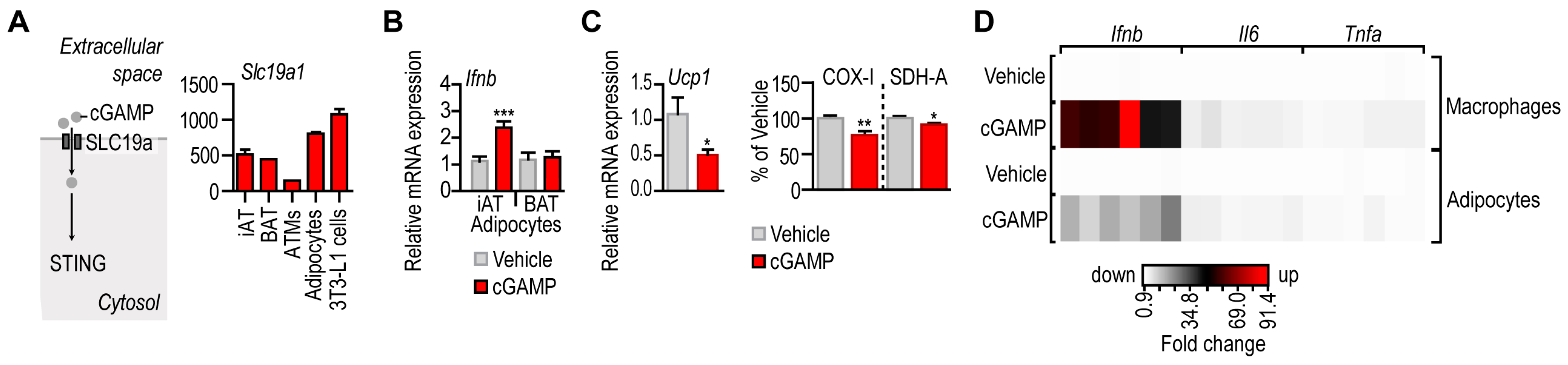
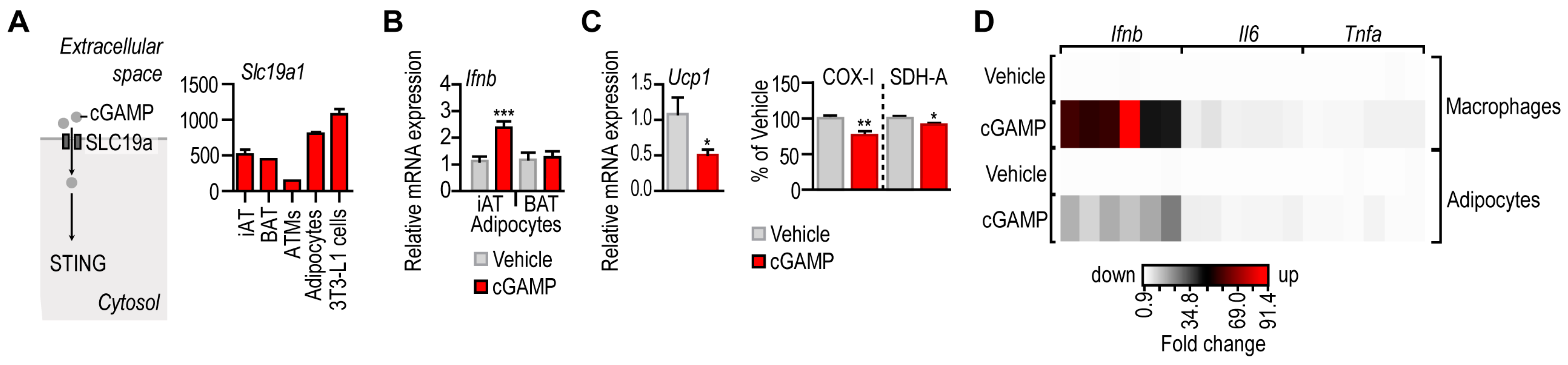
3.2. IFN-I Response following STING Activation in Adipocytes
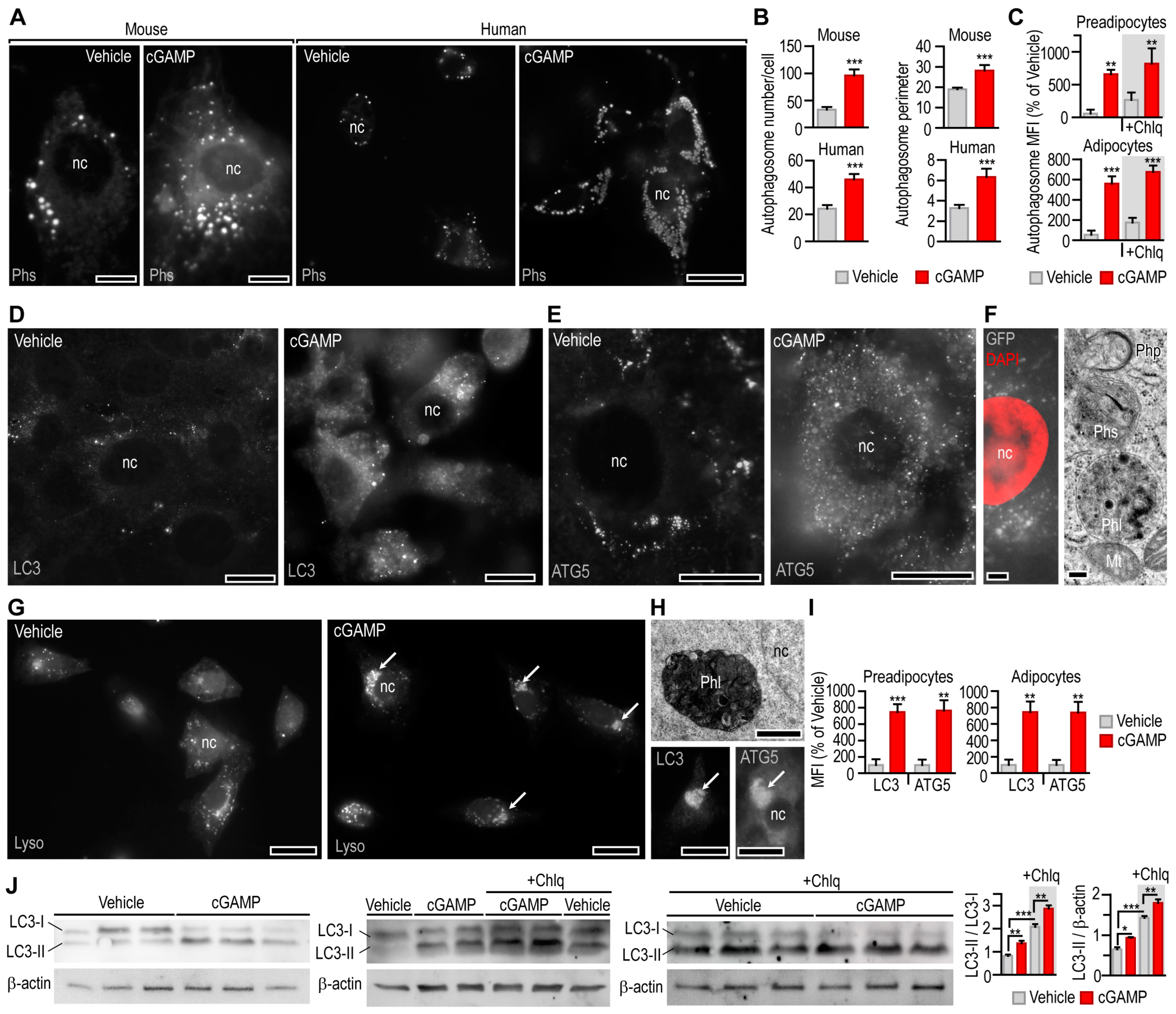
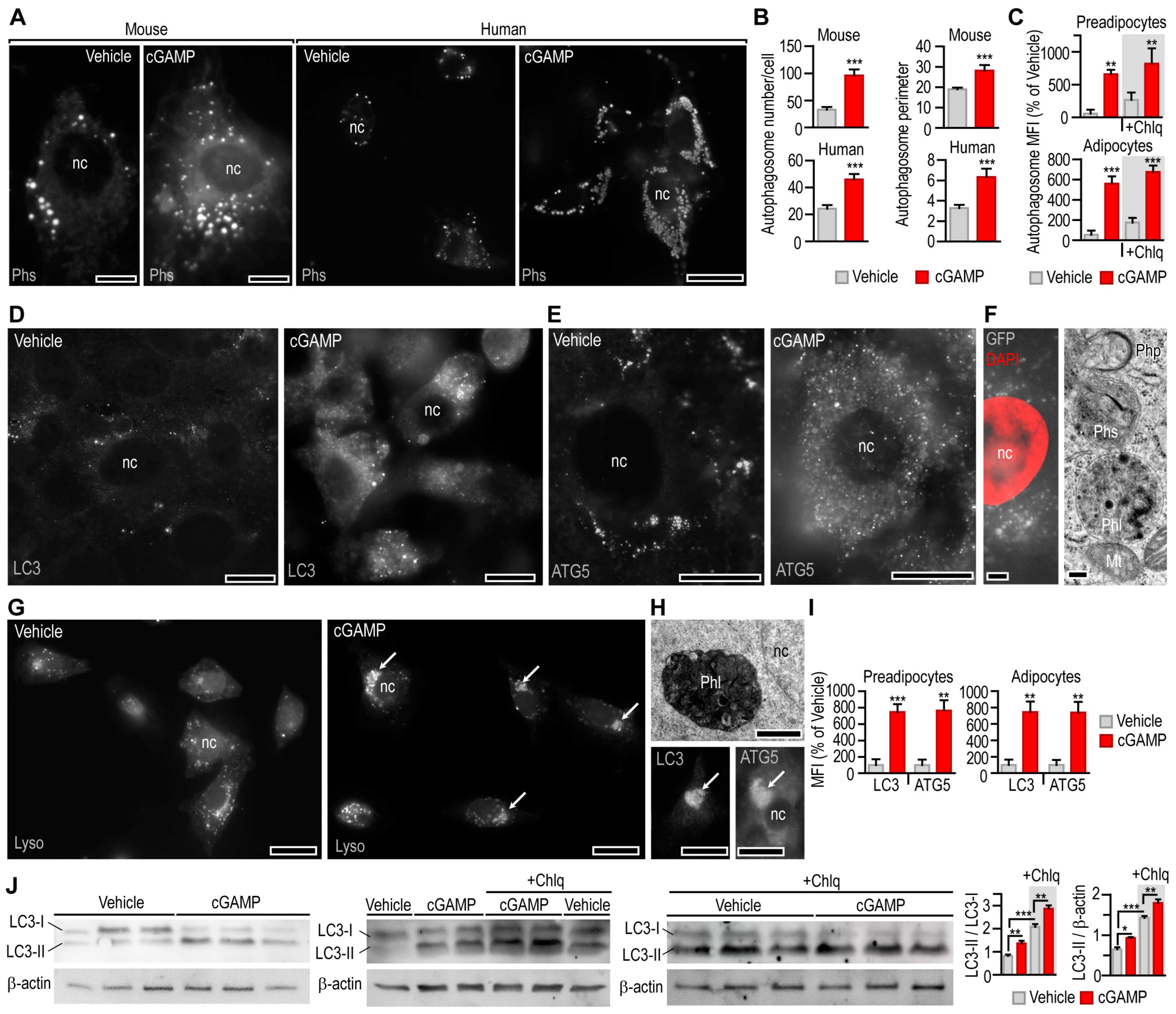
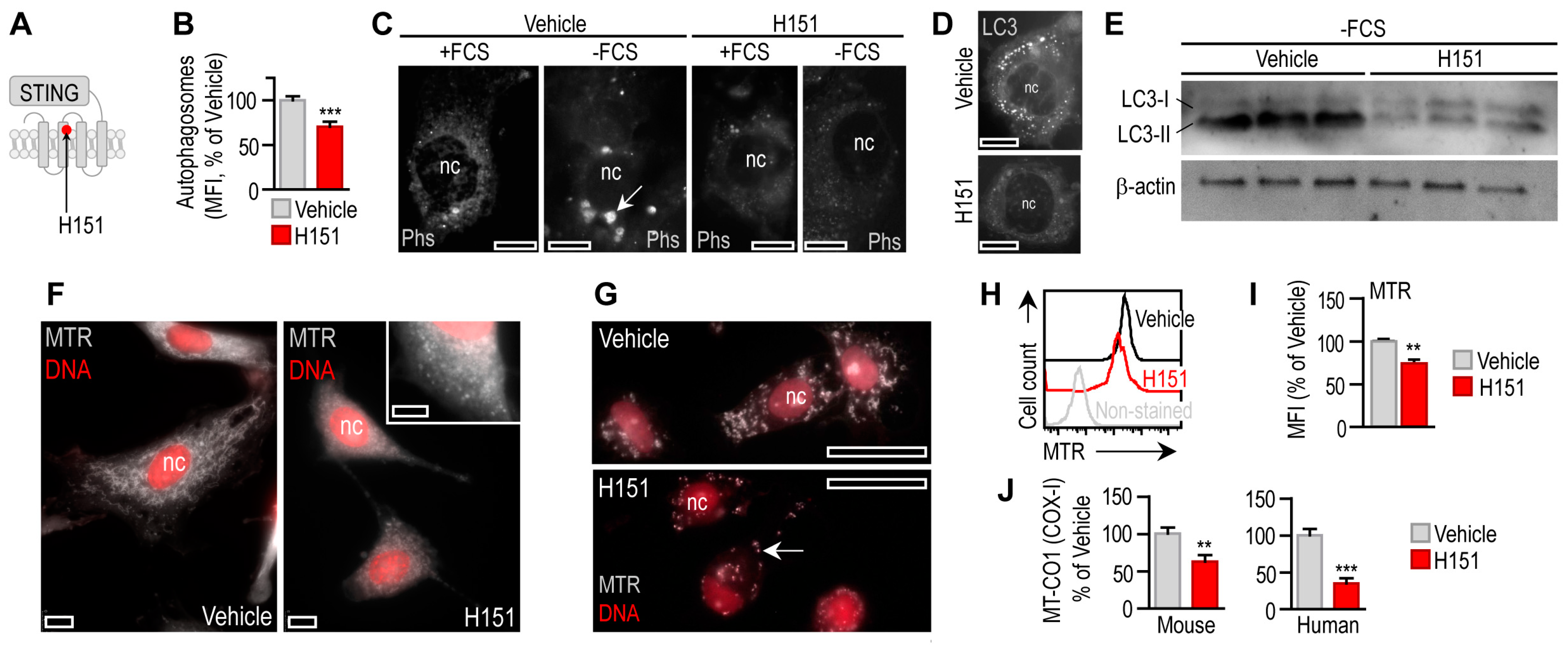
3.3. STING Activation Triggers Mitophagy in Adipocytes
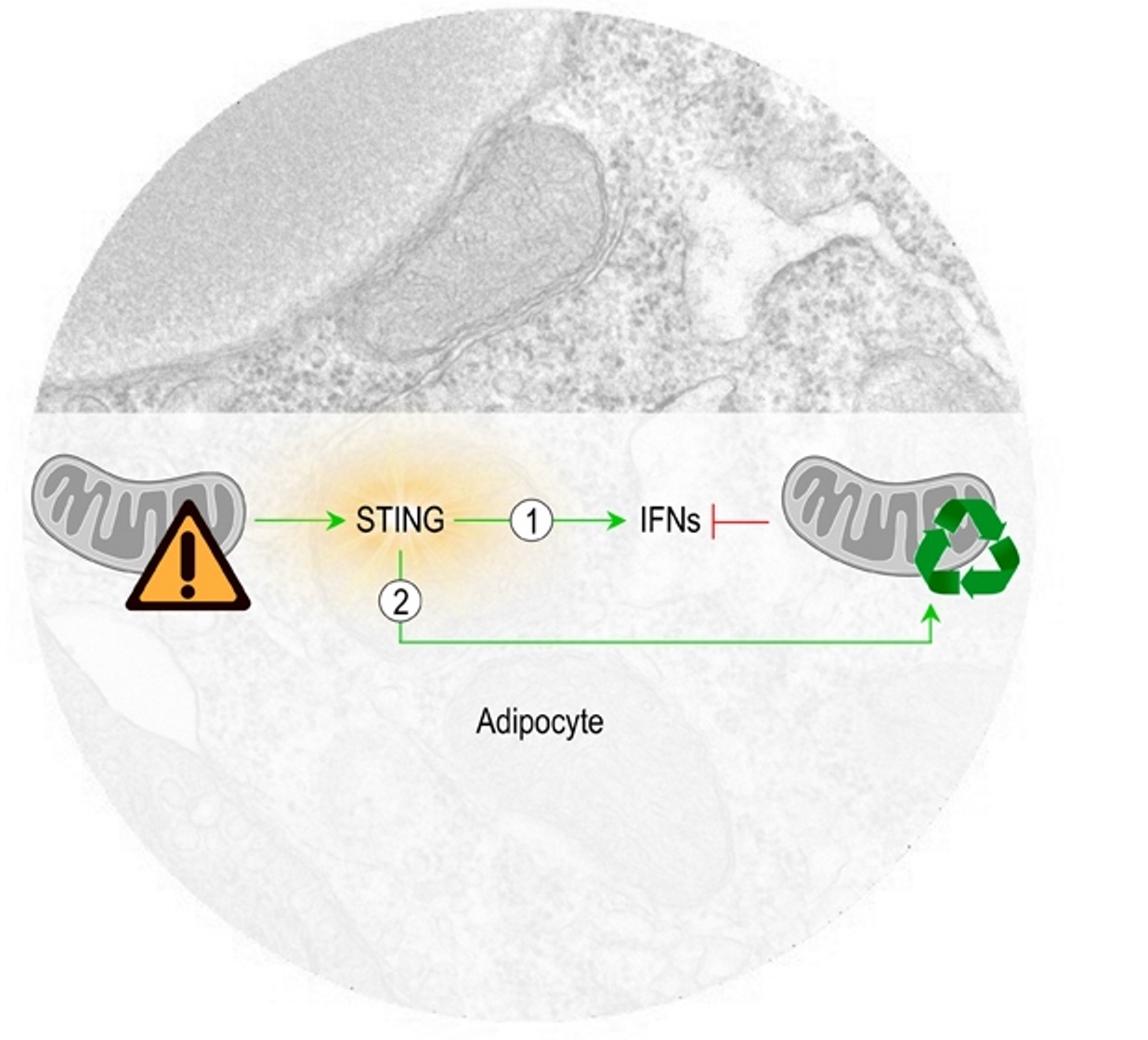
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef]
- Wieser, V.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Moser, P.; Moschen, A.R.; Kaser, S.; Tilg, H. Adipose type I interferon signalling protects against metabolic dysfunction. Gut 2018, 67, 157–165. [Google Scholar] [CrossRef]
- Birk, R.Z.; Rubinstein, M. IFN-alpha induces apoptosis of adipose tissue cells. Biochem. Biophys. Res. Commun. 2006, 345, 669–674. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.R.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef]
- Kissig, M.; Ishibashi, J.; Harms, M.J.; Lim, H.-W.; Stine, R.R.; Won, K.-J.; Seale, P. PRDM16 represses the type I interferon response in adipocytes to promote mitochondrial and thermogenic programing. EMBO J. 2017, 36, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.C.; Damen, M.S.M.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Alarcon, P.C.; Oates, J.R.; Doll, J.R.; Mukherjee, R.; Chen, X.; et al. Type I interferon sensing unlocks dormant adipocyte inflammatory potential. Nat. Commun. 2020, 11, 2745. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Allenbach, Y.; Grelet, E.; Ueberschlag, V.; Echaniz-Laguna, A.; Lannes, B.; Alsaleh, G.; Charles, A.L.; Singh, F.; et al. IFN-β-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017, 134, 655–666. [Google Scholar] [CrossRef]
