
TRIMming Down Hormone-Driven Cancers: The Biological Impact of TRIM Proteins on Tumor Development, Progression and Prognostication

 , and
, and 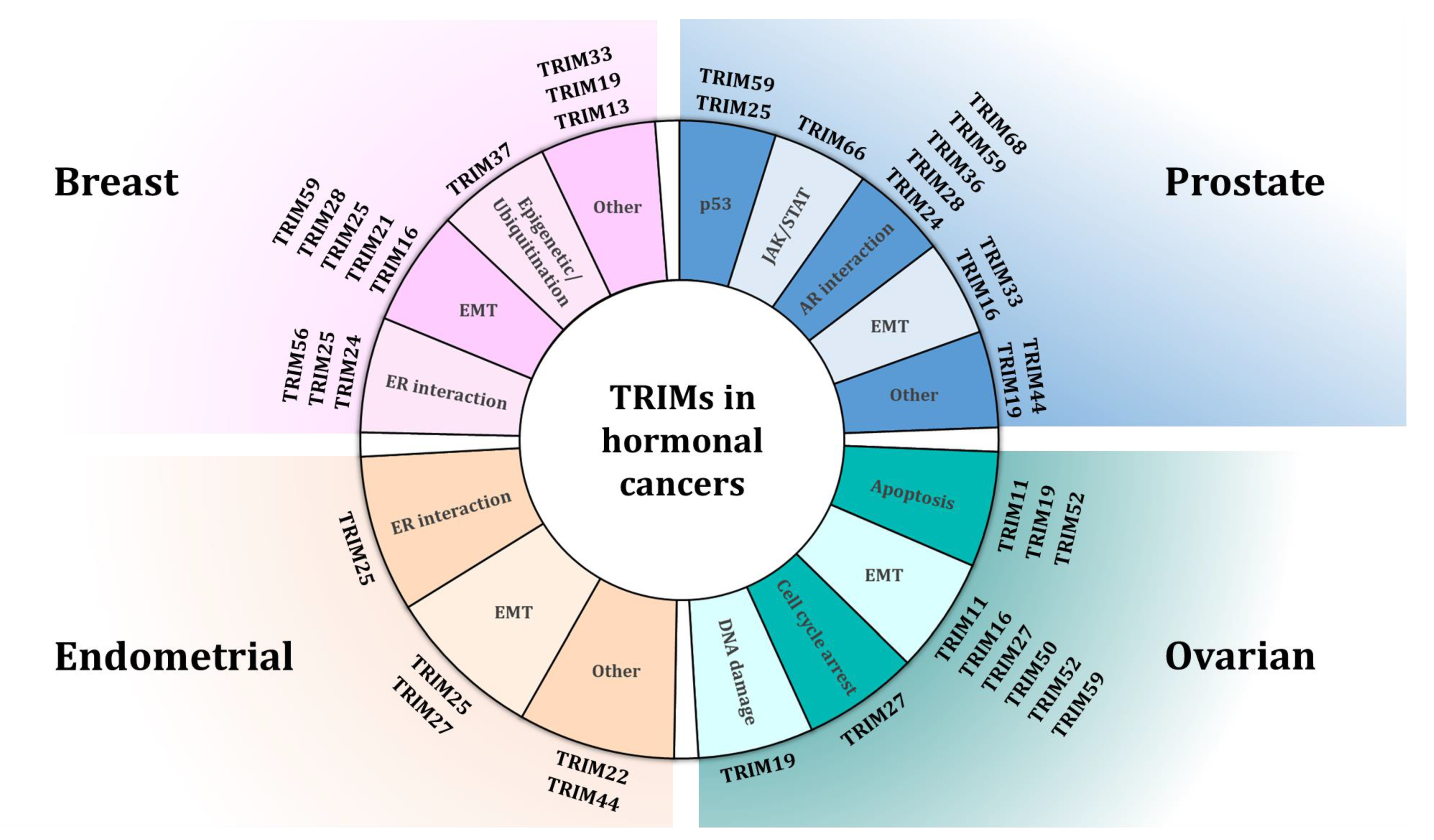{kind=link}
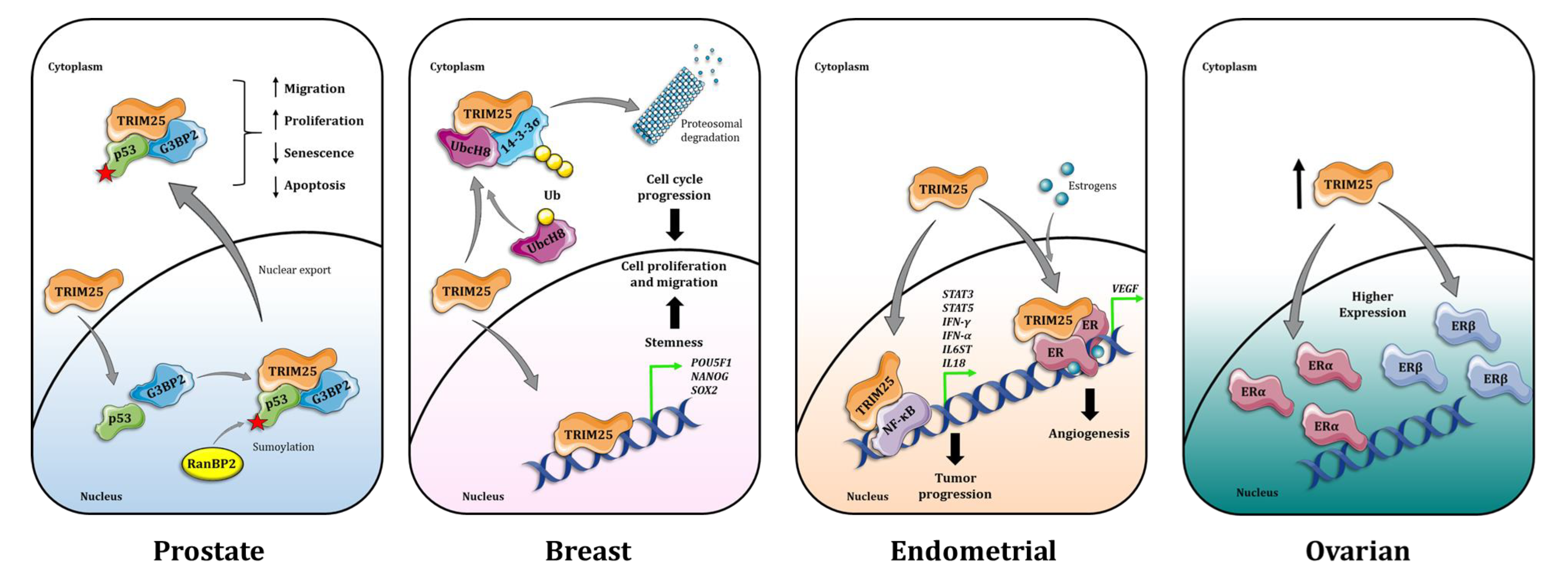{kind=link}
Abstract
1. Introduction
2. TRIMs: Cellular Processes and Mechanisms
3. Steroid Hormone Receptors
4. Prostate Cancer
4.1. TRIMs Involved in Androgen Receptor Biology
4.2. TRIMs and AR-Independent Signaling Pathways
5. Breast Cancer
5.1. The Role of TRIMs in BC Tumor Growth and Proliferation
5.2. TRIMs in BC Development, Progression and Prognosis
6. Ovarian Cancer
6.1. Tumor Suppressive TRIMs
6.2. TRIMs That Promote Proliferation and Metastasis of OC
6.3. Other TRIMs
7. Endometrial Cancer
TRIMs in EC
8. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Meroni, G. Genomics and Evolution of the TRIM Gene Family. In TRIM/RBCC Proteins; Meroni, G., Ed.; Springer: New York, NY, USA, 2012; pp. 1–9. [Google Scholar]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Freemont, P.S.; Hanson, I.M.; Trowsdale, J. A novel cysteine-rich sequence motif. Cell 1991, 64, 483–484. [Google Scholar] [CrossRef]
- Reddy, B.A.; Etkin, L.D.; Freemont, P.S. A novel zinc finger coiled-coil domain in a family of nuclear proteins. Trends Biochem. Sci. 1992, 17, 344–345. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Hage, A.; van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weinert, C.; Morger, D.; Djekic, A.; Grütter, M.G.; Mittl, P.R.E. Crystal structure of TRIM20 C-terminal coiled-coil/B30.2 fragment: Implications for the recognition of higher order oligomers. Sci. Rep. 2015, 5, 10819. [Google Scholar] [CrossRef] [PubMed]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef]
- Sugiura, T.; Miyamoto, K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J. Cell Biochem. 2008, 105, 1081–1091. [Google Scholar] [CrossRef]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Koepke, L.; Gack, M.U.; Sparrer, K.M. The antiviral activities of TRIM proteins. Curr. Opin. Microbiol. 2021, 59, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Petrera, F.; Meroni, G. TRIM proteins in development. Adv. Exp. Med. Biol. 2012, 770, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.; Czerwinska, P. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem Cells 2020, 38, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Nenasheva, V.V.; Tarantul, V.Z. Many Faces of TRIM Proteins on the Road from Pluripotency to Neurogenesis. Stem Cells Dev. 2020, 29, 1–14. [Google Scholar] [CrossRef]
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M. All roads lead to ubiquitin. Cell Death Differ. 2017, 24, 1135–1136. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Mandell, M.A.; Saha, B.; Thompson, T.A. The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharm. 2020, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef]
- Hsu, K.S.; Zhao, X.; Cheng, X.; Guan, D.; Mahabeleshwar, G.H.; Liu, Y.; Borden, E.; Jain, M.K.; Kao, H.Y. Dual regulation of Stat1 and Stat3 by the tumor suppressor protein PML contributes to interferon α-mediated inhibition of angiogenesis. J. Biol. Chem. 2017, 292, 10048–10060. [Google Scholar] [CrossRef]
- Noguchi, K.; Okumura, F.; Takahashi, N.; Kataoka, A.; Kamiyama, T.; Todo, S.; Hatakeyama, S. TRIM40 promotes neddylation of IKKγ and is downregulated in gastrointestinal cancers. Carcinogenesis 2011, 32, 995–1004. [Google Scholar] [CrossRef]
