
To Ubiquitinate or Not to Ubiquitinate: TRIM17 in Cell Life and Death



Abstract
1. Introduction

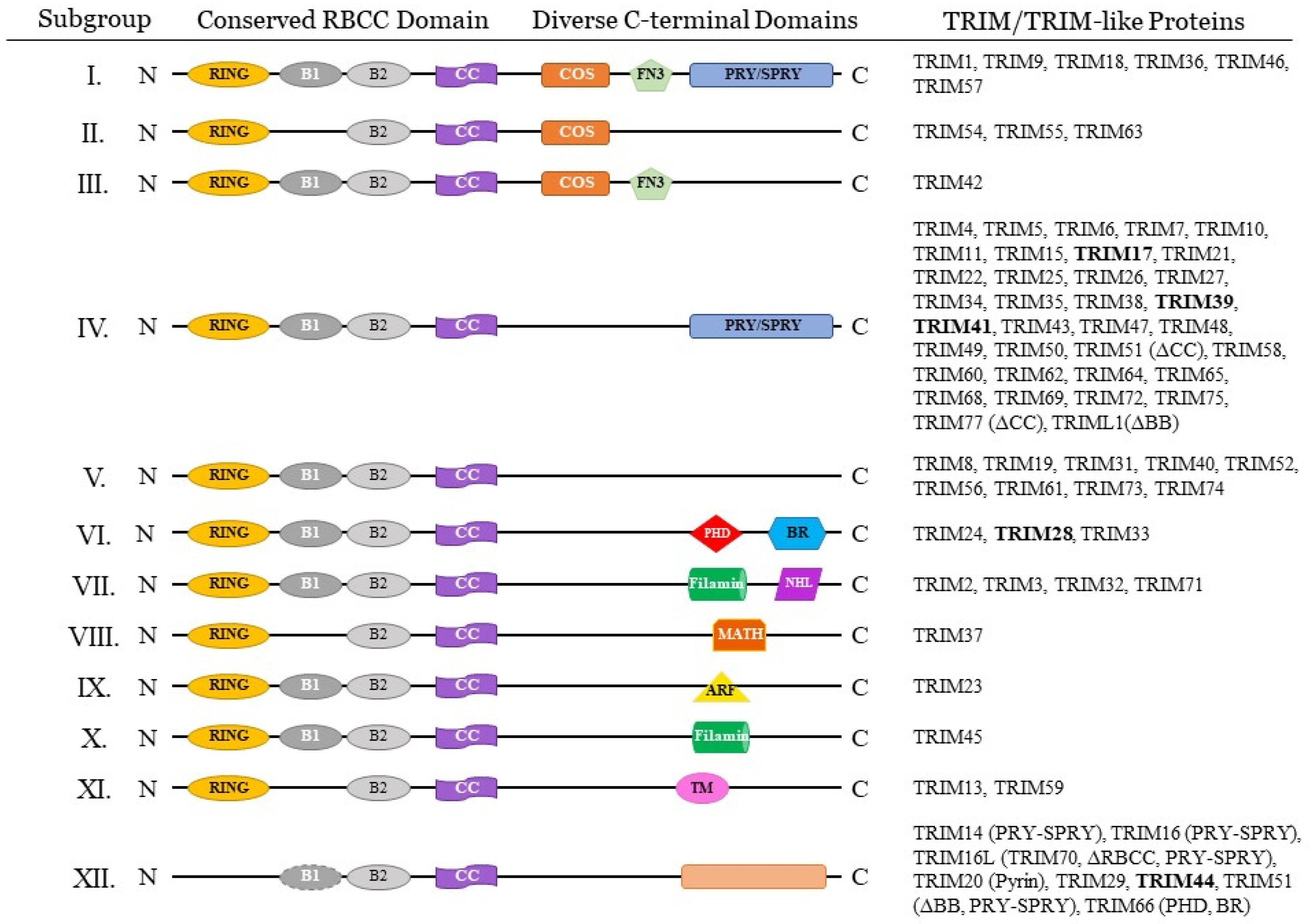{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Structure and Regulation of the TRIM17 Gene
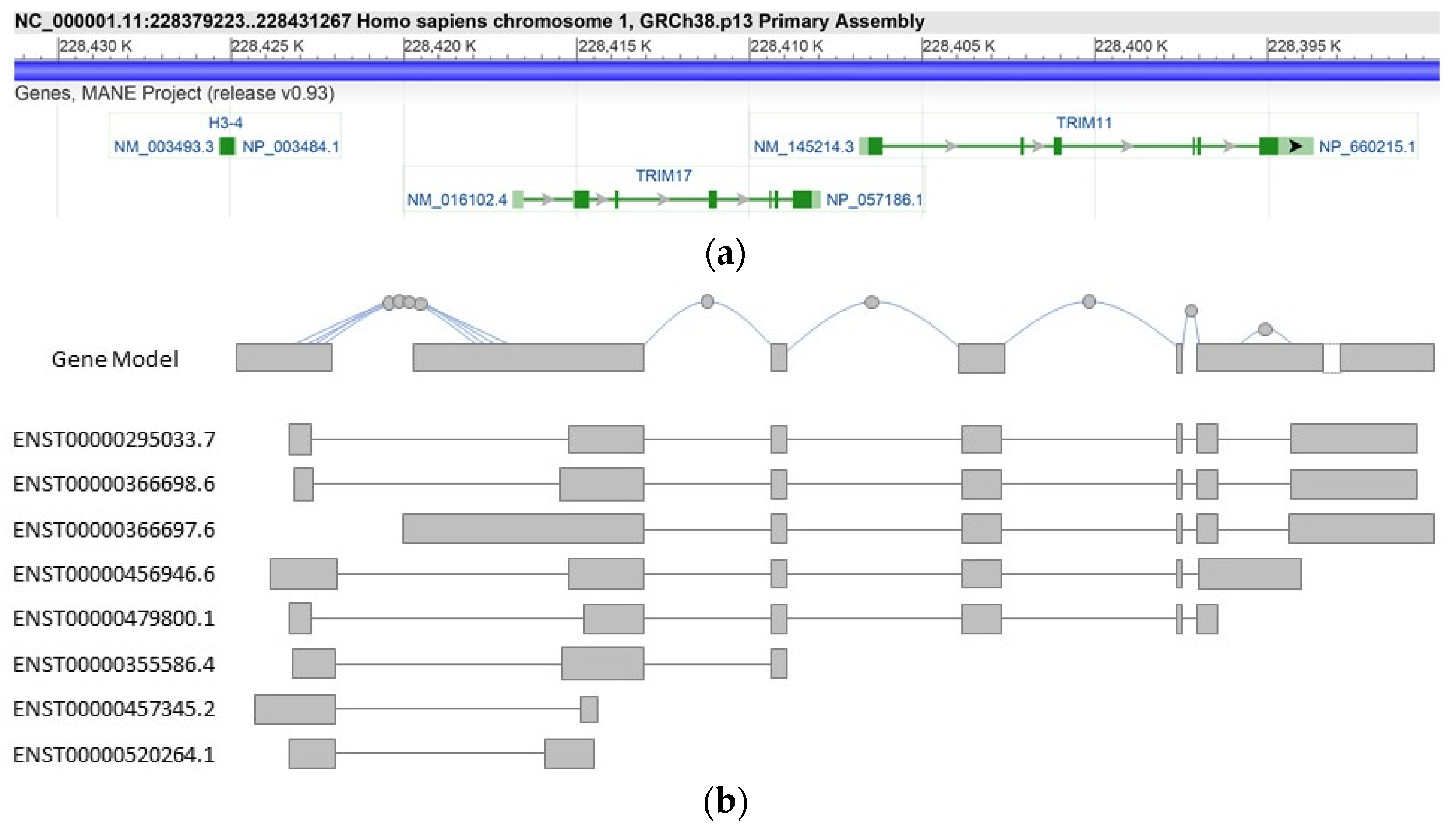
2.1. Structure of the TRIM17 Gene
2.2. Transcription Factors and Regulatory Elements of the TRIM17 Gene
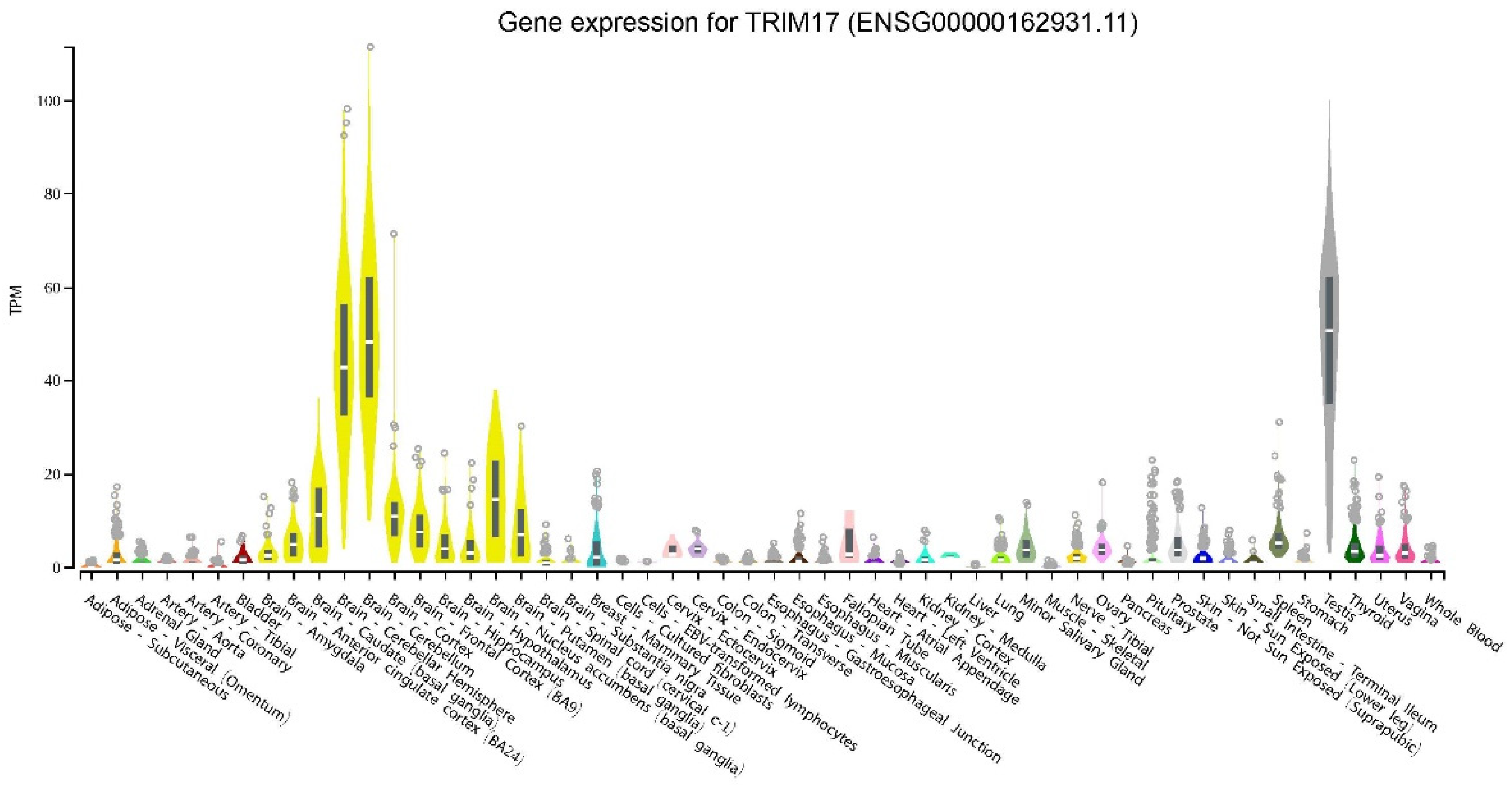
2.3. Induction of TRIM17 Expression and Regulatory Pathways: TRIM17 as A Stress Response Gene
3. TRIM17 Protein Structure and Molecular Function
3.1. TRIM17 Domain Composition and Species Conservation
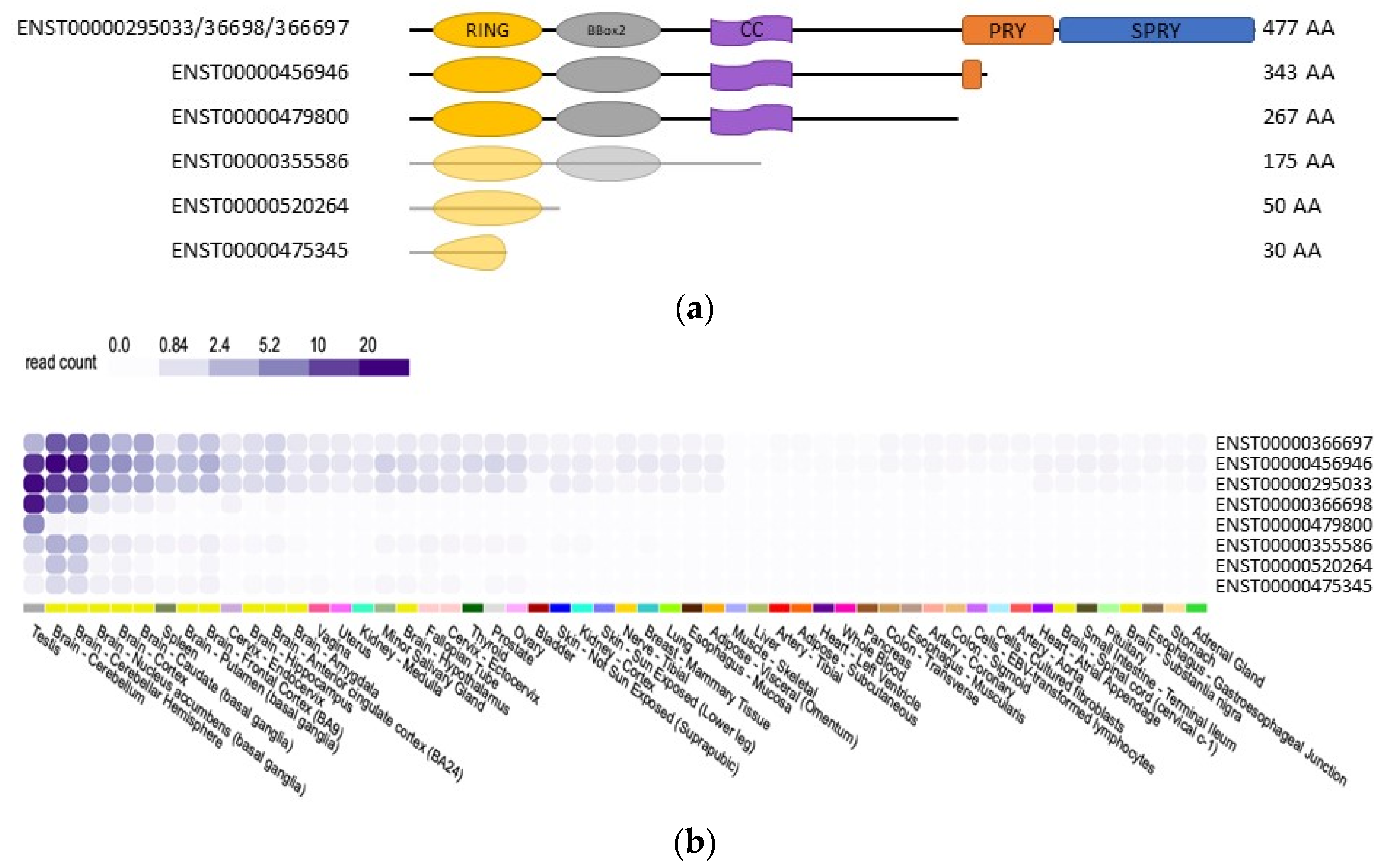
3.2. TRIM17 Isoforms
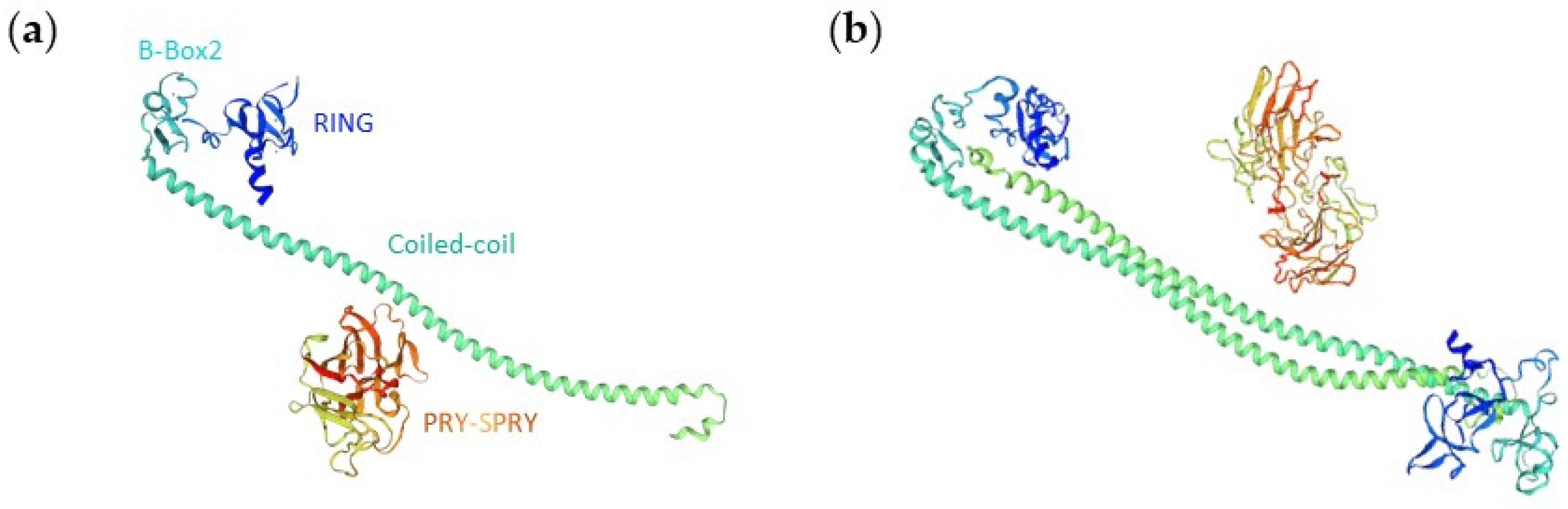
3.3. Secondary and Higher Order Structures of TRIM17
3.3.1. Monomer 3D Structure Model
3.3.2. Homodimerization of TRIM17
3.3.3. Multimerization
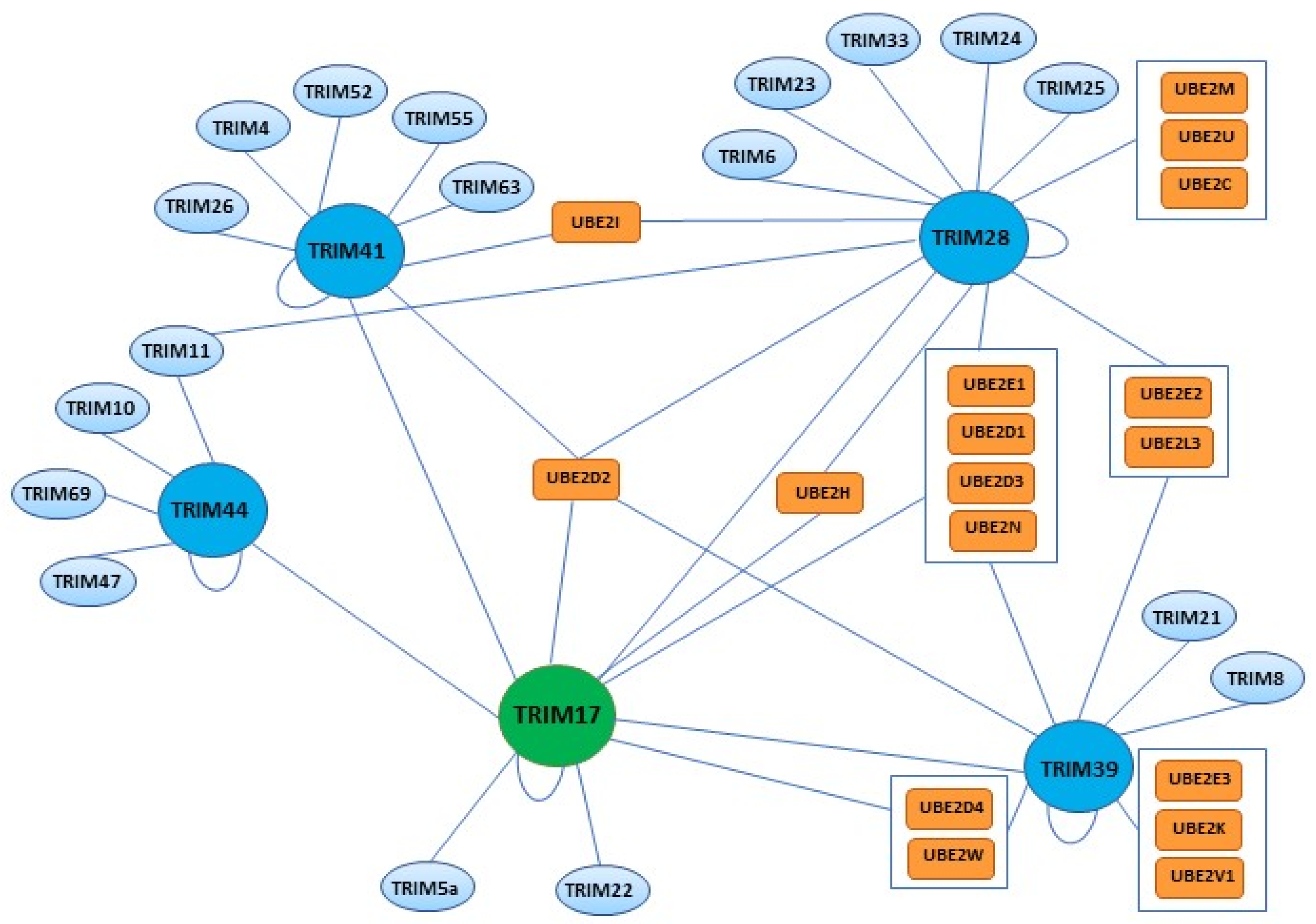
3.4. Hetero-Interactions of TRIM17 with Other TRIM Proteins
3.5. Ubiquitin Ligase Activity of TRIM17 and Interactions with E2 Enzymes
3.6. Inhibition of Other TRIMs by TRIM17
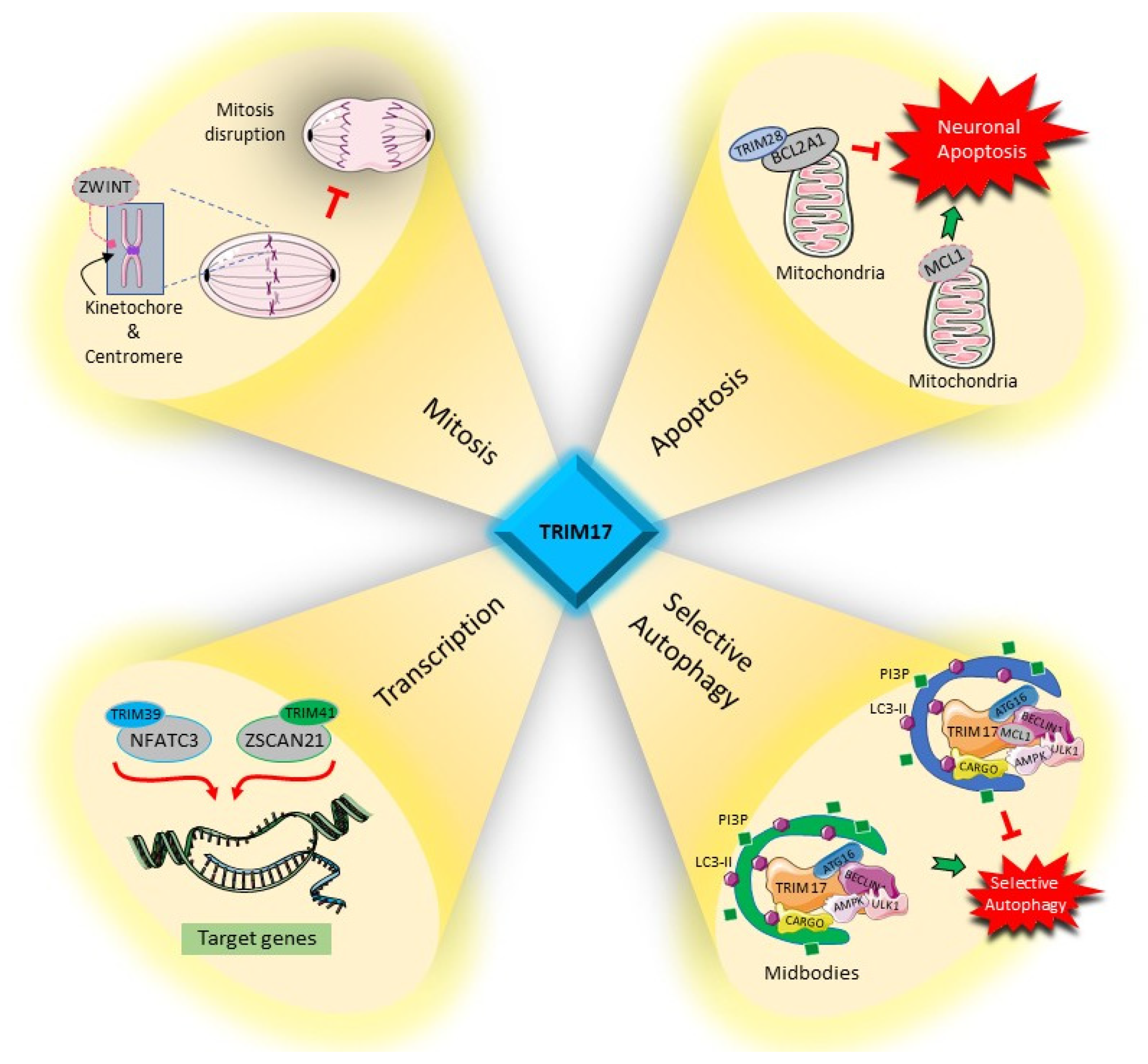
4. Cellular Functions of TRIM17
4.1. Regulation of Transcription Factors
4.1.1. NFATc3 and NFATc4
4.1.2. ZSCAN21
4.2. Apoptosis
4.2.1. MCL1
4.2.2. BCL2A1
4.2.3. NFATc3 and NFATc4
4.3. Autophagy
4.4. Cell Proliferation and Mitosis
4.5. TRIM17 Knockout Mice
5. TRIM17 and Diseases
5.1. Parkinson’s Disease
5.2. Autism
5.3. Cancer and Chemoresistance
5.4. Other Pathologies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a Novel Class of “single Protein RING Finger” E3 Ubiquitin Ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic Analysis of the TRIM Family Reveals Two Groups of Genes with Distinct Evolutionary Properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Liu, H.; Jian, Z.; Fan, Z.; Liu, S.; Xing, J.; Li, J. Characterization of the Primate TRIM Gene Family Reveals the Recent Evolution in Primates. Mol. Genet. Genom. 2020, 295, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM Family Proteins and Their Emerging Roles in Innate Immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Wang, H.-T.; Hur, S. Substrate Recognition by TRIM and TRIM-like Proteins in Innate Immunity. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural Determinants of TRIM Protein Function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef]
- Meroni, G. Genomics and Evolution of the TRIM Gene Family. Adv. Exp. Med. Biol. 2012, 770, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Pérez Berrocal, D.A.; Witting, K.F.; Ovaa, H.; Mulder, M.P.C. Hybrid Chains: A Collaboration of Ubiquitin and Ubiquitin-Like Modifiers Introducing Cross-Functionality to the Ubiquitin Code. Front. Chem. 2020, 7. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The Recognition of Ubiquitinated Proteins by the Proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin Chain-Induced P62 Phase Separation Drives Autophagic Cargo Segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Ogawa, S.; Goto, W.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M.; Inoue, S. Molecular Cloning of a Novel RING Finger-B Box-Coiled Coil (RBCC) Protein, Terf, Expressed in the Testis. Biochem. Biophys. Res. Commun. 1998, 251, 515–519. [Google Scholar] [CrossRef]