- Derecka, M.; Gornicka, A.; Koralov, S.B.; Szczepanek, K.; Morgan, M.; Raje, V.; Sisler, J.; Zhang, Q.; Otero, D.; Cichy, J.; et al. Tyk2 and Stat3 Regulate Brown Adipose Tissue Differentiation and Obesity. Cell Metab. 2012, 16, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dilbaz, S.; Coßmann, J.; Hoang, A.C.; Diedrich, V.; Herwig, A.; Harauma, A.; Hoshi, Y.; Moriguchi, T.; Landgraf, K.; et al. Breast milk alkylglycerols sustain beige adipocytes through adipose tissue macrophages. J. Clin. Investig. 2019, 129, 2485–2499. [Google Scholar] [CrossRef]
- Babaei, R.; Schuster, M.; Meln, I.; Lerch, S.; Ghandour, R.A.; Pisani, D.F.; Bayindir-Buchhalter, I.; Marx, J.; Wu, S.; Schoiswohl, G.; et al. Jak-TGFβ cross-talk links transient adipose tissue inflammation to beige adipogenesis. Sci. Signal. 2018, 11, eaai7838. [Google Scholar] [CrossRef]
- Asterholm, I.W.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte Inflammation Is Essential for Healthy Adipose Tissue Expansion and Remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef]
- Alsaggar, M.; Mills, M.; Liu, D. Interferon beta overexpression attenuates adipose tissue inflammation and high-fat diet-induced obesity and maintains glucose homeostasis. Gene Ther. 2017, 24, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 Mitogen-Activated Protein Kinase Is the Central Regulator of Cyclic AMP-Dependent Transcription of the Brown Fat Uncoupling Protein 1 Gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. The cGAS-cGAMP-STING Pathway: A Molecular Link between Immunity and Metabolism. Diabetes 2019, 68, 1099–1108. [Google Scholar] [CrossRef]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.-S.; Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cervantes, C.; He, S.; He, J.; Plasko, G.R.; Wen, J.; Li, Z.; Yin, D.; Zhang, C.; Liu, M.; et al. Mitochondrial stress-activated cGAS-STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun. Biol. 2020, 3, 257. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. cGAS–STING signaling and function in metabolism and kidney diseases. J. Mol. Cell Biol. 2021, 13, 728–738. [Google Scholar] [CrossRef]
- Guo, X.; Shu, C.; Li, H.; Pei, Y.; Woo, S.-L.; Zheng, J.; Liu, M.; Xu, H.; Botchlett, R.; Guo, T.; et al. Cyclic GMP-AMP Ameliorates Diet-induced Metabolic Dysregulation and Regulates Proinflammatory Responses Distinctly from STING Activation. Sci. Rep. 2017, 7, 6355. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Goldstein, I. Protocol for Primary Mouse Hepatocyte Isolation. STAR Protoc. 2020, 1, 100086. [Google Scholar] [CrossRef] [PubMed]
- Keire, P.; Shearer, A.; Shefer, G.; Yablonka-Reuveni, Z. Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol. Biol. 2013, 946, 431–468. [Google Scholar]
- Hoang, A.C.; Sasi-Szabó, L.; Pál, T.; Szabó, T.; Diedrich, V.; Herwig, A.; Landgraf, K.; Körner, A.; Röszer, T. Mitochondrial RNA stimulates beige adipocyte development in young mice. Nat. Metab. 2022, 4, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Must, A.; Anderson, S.E. Body mass index in children and adolescents: Considerations for population-based applications. Int. J. Obes. 2006, 30, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Geserick, M.; Vogel, M.; Gausche, R.; Lipek, T.; Spielau, U.; Keller, E.; Pfäffle, R.; Kiess, W.; Körner, A. Acceleration of BMI in Early Childhood and Risk of Sustained Obesity. N. Engl. J. Med. 2018, 379, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Hoang, A.C.; Yu, H.; Röszer, T. Transcriptional Landscaping Identifies a Beige Adipocyte Depot in the Newborn Mouse. Cells 2021, 10, 2368. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Butler, T.A.J.; Paul, J.W.; Chan, E.C.; Smith, R.; Tolosa, J.M. Misleading Westerns: Common Quantification Mistakes in Western Blot Densitometry and Proposed Corrective Measures. Biomed. Res. Int. 2019, 2019, 5214821. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium, Overall Coordination, Logistical Coordination, Organ Collection and Processing, Library Preparation and Sequencing, Computational Data Analysis, Cell Type Annotation, Writing Group, Supplemental Text Writing Group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, R.X.; Xu, H.G. STING and liver disease. J. Gastroenterol. 2021, 56, 704–712. [Google Scholar] [CrossRef]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381.e375. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Song, Y.; Zhou, Y.; Zhou, X. The role of mitophagy in innate immune responses triggered by mitochondrial stress. Cell Commun. Signal. 2020, 18, 186. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Deen, N.; Zamani, S.; Hasnat, M.A. Mitophagy and the release of inflammatory cytokines. Mitochondrion 2018, 41, 2–8. [Google Scholar] [CrossRef]
- Kwon, D.; Park, E.; Kang, S.-J. Stimulator of IFN genes–mediated DNA-sensing pathway is suppressed by NLRP3 agonists and regulated by mitofusin 1 and TBC1D15, mitochondrial dynamics mediators. FASEB J. 2017, 31, 4866–4878. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, H.; Wang, C.; Li, Y.; Tian, H.; Siraj, S.; Sehgal, S.A.; Wang, X.; Wang, J.; Shang, Y.; et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019, 26, 1735–1749. [Google Scholar] [CrossRef]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, bio038521. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Deretic, V., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 77–88. [Google Scholar]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, M.; Karim, M.R. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzym. 2009, 452, 199–213. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef]