- Zhang, L.; Afolabi, L.O.; Wan, X.; Li, Y.; Chen, L.; Zhang, L.; Afolabi, L.O.; Wan, X.; Li, Y.; Chen, L. Emerging Roles of Tripartite Motif-Containing Family Proteins (TRIMs) in Eliminating Misfolded Proteins. Front. Cell Dev. Biol. 2020, 8, 802. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharm. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Ko, Y.J.; Balk, S.P. Targeting steroid hormone receptor pathways in the treatment of hormone dependent cancers. Curr. Pharm. Biotechnol. 2004, 5, 459–470. [Google Scholar] [CrossRef]
- Truong, T.H.; Lange, C.A. Deciphering Steroid Receptor Crosstalk in Hormone-Driven Cancers. Endocrinology 2018, 159, 3897–3907. [Google Scholar] [CrossRef]
- Palmieri, C.; Cheng, G.J.; Saji, S.; Zelada-Hedman, M.; Wärri, A.; Weihua, Z.; Van Noorden, S.; Wahlstrom, T.; Coombes, R.C.; Warner, M.; et al. Estrogen receptor beta in breast cancer. Endocr. Relat. Cancer 2002, 9, 1–13. [Google Scholar] [CrossRef]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Thompson, E.B. Transactivation functions of the N-terminal domains of nuclear hormone receptors: Protein folding and coactivator interactions. Mol. Endocrinol. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Zilliacus, J.; Wright, A.P.; Carlstedt-Duke, J.; Gustafsson, J.A. Structural determinants of DNA-binding specificity by steroid receptors. Mol. Endocrinol. 1995, 9, 389–400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Hill, K.K.; Roemer, S.C.; Churchill, M.E.; Edwards, D.P. Structural and functional analysis of domains of the progesterone receptor. Mol. Cell. Endocrinol. 2012, 348, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharm. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Oren, I.; Fleishman, S.J.; Kessel, A.; Ben-Tal, N. Free diffusion of steroid hormones across biomembranes: A simplex search with implicit solvent model calculations. Biophys. J. 2004, 87, 768–779. [Google Scholar] [CrossRef]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Home Page. Available online: https://www.cancer.org/cancer/prostate-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 20 April 2021).
- Kanno, Y.; Watanabe, M.; Kimura, T.; Nonomura, K.; Tanaka, S.; Hatakeyama, S. TRIM29 as a novel prostate basal cell marker for diagnosis of prostate cancer. Acta Histochem. 2014, 116, 708–712. [Google Scholar] [CrossRef]
- Qi, L.; Lu, Z.; Sun, Y.H.; Song, H.T.; Xu, W.K. TRIM16 suppresses the progression of prostate tumors by inhibiting the Snail signaling pathway. Int. J. Mol. Med. 2016, 38, 1734–1742. [Google Scholar] [CrossRef]
- Kikuchi, M.; Okumura, F.; Tsukiyama, T.; Watanabe, M.; Miyajima, N.; Tanaka, J.; Imamura, M.; Hatakeyama, S. TRIM24 mediates ligand-dependent activation of androgen receptor and is repressed by a bromodomain-containing protein, BRD7, in prostate cancer cells. Biochim. Biophys. Acta 2009, 1793, 1828–1836. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Hoekman, L.; Bleijerveld, O.B.; Mirza, T.; Wessels, L.F.A.; van Weerden, W.M.; Altelaar, A.F.M.; Bergman, A.M.; et al. Endogenous androgen receptor proteomic profiling reveals genomic subcomplex involved in prostate tumorigenesis. Oncogene 2018, 37, 313–322. [Google Scholar] [CrossRef]
- Miyajima, N.; Maruyama, S.; Bohgaki, M.; Kano, S.; Shigemura, M.; Shinohara, N.; Nonomura, K.; Hatakeyama, S. TRIM68 regulates ligand-dependent transcription of androgen receptor in prostate cancer cells. Cancer Res. 2008, 68, 3486–3494. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- vom Baur, E.; Zechel, C.; Heery, D.; Heine, M.J.; Garnier, J.M.; Vivat, V.; Le Douarin, B.; Gronemeyer, H.; Chambon, P.; Losson, R. Differential ligand-dependent interactions between the AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J. 1996, 15, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, B.; Nielsen, A.L.; Garnier, J.M.; Ichinose, H.; Jeanmougin, F.; Losson, R.; Chambon, P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996, 15, 6701–6715. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Fong, K.W.; Zhao, J.C.; Song, B.; Zheng, B.; Yu, J. TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat. Commun. 2018, 9, 5007. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Guan, X.; An, H.; Baihetiya, A.; Wang, W.; Shao, W.; Yang, H.; Wang, Y. Epigenetic silencing of miR-137 induces resistance to bicalutamide by targeting TRIM24 in prostate cancer cells. Am. J. Transl. Res. 2019, 11, 3226–3237. [Google Scholar]
- Theurillat, J.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Burleson, M. SPOP and cancer: A systematic review. Am. J. Cancer Res. 2020, 10, 704–726. [Google Scholar] [PubMed]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.; Burke, L.J.; Barreto, G.; Goeman, F.; Greb, H.; Arnold, R.; Schultheiss, H.; Brehm, A.; Kouzarides, T.; Lobanenkov, V.; et al. Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic Acids Res. 2000, 28, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborgh, N.; Spans, L.; Helsen, C.; Clinckemalie, L.; Dubois, V.; Lerut, E.; Boonen, S.; Vanderschueren, D.; Claessens, F. The transcription intermediary factor 1beta coactivates the androgen receptor. J. Endocrinol. Investig. 2013, 36, 699–706. [Google Scholar] [CrossRef]
- Czerwinska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Balint, I.; Muller, A.; Nagy, A.; Kovacs, G. Cloning and characterisation of the RBCC728/TRIM36 zinc-binding protein from the tumor suppressor gene region at chromosome 5q22.3. Gene 2004, 332, 45–50. [Google Scholar] [CrossRef]
- Kimura, N.; Yamada, Y.; Takayama, K.I.; Fujimura, T.; Takahashi, S.; Kume, H.; Inoue, S. Androgen-responsive tripartite motif 36 enhances tumor-suppressive effect by regulating apoptosis-related pathway in prostate cancer. Cancer Sci. 2018, 109, 3840–3852. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Liang, C.; Wang, S.; Qin, C.; Bao, M.; Cheng, G.; Liu, B.; Shao, P.; Lv, Q.; Song, N.; Hua, L.; et al. TRIM36, a novel androgen-responsive gene, enhances anti-androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis. 2018, 9, 155. [Google Scholar] [CrossRef]
- Ruiz-Hernandez, A.; Kuo, C.C.; Rentero-Garrido, P.; Tang, W.Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenetics 2015, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Beck, S.; Kogner, P.; Martinsson, T.; Caren, H. Genome-wide methylation profiling identifies novel methylated genes in neuroblastoma tumors. Epigenetics 2016, 11, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Gong, Y.; He, Y.; Gao, W.-Q.; Dong, B.; Zhu, H.H.; Xue, W. TRIM59 is Suppressed by Androgen Receptor and Acts to Promote Lineage Plasticity and Neuroendocrine Differentiation in Prostate Cancer. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Lin, W.Y.; Wang, H.; Song, X.; Zhang, S.X.; Zhou, P.S.; Sun, J.M.; Li, J.S. Knockdown of tripartite motif 59 (TRIM59) inhibits tumor growth in prostate cancer. Eur. Rev. Med. Pharm. Sci. 2016, 20, 4864–4873. [Google Scholar]
- Tan, Y.; Yao, H.; Hu, J.; Liu, L. Knockdown of TRIM44 Inhibits the Proliferation and Invasion in Prostate Cancer Cells. Oncol. Res. 2017, 25, 1253–1259. [Google Scholar] [CrossRef]
- Cao, H.; Gao, R.; Chen, L.; Feng, Y. TRIM66 promotes malignant progression of prostate carcinoma through the JAK/STAT pathway. FEBS Open Bio 2020, 10, 515–524. [Google Scholar] [CrossRef]