- Urano, T.; Usui, T.; Takeda, S.; Ikeda, K.; Okada, A.; Ishida, Y.; Iwayanagi, T.; Otomo, J.; Ouchi, Y.; Inoue, S. TRIM44 Interacts with and Stabilizes Terf, a TRIM Ubiquitin E3 Ligase. Biochem. Biophys. Res. Commun. 2009, 383, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lassot, I.; Robbins, I.; Kristiansen, M.; Rahmeh, R.; Jaudon, F.; Magiera, M.M.; Mora, S.; Vanhille, L.; Lipkin, A.; Pettmann, B.; et al. Trim17, a Novel E3 Ubiquitin-Ligase, Initiates Neuronal Apoptosis. Cell Death Differ. 2010, 17, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, E.; Woodside, J.; Tan, M.M.X.; Shoai, M.; Pittman, A.; Ferrari, R.; Mok, K.Y.; Zhang, D.; Reynolds, R.H.; de Silva, R.; et al. Variation at the TRIM11 Locus Modifies Progressive Supranuclear Palsy Phenotype. Ann. Neurol. 2018, 84, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Oura, S.; Matsumura, T.; Oji, A.; Sakurai, N.; Fujihara, Y.; Shimada, K.; Miyata, H.; Tobita, T.; Noda, T.; et al. CRISPR/Cas9-Mediated Genome Editing Reveals 30 Testis-Enriched Genes Dispensable for Male Fertility in Mice†. Biol. Reprod. 2019, 101, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM Gene Expression in Response to Interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef]
- Yang, W.; Gu, Z.; Zhang, H.; Hu, H. To TRIM the Immunity: From Innate to Adaptive Immunity. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic. Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Song, K.-H.; Choi, C.H.; Lee, H.-J.; Oh, S.J.; Woo, S.R.; Hong, S.-O.; Noh, K.H.; Cho, H.; Chung, E.J.; Kim, J.-H.; et al. HDAC1 Upregulation by NANOG Promotes Multidrug Resistance and a Stem-like Phenotype in Immune Edited Tumor Cells. Cancer Res. 2017, 77, 5039–5053. [Google Scholar] [CrossRef]
- Mojsa, B.; Mora, S.; Bossowski, J.P.; Lassot, I.; Desagher, S. Control of Neuronal Apoptosis by Reciprocal Regulation of NFATc3 and Trim17. Cell Death Differ. 2015, 22, 274–286. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef]
- Desagher, S.; Severac, D.; Lipkin, A.; Bernis, C.; Ritchie, W.; Le Digarcher, A.; Journot, L. Genes Regulated in Neurons Undergoing Transcription-Dependent Apoptosis Belong to Signaling Pathways Rather than the Apoptotic Machinery. J. Biol. Chem. 2005, 280, 5693–5702. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic Cell Death Regulation in Neurons. FEBS J. 2019, 286, 3276–3298. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho Family GTPases: Key Players in Neuronal Development, Neuronal Survival, and Neurodegeneration. Front. Cell Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, T.J.; Chen, S.S.; Hsiao, P.C.; Yang, R.C. Activation of Trim17 by PPARgamma Is Involved in Di(2-Ethylhexyl) Phthalate (DEHP)-Induced Apoptosis on Neuro-2a Cells. Toxicol. Lett. 2011, 206, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Lassot, I.; Mora, S.; Lesage, S.; Zieba, B.A.; Coque, E.; Condroyer, C.; Bossowski, J.P.; Mojsa, B.; Marelli, C.; Soulet, C.; et al. The E3 Ubiquitin Ligases TRIM17 and TRIM41 Modulate α-Synuclein Expression by Regulating ZSCAN21. Cell Rep. 2018, 25, 2484–2496.e9. [Google Scholar] [CrossRef]
- Pierre, W.C.; Legault, L.; Londono, I.; McGraw, S.; Lodygensky, G.A. Alteration of the Brain Methylation Landscape Following Postnatal Inflammatory Injury in Rat Pups. FASEB J. 2020, 34, 432–445. [Google Scholar] [CrossRef]
- Joazeiro, C.A.; Weissman, A.M. RING Finger Proteins: Mediators of Ubiquitin Ligase Activity. Cell 2000, 102, 549–552. [Google Scholar] [CrossRef]
- Chu, Y.; Yang, X. SUMO E3 Ligase Activity of TRIM Proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Okumura, F.; Takahashi, N.; Kataoka, A.; Kamiyama, T.; Todo, S.; Hatakeyama, S. TRIM40 Promotes Neddylation of IKKγ and Is Downregulated in Gastrointestinal Cancers. Carcinogenesis 2011, 32, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Bellon, S.F.; Rodgers, K.K.; Schatz, D.G.; Coleman, J.E.; Steitz, T.A. Crystal Structure of the RAG1 Dimerization Domain Reveals Multiple Zinc-Binding Motifs Including a Novel Zinc Binuclear Cluster. Nat. Struct. Biol. 1997, 4, 586–591. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Meza, J.E.; King, M.C.; Klevit, R.E. BRCA1 RING Domain Cancer-Predisposing Mutations. Structural Consequences and Effects on Protein-Protein Interactions. J. Biol. Chem. 2001, 276, 41399–41406. [Google Scholar] [CrossRef]