- Oliva, A.; Meroño, C.; Traba, J. Mitochondrial function and dysfunction in innate immunity. Curr. Opin. Physiol. 2022, 28, 100571. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front. Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef]
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci. Rep. 2015, 5, 15701. [Google Scholar] [CrossRef]
- Jang, C.-H.; Choi, J.-H.; Byun, M.-S.; Jue, D.-M. Chloroquine inhibits production of TNF-α, IL-1β and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology 2006, 45, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [PubMed]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef]
- Gkirtzimanaki, K.; Kabrani, E.; Nikoleri, D.; Polyzos, A.; Blanas, A.; Sidiropoulos, P.; Makrigiannakis, A.; Bertsias, G.; Boumpas, D.T.; Verginis, P. IFNα Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018, 25, 921–933.e5. [Google Scholar] [CrossRef]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef]
- Harkins, J.M.; Moustaid-Moussa, N.; Chung, Y.-J.; Penner, K.M.; Pestka, J.J.; North, C.M.; Claycombe, K.J. Expression of Interleukin-6 Is Greater in Preadipocytes than in Adipocytes of 3T3-L1 Cells and C57BL/6J and ob/ob Mice. J. Nutr. 2004, 134, 2673–2677. [Google Scholar]
- Jing, X.; Wu, J.; Dong, C.; Gao, J.; Seki, T.; Kim, C.; Urgard, E.; Hosaka, K.; Yang, Y.; Long, S.; et al. COVID-19 instigates adipose browning and atrophy through VEGF in small mammals. Nat. Metab. 2022, 4, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Molocea, C.-E.; Tsokanos, F.