- Qi, G.; Lu, G.; Yu, J.; Zhao, Y.; Wang, C.; Zhang, H.; Xia, Q. Up-regulation of TIF1gamma by valproic acid inhibits the epithelial mesenchymal transition in prostate carcinoma through TGF-beta/Smad signaling pathway. Eur. J. Pharm. 2019, 860, 172551. [Google Scholar] [CrossRef]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef]
- He, D.; Mu, Z.M.; Le, X.; Hsieh, J.T.; Pong, R.C.; Chung, L.W.; Chang, K.S. Adenovirus-mediated expression of PML suppresses growth and tumorigenicity of prostate cancer cells. Cancer Res. 1997, 57, 1868–1872. [Google Scholar] [PubMed]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Carracedo, A.; Egia, A.; Lo-Coco, F.; Teruya-Feldstein, J.; Pandolfi, P.P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008, 455, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef]
- Yuan, W.C.; Lee, Y.R.; Huang, S.F.; Lin, Y.M.; Chen, T.Y.; Chung, H.C.; Tsai, C.H.; Chen, H.Y.; Chiang, C.T.; Lai, C.K.; et al. A Cullin3-KLHL20 Ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell 2011, 20, 214–228. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chatterjee, U.; Ghosh, M.K. Activation of protein kinase CK2 attenuates FOXO3a functioning in a PML-dependent manner: Implications in human prostate cancer. Cell Death Dis. 2013, 4, e543. [Google Scholar] [CrossRef]
- Birch, S.E.; Kench, J.G.; Takano, E.; Chan, P.; Chan, A.L.; Chiam, K.; Veillard, A.S.; Stricker, P.; Haupt, S.; Haupt, Y.; et al. Expression of E6AP and PML predicts for prostate cancer progression and cancer-specific death. Ann. Oncol. 2014, 25, 2392–2397. [Google Scholar] [CrossRef]
- Kalathur, M.; Toso, A.; Chen, J.; Revandkar, A.; Danzer-Baltzer, C.; Guccini, I.; Alajati, A.; Sarti, M.; Pinton, S.; Brambilla, L.; et al. A chemogenomic screening identifies CK2 as a target for pro-senescence therapy in PTEN-deficient tumours. Nat. Commun. 2015, 6, 7227. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.J.; Raghu, D.; Chan, A.L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yeh, S.D.; Xie, S.; Altuwaijri, S.; Ni, J.; Hu, Y.C.; Chen, Y.T.; Bao, B.Y.; Su, C.H.; Chang, C. Androgen suppresses PML protein expression in prostate cancer CWR22R cells. Biochem. Biophys. Res. Commun. 2004, 314, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Mangosh, T.L.; Awadallah, W.N.; Grabowska, M.M.; Taylor, D.J. SLX4IP promotes telomere maintenance in androgen receptor-independent castration-resistant prostate cancer through ALT-like telomeric PML localization. Mol. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef]
- Addison, J.B.; Koontz, C.; Fugett, J.H.; Creighton, C.J.; Chen, D.; Farrugia, M.K.; Padon, R.R.; Voronkova, M.A.; McLaughlin, S.L.; Livengood, R.H.; et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer Res. 2015, 75, 344–355. [Google Scholar] [CrossRef]
- Xue, M.; Zhang, K.; Mu, K.; Xu, J.; Yang, H.; Liu, Y.; Wang, B.; Wang, Z.; Li, Z.; Kong, Q.; et al. Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis 2019, 8, 30. [Google Scholar] [CrossRef]
- Kawabata, H.; Azuma, K.; Ikeda, K.; Sugitani, I.; Kinowaki, K.; Fujii, T.; Osaki, A.; Saeki, T.; Horie-Inoue, K.; Inoue, S. TRIM44 Is a Poor Prognostic Factor for Breast Cancer Patients as a Modulator of NF-κB Signaling. Int. J. Mol. Sci. 2017, 18, 1931. [Google Scholar] [CrossRef]
- Chen, W.-x.; Cheng, L.; Xu, L.-y.; Qian, Q.; Zhu, Y.-l. Bioinformatics analysis of prognostic value of TRIM13 gene in breast cancer. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Tan, P.; Ye, Y.; He, L.; Xie, J.; Jing, J.; Ma, G.; Pan, H.; Han, L.; Han, W.; Zhou, Y. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018, 16, e3000051. [Google Scholar] [CrossRef]