- Liu, J.; Deng, Y.; Zheng, Q.; Cheng, C.-S.; Kallenbach, N.R.; Lu, M. A Parallel Coiled-Coil Tetramer with Offset Helices. Biochemistry 2006, 45, 15224–15231. [Google Scholar] [CrossRef]
- Micale, L.; Chaignat, E.; Fusco, C.; Reymond, A.; Merla, G. The Tripartite Motif: Structure and Function. Adv. Exp. Med. Biol. 2012, 770, 11–25. [Google Scholar]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The Tripartite Motif Family Identifies Cell Compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML Protein Isoforms and the RBCC/TRIM Motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Battivelli, E.; Migraine, J.; Lecossier, D.; Matsuoka, S.; Perez-Bercoff, D.; Saragosti, S.; Clavel, F.; Hance, A.J. Modulation of TRIM5alpha Activity in Human Cells by Alternatively Spliced TRIM5 Isoforms. J. Virol. 2011, 85, 7828–7835. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Begg, G.E.; Schultz, D.C.; Friedman, J.R.; Jensen, D.E.; Speicher, D.W.; Rauscher, F.J. Reconstitution of the KRAB-KAP-1 Repressor Complex: A Model System for Defining the Molecular Anatomy of RING-B Box-Coiled-Coil Domain-Mediated Protein-Protein Interactions. J. Mol. Biol. 2000, 295, 1139–1162. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.-G.; Gao, N.; Yu, H.; et al. Structural Insights into the TRIM Family of Ubiquitin E3 Ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional Role of TRIM E3 Ligase Oligomerization and Regulation of Catalytic Activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Meroni, G. TRIM Family: Pleiotropy and Diversification through Homomultimer and Heteromultimer Formation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The Tripartite Motif Coiled-Coil Is an Elongated Antiparallel Hairpin Dimer. Proc. Sci. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking Structural Similarities between Macromolecular Complexes. Nucleic. Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef]
- Biris, N.; Yang, Y.; Taylor, A.B.; Tomashevski, A.; Guo, M.; Hart, P.J.; Diaz-Griffero, F.; Ivanov, D.N. Structure of the Rhesus Monkey TRIM5α PRYSPRY Domain, the HIV Capsid Recognition Module. Proc. Sci. Natl. Acad. Sci. USA 2012, 109, 13278–13283. [Google Scholar] [CrossRef]
- Stoll, G.A.; Oda, S.; Chong, Z.-S.; Yu, M.; McLaughlin, S.H.; Modis, Y. Structure of KAP1 Tripartite Motif Identifies Molecular Interfaces Required for Retroelement Silencing. Proc. Sci. Natl. Acad. Sci. USA 2019, 116, 15042–15051. [Google Scholar] [CrossRef]
- Weinert, C.; Grütter, C.; Roschitzki-Voser, H.; Mittl, P.R.E.; Grütter, M.G. The Crystal Structure of Human Pyrin B30.2 Domain: Implications for Mutations Associated with Familial Mediterranean Fever. J. Mol. Biol. 2009, 394, 226–236. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Chen, Z.; Wu, H.; Wang, P.; Wu, W.; Cheng, N.; Zeng, L.; Zhang, H.; Cai, X.; et al. B1 Oligomerization Regulates PML Nuclear Body Biogenesis and Leukemogenesis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Chandrasekaran, V.; Carter, S.D.; Woodward, C.L.; Christensen, D.E.; Dryden, K.A.; Pornillos, O.; Yeager, M.; Ganser-Pornillos, B.K.; Jensen, G.J.; et al. Primate TRIM5 Proteins Form Hexagonal Nets on HIV-1 Capsids. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Benhenda, S.; Wu, H.; Lallemand-Breitenbach, V.; Zhen, T.; Jollivet, F.; Peres, L.; Li, Y.; Chen, S.-J.; Chen, Z.; et al. RING Tetramerization Is Required for Nuclear Body Biogenesis and PML Sumoylation. Nat. Commun. 2018, 9, 1277. [Google Scholar] [CrossRef]