-F.; Herzig, S. Exploiting common aspects of obesity and cancer cachexia for future therapeutic strategies. Curr. Opin. Pharmacol. 2020, 53, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Röszer, T. Adipose Tissue Immunometabolism and Apoptotic Cell Clearance. Cells 2021, 10, 2288. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Ahmad, A.; Montalbano, M.; Brunyánszki, A.; Bibli, S.-I.; Papapetropoulos, A.; Szabo, C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, S.; Sun, Y.; Li, L.; Ding, S.; Wang, X. When STING Meets Viruses: Sensing, Trafficking and Response. Front. Immunol. 2020, 11, 2064. [Google Scholar] [CrossRef]
- Rong, Y.; Zhang, S.; Nandi, N.; Wu, Z.; Li, L.; Liu, Y.; Wei, Y.; Zhao, Y.; Yuan, W.; Zhou, C.; et al. STING controls energy stress-induced autophagy and energy metabolism via STX17. J. Cell Biol. 2022, 221. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Wan, W.; Qian, C.; Wang, Q.; Li, J.; Zhang, H.; Wang, L.; Pu, M.; Huang, Y.; He, Z.; Zhou, T.; et al. STING directly recruits WIPI2 for autophagosome formation during STING-induced autophagy. EMBO J. 2023, 42, e112387. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Bárcena, C.; Martínez-García, G.G.; Tamargo-Gómez, I.; Suárez, M.F.; Pietrocola, F.; Castoldi, F.; Esteban, L.; Sierra-Filardi, E.; Boya, P.; et al. Autophagy couteracts weight gain, lipotoxicity and pancreatic β-cell death upon hypercaloric pro-diabetic regimens. Cell Death Dis. 2017, 8, e2970. [Google Scholar] [CrossRef]
- MacFarlane Burnet, F.; White, D.O. Natural History of Infectious Disease; Cambridge University Press: Cambridge, UK, 1971. [Google Scholar]
- Röszer, T. Co-Evolution of Breast Milk Lipid Signaling and Thermogenic Adipose Tissue. Biomolecules 2021, 11, 1705. [Google Scholar] [CrossRef]