- Fowler, A.M.; Solodin, N.M.; Valley, C.C.; Alarid, E.T. Altered target gene regulation controlled by estrogen receptor-alpha concentration. Mol. Endocrinol. 2006, 20, 291–301. [Google Scholar] [CrossRef][Green Version]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Orimo, A.; Hosoi, T.; Kondo, S.; Toyoshima, H.; Kondo, T.; Ikegami, A.; Ouchi, Y.; Orimo, H.; Muramatsu, M. Genomic binding-site cloning reveals an estrogen-responsive gene that encodes a RING finger protein. Proc. Natl. Acad. Sci. USA 1993, 90, 11117–11121. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Orimo, A.; Higashi, Y.; Muramatsu, M.; Inoue, S. Efp as a primary estrogen-responsive gene in human breast cancer. FEBS Lett. 2000, 472, 9–13. [Google Scholar] [CrossRef]
- Orimo, A.; Inoue, S.; Minowa, O.; Tominaga, N.; Tomioka, Y.; Sato, M.; Kuno, J.; Hiroi, H.; Shimizu, Y.; Suzuki, M.; et al. Underdeveloped uterus and reduced estrogen responsiveness in mice with disruption of the estrogen-responsive finger protein gene, which is a direct target of estrogen receptor alpha. Proc. Natl. Acad. Sci. USA 1999, 96, 12027–12032. [Google Scholar] [CrossRef]
- Suzuki, T.; Urano, T.; Tsukui, T.; Horie-Inoue, K.; Moriya, T.; Ishida, T.; Muramatsu, M.; Ouchi, Y.; Sasano, H.; Inoue, S. Estrogen-responsive finger protein as a new potential biomarker for breast cancer. Clin. Cancer Res. 2005, 11, 6148–6154. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Alvarez, M.J.; Sabio, E.Y.; Reyngold, M.; Makarov, V.; Mukherjee, S.; Lee, K.W.; Desrichard, A.; Turcan, Ş.; Dalin, M.G.; et al. An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep. 2017, 20, 1623–1640. [Google Scholar] [CrossRef]
- Urano, T.; Saito, T.; Tsukui, T.; Fujita, M.; Hosoi, T.; Muramatsu, M.; Ouchi, Y.; Inoue, S. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature 2002, 417, 871–875. [Google Scholar] [CrossRef]
- Akrap, N.; Andersson, D.; Bom, E.; Gregersson, P.; Ståhlberg, A.; Landberg, G. Identification of Distinct Breast Cancer Stem Cell Populations Based on Single-Cell Analyses of Functionally Enriched Stem and Progenitor Pools. Stem Cell Rep. 2016, 6, 121–136. [Google Scholar] [CrossRef]
- Wei, C.; Cheng, J.; Zhou, B.; Zhu, L.; Khan, M.A.; He, T.; Zhou, S.; He, J.; Lu, X.; Chen, H.; et al. Tripartite motif containing 28 (TRIM28) promotes breast cancer metastasis by stabilizing TWIST1 protein. Sci. Rep. 2016, 6, 29822. [Google Scholar] [CrossRef]
- Czerwińska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozańska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, Y.; Zhong, C.; Hu, J.; Hu, H.; Zhou, D.; Cao, M. Decreased expression of TRIM21 indicates unfavorable outcome and promotes cell growth in breast cancer. Cancer Manag. Res. 2018, 10, 3687–3696. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Y.; Li, B.; Zhang, J.; Dong, Z.; Hu, X.; Wan, Y. TRIM21 mediates ubiquitination of Snail and modulates epithelial to mesenchymal transition in breast cancer cells. Int. J. Biol. Macromol. 2019, 124, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Xu, T.; Tian, T.; Fu, X.; Wang, W.; Li, S.; Shi, T.; Suo, A.; Ruan, Z.; Guo, H.; et al. Tripartite motif 16 suppresses breast cancer stem cell properties through regulation of Gli-1 degradation via the ubiquitin-proteasome pathway. Oncol. Rep. 2016, 35, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Sachini, N.; Arampatzi, P.; Klonizakis, A.; Nikolaou, C.; Makatounakis, T.; Lam, E.W.; Kretsovali, A.; Papamatheakis, J. Promyelocytic leukemia protein (PML) controls breast cancer cell proliferation by modulating Forkhead transcription factors. Mol. Oncol. 2019, 13, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Arreal, L.; Piva, M.; Fernández, S.; Revandkar, A.; Schaub- Clerigué, A.; Villanueva, J.; Zabala-Letona, A.; Pujana, M.; Astobiza, I.; Cortazar, A.R.; et al. Targeting PML in triple negative breast cancer elicits growth suppression and senescence. Cell Death Differ. 2020, 27, 1186–1199. [Google Scholar] [CrossRef]