- Skorupka, K.A.; Roganowicz, M.D.; Christensen, D.E.; Wan, Y.; Pornillos, O.; Ganser-Pornillos, B.K. Hierarchical Assembly Governs TRIM5α Recognition of HIV-1 and Retroviral Capsids. Sci. Adv. 2019, 5, eaaw3631. [Google Scholar] [CrossRef]
- Carter, S.D.; Mamede, J.I.; Hope, T.J.; Jensen, G.J. Correlated Cryogenic Fluorescence Microscopy and Electron Cryo-Tomography Shows That Exogenous TRIM5α Can Form Hexagonal Lattices or Autophagy Aggregates in Vivo. Proc. Sci. Natl. Acad. Sci. USA 2020, 117, 29702–29711. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yeung, D.F.; Fiegen, A.M.; Sodroski, J. Determinants of the Higher Order Association of the Restriction Factor TRIM5alpha and Other Tripartite Motif (TRIM) Proteins. J. Biol. Chem. 2011, 286, 27959–27970. [Google Scholar] [CrossRef]
- Streich, F.C.; Ronchi, V.P.; Connick, J.P.; Haas, A.L. Tripartite Motif Ligases Catalyze Polyubiquitin Chain Formation through a Cooperative Allosteric Mechanism. J. Biol. Chem. 2013, 288, 8209–8221. [Google Scholar] [CrossRef]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. RING Dimerization Links Higher-Order Assembly of TRIM5α to Synthesis of K63-Linked Polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef]
- Yamauchi, K.; Wada, K.; Tanji, K.; Tanaka, M.; Kamitani, T. Ubiquitination of E3 Ubiquitin Ligase TRIM5 Alpha and Its Potential Role. FEBS J. 2008, 275, 1540–1555. [Google Scholar] [CrossRef]
- Li, X.; Gold, B.; O’hUigin, C.; Diaz-Griffero, F.; Song, B.; Si, Z.; Li, Y.; Yuan, W.; Stremlau, M.; Mische, C.; et al. Unique Features of TRIM5alpha among Closely Related Human TRIM Family Members. Virology 2007, 360, 419–433. [Google Scholar] [CrossRef]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A Proteome-Scale Map of the Human Interactome Network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a Proteome-Scale Map of the Human Protein-Protein Interaction Network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Woodsmith, J.; Jenn, R.C.; Sanderson, C.M. Systematic Analysis of Dimeric E3-RING Interactions Reveals Increased Combinatorial Complexity in Human Ubiquitination Networks. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Shrivastava, M.B.; Mojsa, B.; Mora, S.; Robbins, I.; Bossis, G.; Lassot, I.; Desagher, S. Trim39 Regulates Neuronal Apoptosis by Acting as a SUMO-Targeted E3 Ubiquitin-Ligase for the Transcription Factor NFATc3. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, S.; Fragoza, R.; Klei, L.; Liu, Y.; Wang, J.; Roeder, K.; Devlin, B.; Yu, H. An Interactome Perturbation Framework Prioritizes Damaging Missense Mutations for Developmental Disorders. Nat. Genet. 2018, 50, 1032–1040. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A Reference Map of the Human Binary Protein Interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Mandell, M.A.; Jain, A.; Kumar, S.; Castleman, M.J.; Anwar, T.; Eskelinen, E.-L.; Johansen, T.; Prekeris, R.; Deretic, V. TRIM17 Contributes to Autophagy of Midbodies While Actively Sparing Other Targets from Degradation. J. Cell Sci. 2016, 129, 3562–3573. [Google Scholar] [CrossRef]
- Lionnard, L.; Duc, P.; Brennan, M.S.; Kueh, A.J.; Pal, M.; Guardia, F.; Mojsa, B.; Damiano, M.-A.; Mora, S.; Lassot, I.; et al. TRIM17 and TRIM28 Antagonistically Regulate the Ubiquitination and Anti-Apoptotic Activity of BCL2A1. Cell Death Differ. 2019, 26, 902–917. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the Human Interactome Defines Protein Communities and Disease Networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Pickart, C.M. Back to the Future with Ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef]