- Bian, X.; Wallstrom, G.; Davis, A.; Wang, J.; Park, J.; Throop, A.; Steel, J.; Yu, X.; Wasserfall, C.; Schatz, D.; et al. Immunoproteomic Profiling of Antiviral Antibodies in New-Onset Type 1 Diabetes Using Protein Arrays. Diabetes 2015, 65, 285–296. [Google Scholar] [CrossRef]
- Alba, A.; Puertas, M.C.; Carrillo, J.; Planas, R.; Ampudia, R.; Pastor, X.; Bosch, F.; Pujol-Borrell, R.; Verdaguer, J.; Vives-Pi, M. IFNβ Accelerates Autoimmune Type 1 Diabetes in Nonobese Diabetic Mice and Breaks the Tolerance to β Cells in Nondiabetes-Prone Mice. J. Immunol. 2004, 173, 6667–6675. [Google Scholar] [CrossRef]
- Assmann, T.S.; Brondani Lde, A.; Boucas, A.P.; Canani, L.H.; Crispim, D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.-M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Basu, T.; Kim, C. Lower Incidence Rate of Type 1 Diabetes after Receipt of the Rotavirus Vaccine in the United States, 2001–2017. Sci. Rep. 2019, 9, 7727. [Google Scholar] [CrossRef]
- Wu, X.; Yang, J.; Na, T.; Zhang, K.; Davidoff, A.M.; Yuan, B.-Z.; Wang, Y. RIG-I and IL-6 are negative-feedback regulators of STING induced by double-stranded DNA. PLoS ONE 2017, 12, e0182961. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, A.; Yoshinaga, M.; Takeuchi, O. TANK prevents IFN-dependent fatal diffuse alveolar hemorrhage by suppressing DNA-cGAS aggregation. Life Sci. Alliance 2022, 5, e202101067. [Google Scholar] [CrossRef]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. Autophagy in autoimmune disease. J. Mol. Med. 2015, 93, 707–717. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Actinb | fw | GCACCAGGGTGTGATGGTG |
| rev | CCAGATCTTCTCCATGTCGTCC | |
| Ppia | fw | ATTTCTTTTGACTTGCGGGC |
| rev | AGACTTGAAGGGGAATG | |
| Gapdh | fw | TGACGTGCCGCCTGGAGAAA |
| rev | AGTGTAGCCCAAGATGCCCTTCAG | |
| Cgas/Mb21d1 | fw | AGGAAGCCCTGCTGTAACACTTCT |