- Papachristou, E.K.; Kishore, K.; Holding, A.N.; Harvey, K.; Roumeliotis, T.I.; Chilamakuri, C.S.R.; Omarjee, S.; Chia, K.M.; Swarbrick, A.; Lim, E.; et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018, 9, 2311. [Google Scholar] [CrossRef]
- Song, W.; Wang, Z.; Gu, X.; Wang, A.; Chen, X.; Miao, H.; Chu, J.; Tian, Y. TRIM11 promotes proliferation and glycolysis of breast cancer cells via targeting AKT/GLUT1 pathway. OncoTargets Ther. 2019, 12, 4975–4984. [Google Scholar] [CrossRef]
- Zhao, T.T.; Jin, F.; Li, J.G.; Xu, Y.Y.; Dong, H.T.; Liu, Q.; Xing, P.; Zhu, G.L.; Xu, H.; Yin, S.C.; et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway. J. Cancer 2018, 9, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Gazin, C.; Chamberlain, L.; Ou, J.; Zhu, X.; Tushir, J.S.; Virbasius, C.M.; Lin, L.; Zhu, L.J.; Wajapeyee, N.; et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014, 516, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Azuma, K.; Sato, J.; Kinowaki, K.; Ikeda, K.; Kawabata, H.; Inoue, S. Abstract P3-06-11: TRIM39 is a poor prognostic factor for breast cancer patients and promotes proliferation and migration of breast cancer cells. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Xie, Z.; Lu, H. Knockdown of TRIM47 inhibits breast cancer tumorigenesis and progression through the inactivation of PI3K/Akt pathway. Chem. Biol. Interact. 2020, 317, 108960. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Pan, W.; Ma, Y.; Xu, X.; Gao, Y.; He, Y.; Wei, L.; Zhang, J. A novel oncogene TRIM63 promotes cell proliferation and migration via activating Wnt/β-catenin signaling pathway in breast cancer. Pathol. Res. Pract. 2019, 215, 152573. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J. Clin. Investig. 2014, 124, 564–579. [Google Scholar] [CrossRef]
- Kassem, L.; Deygas, M.; Fattet, L.; Lopez, J.; Goulvent, T.; Lavergne, E.; Chabaud, S.; Carrabin, N.; Chopin, N.; Bachelot, T.; et al. TIF1γ interferes with TGFβ1/SMAD4 signaling to promote poor outcome in operable breast cancer patients. BMC Cancer 2015, 15, 453. [Google Scholar] [CrossRef] [PubMed]
- Ottevanger, P.B. Ovarian cancer stem cells more questions than answers. Semin. Cancer Biol. 2017, 44, 67–71. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Rao, B.R.; Slotman, B.J. Endocrine factors in common epithelial ovarian cancer. Endocr. Rev. 1991, 12, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Modugno, F.; Laskey, R.; Smith, A.L.; Andersen, C.L.; Haluska, P.; Oesterreich, S. Hormone response in ovarian cancer: Time to reconsider as a clinical target? Endocr. Relat. Cancer 2012, 19, R255–R279. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Zhan, F.; Bellone, S.; Palmieri, M.; Cane, S.; Bignotti, E.; Anfossi, S.; Gokden, M.; Dunn, D.; Roman, J.J.; et al. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: Identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int. J. Cancer 2004, 112, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Qi, J.; Chu, G.; Liu, Z. Tripartite Motif 16 Inhibits the Migration and Invasion in Ovarian Cancer Cells. Oncol. Res. 2017, 25, 551–558. [Google Scholar] [CrossRef]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Liu, P.; Ma, X.; Ma, X.; Zhu, L.; Lin, Y.; You, Y.; Yu, W.; Ma, D.; Sun, C.; et al. TRIM50 acts as a novel Src suppressor and inhibits ovarian cancer progression. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1412–1420. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Wang, X.; Zhang, X.; Chen, Y.; Bai, J.; Di, W. TRIM59 Is a Novel Marker of Poor Prognosis and Promotes Malignant Progression of Ovarian Cancer by Inducing Annexin A2 Expression. Int. J. Biol. Sci. 2018, 14, 2073–2082. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, H.; Wang, Y.; Zhang, P.; Qi, Y. Tripartite Motif-Containing Protein 59 (TRIM59) Promotes Epithelial Ovarian Cancer Progression via the Focal Adhesion Kinase (FAK)/AKT/Matrix Metalloproteinase (MMP) Pathway. Med. Sci. Monit. 2019, 25, 3366–3373. [Google Scholar] [CrossRef]
- Horio, M.; Kato, T.; Mii, S.; Enomoto, A.; Asai, M.; Asai, N.; Murakumo, Y.; Shibata, K.; Kikkawa, F.; Takahashi, M. Expression of RET finger protein predicts chemoresistance in epithelial ovarian cancer. Cancer Med. 2012, 1, 218–229. [Google Scholar] [CrossRef]
- Jiang, J.; Xie, C.; Liu, Y.; Shi, Q.; Chen, Y. Up-regulation of miR-383-5p suppresses proliferation and enhances chemosensitivity in ovarian cancer cells by targeting TRIM27. Biomed. Pharm. 2019, 109, 595–601. [Google Scholar] [CrossRef]
- Ma, Y.; Wei, Z.; Bast, R.C., Jr.; Wang, Z.; Li, Y.; Gao, M.; Liu, Y.; Wang, X.; Guo, C.; Zhang, L.; et al. Downregulation of TRIM27 expression inhibits the proliferation of ovarian cancer cells in vitro and in vivo. Lab. Investig. 2016, 96, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, L.; Li, C.; Luo, N.; Chen, R.; Li, L.; Yu, F.; Cheng, Z. TRIM52 plays an oncogenic role in ovarian cancer associated with NF-kB pathway. Cell Death Dis. 2018, 9, 908. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, M.; Akahira, J.; Suzuki, T.; Inoue, S.; Ito, K.; Moriya, T.; Sasano, H.; Okamura, K.; Yaegashi, N. Expression of estrogen-responsive finger protein (Efp) is associated with advanced disease in human epithelial ovarian cancer. Gynecol. Oncol. 2005, 99, 664–670. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Leber, M.F.; Efferth, T. Molecular principles of cancer invasion and metastasis (review). Int. J. Oncol. 2009, 34, 881–895. [Google Scholar] [CrossRef]
- Wang, Y.H.; Sui, X.M.; Sui, Y.N.; Zhu, Q.W.; Yan, K.; Wang, L.S.; Wang, F.; Zhou, J.H. BRD4 induces cell migration and invasion in HCC cells through MMP-2 and MMP-9 activation mediated by the Sonic hedgehog signaling pathway. Oncol. Lett. 2015, 10, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Suyama, K.; Onishi, H.; Imaizumi, A.; Shinkai, K.; Umebayashi, M.; Kubo, M.; Mizuuchi, Y.; Oda, Y.; Tanaka, M.; Nakamura, M.; et al. CD24 suppresses malignant phenotype by downregulation of SHH transcription through STAT1 inhibition in breast cancer cells. Cancer Lett. 2016, 374, 44–53. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef]
- Pichierri, P.; Rosselli, F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004, 23, 1178–1187. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Pardee, A.B. Exploiting Cancer Cell Cycling for Selective Protection of Normal Cells. Cancer Res. 2001, 61, 4301–4305. [Google Scholar]