- Magiera, M.M.; Mora, S.; Mojsa, B.; Robbins, I.; Lassot, I.; Desagher, S. Trim17-Mediated Ubiquitination and Degradation of Mcl-1 Initiate Apoptosis in Neurons. Cell Death Differ. 2013, 20, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Inoue, S. Terf/TRIM17 Stimulates Degradation of Kinetochore Protein ZWINT and Regulates Cell Proliferation. J. Biochem. 2012, 151, 139–144. [Google Scholar] [CrossRef]
- Markson, G.; Kiel, C.; Hyde, R.; Brown, S.; Charalabous, P.; Bremm, A.; Semple, J.; Woodsmith, J.; Duley, S.; Salehi-Ashtiani, K.; et al. Analysis of the Human E2 Ubiquitin Conjugating Enzyme Protein Interaction Network. Genome Res. 2009, 19, 1905–1911. [Google Scholar] [CrossRef]
- van Wijk, S.J.L.; de Vries, S.J.; Kemmeren, P.; Huang, A.; Boelens, R.; Bonvin, A.M.J.J.; Timmers, H.T.M. A Comprehensive Framework of E2-RING E3 Interactions of the Human Ubiquitin-Proteasome System. Mol. Syst. Biol. 2009, 5, 295. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional Interactions between Ubiquitin E2 Enzymes and TRIM Proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef]
- Hao, Y.-H.; Fountain, M.D.; Fon Tacer, K.; Xia, F.; Bi, W.; Kang, S.-H.L.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell 2015, 59, 956–969. [Google Scholar] [CrossRef]
- Nicklas, S.; Hillje, A.-L.; Okawa, S.; Rudolph, I.-M.; Collmann, F.M.; van Wuellen, T.; Del Sol, A.; Schwamborn, J.C. A Complex of the Ubiquitin Ligase TRIM32 and the Deubiquitinase USP7 Balances the Level of C-Myc Ubiquitination and Thereby Determines Neural Stem Cell Fate Specification. Cell Death Differ. 2019, 26, 728–740. [Google Scholar] [CrossRef]
- Borlepawar, A.; Rangrez, A.Y.; Bernt, A.; Christen, L.; Sossalla, S.; Frank, D.; Frey, N. TRIM24 Protein Promotes and TRIM32 Protein Inhibits Cardiomyocyte Hypertrophy via Regulation of Dysbindin Protein Levels. J. Biol. Chem. 2017, 292, 10180–10196. [Google Scholar] [CrossRef]
- Macian, F. NFAT Proteins: Key Regulators of T-Cell Development and Function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Muller, M.R.; Rao, A. NFAT, Immunity and Cancer: A Transcription Factor Comes of Age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional Regulation by Calcium, Calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; Carneiro, F.R.G.; Robbs, B.K.; Faget, D.V.; Viola, J.P.B. Cell Cycle and Apoptosis Regulation by NFAT Transcription Factors: New Roles for an Old Player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Wersinger, C.; Gasser, T. Transcriptional Regulation of the Alpha-Synuclein Gene in Human Brain Tissue. Neurosci. Lett. 2015, 599, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Clough, R.L.; Dermentzaki, G.; Stefanis, L. Functional Dissection of the Alpha-Synuclein Promoter: Transcriptional Regulation by ZSCAN21 and ZNF219. J. Neurochem. 2009, 110, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Dermentzaki, G.; Paschalidis, N.; Politis, P.K.; Stefanis, L. Complex Effects of the ZSCAN21 Transcription Factor on Transcriptional Regulation of α-Synuclein in Primary Neuronal Cultures and in Vivo. J. Biol. Chem. 2016, 291, 8756–8772. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.W.; Zhong, R.; Heintz, N. Granule Cell Specification in the Developing Mouse Brain as Defined by Expression of the Zinc Finger Transcription Factor RU49. Development 1996, 122, 555–566. [Google Scholar] [CrossRef]
- Sunkin, S.M.; Ng, L.; Lau, C.; Dolbeare, T.; Gilbert, T.L.; Thompson, C.L.; Hawrylycz, M.; Dang, C. Allen Brain Atlas: An Integrated Spatio-Temporal Portal for Exploring the Central Nervous System. Nucleic. Acids Res. 2013, 41, D996–D1008. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, R.T.; Higuchi-Sanabria, R.; Shalem, O.; Moehle, E.A.; Webster, B.M.; Joe, L.; Bar-Ziv, R.; Frankino, P.A.; Durieux, J.; Pender, C.; et al. The Hyaluronidase, TMEM2, Promotes ER Homeostasis and Longevity Independent of the UPRER. Cell 2019, 179, 1306–1318.e18. [Google Scholar] [CrossRef]