| rev | AGCCAGCCTTGAATAGGTAGGTAGTCCT | |
| Sting1/Tmem173 | fw | GGGCCCTGTCACTTTTGGTC |
| rev | GAGTATGGCATCAGCAGCCAC | |
| Il6 | fw | GCTACCAAACTGGATATAATCAGGA |
| rev | CCAGGTAGCTATGGTACTCCAGAA | |
| Tnfa | fw | TGCCTATGTCTCAGCCTCTTC |
| rev | GAGGCCATTTGGGAACTTCT | |
| Ifnb | fw | CCAGCTCCAAGAAAGGACGA |
| rev | CGCCCTGTAGGTGAGGTTGAT | |
| CGAS | fw | CATGGCGGCTATCCTTCTCT |
| rev | AAAGCAGAGGCCCAGGTCTT | |
| STING1 | fw | ATATCTGCGGCTGATCCTGC |
| rev | GGTCTGCTGGGGCAGTTTAT | |
| GAPDH | fw | GTCTCCTCTGACTTCAACAGCG |
| rev | ACCACCCTGTTGCTGTAGCCAA | |
| ACTINB | fw | CACCATTGGCAATGAGCGGTTC |
| rev | AGGTCTTTGCGGATGTCCACGT | |
| TrnQ | fw | GATGTCAGAGGGGTGCCTTG |
| rev | AACCCTCGTTCCACAGAAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varga, K.Z.; Gyurina, K.; Radványi, Á.; Pál, T.; Sasi-Szabó, L.; Yu, H.; Felszeghy, E.; Szabó, T.; Röszer, T. Stimulator of Interferon Genes (STING) Triggers Adipocyte Autophagy. Cells 2023, 12, 2345. https://doi.org/10.3390/cells12192345
Varga KZ, Gyurina K, Radványi Á, Pál T, Sasi-Szabó L, Yu H, Felszeghy E, Szabó T, Röszer T. Stimulator of Interferon Genes (STING) Triggers Adipocyte Autophagy. Cells. 2023; 12(19):2345. https://doi.org/10.3390/cells12192345
Chicago/Turabian StyleVarga, Kornél Z., Katalin Gyurina, Ádám Radványi, Tibor Pál, László Sasi-Szabó, Haidong Yu, Enikő Felszeghy, Tamás Szabó, and Tamás Röszer. 2023. "Stimulator of Interferon Genes (STING) Triggers Adipocyte Autophagy" Cells 12, no. 19: 2345. https://doi.org/10.3390/cells12192345
APA StyleVarga, K. Z., Gyurina, K., Radványi, Á., Pál, T., Sasi-Szabó, L., Yu, H., Felszeghy, E., Szabó, T., & Röszer, T. (2023). Stimulator of Interferon Genes (STING) Triggers Adipocyte Autophagy. Cells, 12(19), 2345. https://doi.org/10.3390/cells12192345