- Zhuang, X.; Veltri, D.P.; Long, E.O. Genome-Wide CRISPR Screen Reveals Cancer Cell Resistance to NK Cells Induced by NK-Derived IFN-γ. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from Transcription to Degradation. Cell Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Liu, Z. Ubiquitination and Deubiquitination of MCL1 in Cancer: Deciphering Chemoresistance Mechanisms and Providing Potential Therapeutic Options. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Ko, J.-K.; Choi, K.-H.; Pan, Z.; Lin, P.; Weisleder, N.; Kim, C.-W.; Ma, J. The Tail-Anchoring Domain of Bfl1 and HCCS1 Targets Mitochondrial Membrane Permeability to Induce Apoptosis. J. Cell Sci. 2007, 120, 2912–2923. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-U.; Kim, L.-K.; Choi, J.-M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front. Immunol. 2018, 9, 2747. [Google Scholar] [CrossRef] [PubMed]
- Benedito, A.B.; Lehtinen, M.; Massol, R.; Lopes, U.G.; Kirchhausen, T.; Rao, A.; Bonni, A. The Transcription Factor NFAT3 Mediates Neuronal Survival. J. Biol. Chem. 2005, 280, 2818–2825. [Google Scholar] [CrossRef]
- Vashishta, A.; Habas, A.; Pruunsild, P.; Zheng, J.J.; Timmusk, T.; Hetman, M. Nuclear Factor of Activated T-Cells Isoform C4 (NFATc4/NFAT3) as a Mediator of Antiapoptotic Transcription in NMDA Receptor-Stimulated Cortical Neurons. J. Neurosci. 2009, 29, 15331–15340. [Google Scholar] [CrossRef]
- Quadrato, G.; Benevento, M.; Alber, S.; Jacob, C.; Floriddia, E.M.; Nguyen, T.; Elnaggar, M.Y.; Pedroarena, C.M.; Molkentin, J.D.; Di Giovanni, S. Nuclear Factor of Activated T Cells (NFATc4) Is Required for BDNF-Dependent Survival of Adult-Born Neurons and Spatial Memory Formation in the Hippocampus. Proc. Sci. Natl. Acad. Sci. USA 2012, 109, E1499–E1508. [Google Scholar] [CrossRef]
- Luoma, J.I.; Zirpel, L. Deafferentation-Induced Activation of NFAT (Nuclear Factor of Activated T-Cells) in Cochlear Nucleus Neurons during a Developmental Critical Period: A Role for NFATc4-Dependent Apoptosis in the CNS. J. Neurosci. 2008, 28, 3159–3169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol. Cell 2015, 58, 1053–1066. [Google Scholar] [CrossRef]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 Phosphorylation of P62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM Proteins Regulate Autophagy and Can Target Autophagic Substrates by Direct Recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM Proteins in Autophagy: Selective Sensors in Cell Damage and Innate Immune Responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-Directed Selective Autophagy Regulates Immune Activation. Autophagy 2016, 13, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential Interactions between Beclin 1 and Bcl-2 Family Members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.-A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-Direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F. Chromosomes, Genes, and Cancer. Ca A Cancer J. Clin. 1994, 44, 133–135. [Google Scholar] [CrossRef]
- Hoffmann, I. Centrosomes in Mitotic Spindle Assembly and Orientation. Curr. Opin. Struct. Biol. 2020, 66, 193–198. [Google Scholar] [CrossRef]
- Monda, J.K.; Cheeseman, I.M. The Kinetochore-Microtubule Interface at a Glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Desai, A. A Molecular View of Kinetochore Assembly and Function. Biology 2017, 6, 5. [Google Scholar] [CrossRef]
- Testa, G.; Schaft, J.; van der Hoeven, F.; Glaser, S.; Anastassiadis, K.; Zhang, Y.; Hermann, T.; Stremmel, W.; Stewart, A.F. A Reliable LacZ Expression Reporter Cassette for Multipurpose, Knockout-First Alleles. Genesis 2004, 38, 151–158. [Google Scholar] [CrossRef]
- Gushchina, L.V.; Bodnar, T.A.; Bhattacharya, S.; Weisleder, N.L. Conserved Structural and Functional Aspects of the Tripartite Motif Gene Family Point towards Therapeutic Applications in Multiple Diseases. Pharmacol. Ther. 2018, 185, 12–25. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us about the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-Synuclein for Treatment of Parkinson’s Disease: Mechanistic and Therapeutic Considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Fouka, M.; Mavroeidi, P.; Tsaka, G.; Xilouri, M. In Search of Effective Treatments Targeting α-Synuclein Toxicity in Synucleinopathies: Pros and Cons. Front Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Langston, R.G.; Cookson, M.R. Pathways of Protein Synthesis and Degradation in PD Pathogenesis. Prog. Brain Res. 2020, 252, 217–270. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Farlow, J.L.; Robak, L.A.; Hetrick, K.; Bowling, K.; Boerwinkle, E.; Coban-Akdemir, Z.H.; Gambin, T.; Gibbs, R.A.; Gu, S.; Jain, P.; et al. Whole-Exome Sequencing in Familial Parkinson Disease. JAMA Neurol. 2016, 73, 68–75. [Google Scholar] [CrossRef]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin Determines Toxic versus Beneficial Responses to α-Synuclein. Proc. Sci. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef]
- Luo, J.; Sun, L.; Lin, X.; Liu, G.; Yu, J.; Parisiadou, L.; Xie, C.; Ding, J.; Cai, H. A Calcineurin- and NFAT-Dependent Pathway Is Involved in α-Synuclein-Induced Degeneration of Midbrain Dopaminergic Neurons. Hum. Mol. Genet. 2014, 23, 6567–6574. [Google Scholar] [CrossRef]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The Role of de Novo Mutations in the Genetics of Autism Spectrum Disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, Distribution, and Implications of Post-Zygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.M.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.L.M.; et al. Refining Analyses of Copy Number Variation Identifies Specific Genes Associated with Developmental Delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- Morato Torres, C.A.; Wassouf, Z.; Zafar, F.; Sastre, D.; Outeiro, T.F.; Schüle, B. The Role of Alpha-Synuclein and Other Parkinson’s Genes in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 5724. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-Apoptotic Mcl-1 Is Essential for the Development and Sustained Growth of Acute Myeloid Leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 Ubiquitination: Unique Regulation of an Essential Survival Protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef]
- Mazumder, S.; Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Mcl-1 Phosphorylation Defines ABT-737 Resistance That Can Be Overcome by Increased NOXA Expression in Leukemic B Cells. Cancer Res. 2012, 72, 3069–3079. [Google Scholar] [CrossRef]
- Vogler, M. BCL2A1: The Underdog in the BCL2 Family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef]
- Ottina, E.; Grespi, F.; Tischner, D.; Soratroi, C.; Geley, S.; Ploner, A.; Reichardt, H.M.; Villunger, A.; Herold, M.J. Targeting Antiapoptotic A1/Bfl-1 by in Vivo RNAi Reveals Multiple Roles in Leukocyte Development in Mice. Blood 2012, 119, 6032–6042. [Google Scholar] [CrossRef]
- Haq, R.; Yokoyama, S.; Hawryluk, E.B.; Jönsson, G.B.; Frederick, D.T.; McHenry, K.; Porter, D.; Tran, T.-N.; Love, K.T.; Langer, R.; et al. BCL2A1 Is a Lineage-Specific Antiapoptotic Melanoma Oncogene That Confers Resistance to BRAF Inhibition. Proc. Sci. Natl. Acad. Sci. USA 2013, 110, 4321–4326. [Google Scholar] [CrossRef]
- Brien, G.; Trescol-Biemont, M.-C.; Bonnefoy-Bérard, N. Downregulation of Bfl-1 Protein Expression Sensitizes Malignant B Cells to Apoptosis. Oncogene 2007, 26, 5828–5832. [Google Scholar] [CrossRef]
- Li, H.-N.; Zheng, W.-H.; Du, Y.-Y.; Wang, G.; Dong, M.-L.; Yang, Z.-F.; Li, X.-R. ZW10 Interacting Kinetochore Protein May Serve as a Prognostic Biomarker for Human Breast Cancer: An Integrated Bioinformatics Analysis. Oncol. Lett. 2020, 19, 2163–2174. [Google Scholar] [CrossRef]
- Yang, L.; Han, N.; Zhang, X.; Zhou, Y.; Chen, R.; Zhang, M. ZWINT: A Potential Therapeutic Biomarker in Patients with Glioblastoma Correlates with Cell Proliferation and Invasion. Oncol. Rep. 2020, 43, 1831–1844. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, Z.; Zhu, X.; Qian, G.; Zhou, Y.; Sun, Y.; Yu, W.; Wang, J.; Lu, H.; Lin, F.; et al. N6-Methyladenosine Modification of the TRIM7 Positively Regulates Tumorigenesis and Chemoresistance in Osteosarcoma through Ubiquitination of BRMS1. EBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, K.; Huang, Y.; Xu, L.; Li, X.; Zhang, N. Promoter Hypermethylation of CHODL Contributes to Carcinogenesis and Indicates Poor Survival in Patients with Early-Stage Colorectal Cancer. J. Cancer 2020, 11, 2874–2886. [Google Scholar] [CrossRef]
- Chen, L.; Brewer, M.D.; Guo, L.; Wang, R.; Jiang, P.; Yang, X. Enhanced Degradation of Misfolded Proteins Promotes Tumorigenesis. Cell Rep. 2017, 18, 3143–3154. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Blondel, C.J.; Park, J.S.; Hubbard, T.P.; Pacheco, A.R.; Kuehl, C.J.; Walsh, M.J.; Davis, B.M.; Gewurz, B.E.; Doench, J.G.; Waldor, M.K. CRISPR/Cas Screens Reveal Requirements for Host Cell Sulfation and Fucosylation in Bacterial, Type III Secretion System-Mediated Cytotoxicity. Cell Host Microbe 2016, 20, 226–237. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-Dependent Cancer Cell State Underlies the Clear-Cell Morphology and Confers Sensitivity to Ferroptosis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Aurino, S.; Piluso, G.; Saccone, V.; Cacciottolo, M.; D’Amico, F.; Dionisi, M.; Totaro, A.; Belsito, A.; Di Vicino, U.; Nigro, V. Candidate-Gene Testing for Orphan Limb-Girdle Muscular Dystrophies. Acta Myol. 2008, 27, 90–97. [Google Scholar]
- Zhang, W.; Shi, J.; Zhang, C.; Jiang, X.; Wang, J.; Wang, W.; Wang, D.; Ni, J.; Chen, L.; Lu, W.; et al. Identification of Gene Variants in 130 Han Chinese Patients with Hypospadias by Targeted Next-generation Sequencing. Mol. Genet. Genom. Med. 2019, 7. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial Genome Sequencing and Analysis of Multiple Myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep Sequencing Reveals 50 Novel Genes for Recessive Cognitive Disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basu-Shrivastava, M.; Kozoriz, A.; Desagher, S.; Lassot, I. To Ubiquitinate or Not to Ubiquitinate: TRIM17 in Cell Life and Death. Cells 2021, 10, 1235. https://doi.org/10.3390/cells10051235
Basu-Shrivastava M, Kozoriz A, Desagher S, Lassot I. To Ubiquitinate or Not to Ubiquitinate: TRIM17 in Cell Life and Death. Cells. 2021; 10(5):1235. https://doi.org/10.3390/cells10051235
Chicago/Turabian StyleBasu-Shrivastava, Meenakshi, Alina Kozoriz, Solange Desagher, and Iréna Lassot. 2021. "To Ubiquitinate or Not to Ubiquitinate: TRIM17 in Cell Life and Death" Cells 10, no. 5: 1235. https://doi.org/10.3390/cells10051235
APA StyleBasu-Shrivastava, M., Kozoriz, A., Desagher, S., & Lassot, I. (2021). To Ubiquitinate or Not to Ubiquitinate: TRIM17 in Cell Life and Death. Cells, 10(5), 1235. https://doi.org/10.3390/cells10051235