- Matsuoka, T.; Yashiro, M. The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers 2014, 6, 1441–1463. [Google Scholar] [CrossRef]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Lokman, N.A.; Pyragius, C.E.; Ruszkiewicz, A.; Oehler, M.K.; Ricciardelli, C. Annexin A2 and S100A10 are independent predictors of serous ovarian cancer outcome. Transl. Res. 2016, 171, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Jin, H.; Bian, C.; He, H.; Liu, X.; Zhang, S.; Wang, Y.; Shao, R.G. MR-1 modulates proliferation and migration of human hepatoma HepG2 cells through myosin light chains-2 (MLC2)/focal adhesion kinase (FAK)/Akt signaling pathway. J. Biol. Chem. 2008, 283, 35598–35605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, J.; Ma, J. Proliferation and invasion of ovarian cancer cells are suppressed by knockdown of TRIM11. Oncol. Lett. 2017, 14, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Morisset, J.; Vachon, P.H.; Reed, J.C.; Lainé, J.; Rivard, N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell Biochem. 2000, 79, 355–369. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Punn, A.; Chen, J.; Randeva, H.S. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 2008, 78, 356–365. [Google Scholar] [CrossRef]
- Liu, S.B.; Shen, Z.F.; Guo, Y.J.; Cao, L.X.; Xu, Y. PML silencing inhibits cell proliferation and induces DNA damage in cultured ovarian cancer cells. Biomed. Rep. 2017, 7, 29–35. [Google Scholar] [CrossRef]
- Pan, W.W.; Zhou, J.J.; Liu, X.M.; Xu, Y.; Guo, L.J.; Yu, C.; Shi, Q.H.; Fan, H.Y. Death domain-associated protein DAXX promotes ovarian cancer development and chemoresistance. J. Biol. Chem. 2013, 288, 13620–13630. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Jongen, V.; Briët, J.; de Jong, R.; ten Hoor, K.; Boezen, M.; van der Zee, A.; Nijman, H.; Hollema, H. Expression of estrogen receptor-alpha and -beta and progesterone receptor-A and -B in a large cohort of patients with endometrioid endometrial cancer. Gynecol. Oncol. 2009, 112, 537–542. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, B.; Wei, M.; Xu, Z.; Kong, W.; Deng, K.; Xu, X.; Zhang, L.; Ζhao, X.; Yan, L. TRIM22 inhibits endometrial cancer progression through the NOD2/NF-κB signaling pathway and confers a favorable prognosis. Int. J. Oncol. 2020, 56, 1225–1239. [Google Scholar] [CrossRef]
- Saito-Kanatani, M.; Urano, T.; Hiroi, H.; Momoeda, M.; Ito, M.; Fujii, T.; Inoue, S. Identification of TRIM22 as a progesterone-responsive gene in Ishikawa endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2015, 154, 217–225. [Google Scholar] [CrossRef]
- Nakayama, H.; Sano, T.; Motegi, A.; Oyama, T.; Nakajima, T. Increasing 14-3-3 sigma expression with declining estrogen receptor alpha and estrogen-responsive finger protein expression defines malignant progression of endometrial carcinoma. Pathol. Int. 2005, 55, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zhang, P.; Zhao, S.; Zhang, J.; Wang, B. Regulation of the vascular endothelial growth factor and growth by estrogen and antiestrogens through Efp in Ishikawa endometrial carcinoma cells. Oncol. Rep. 2009, 21, 395–401. [Google Scholar] [PubMed]
- Sato, W.; Ikeda, K.; Urano, T.; Abe, Y.; Nakasato, N.; Horie-Inoue, K.; Takeda, S.; Inoue, S. Efp promotes in vitro and in vivo growth of endometrial cancer cells along with the activation of nuclear factor-κB signaling. PLoS ONE 2018, 13, e0208351. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yin, H.; Meng, F.; Liu, S.; Liu, H.; Ma, R. High TRIM44 expression in endometrial carcinoma is associated with a poorer patient outcome. Pathol. Res. Pract. 2018, 214, 727–731. [Google Scholar] [CrossRef]
- Nabergoj, S.; Mlinarič-Raščan, I.; Jakopin, Ž. Harnessing the untapped potential of nucleotide-binding oligomerization domain ligands for cancer immunotherapy. Med. Res. Rev. 2019, 39, 1447–1484. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Sivaramakrishnan, G.; Sun, Y.; Tan, S.K.; Lin, V.C. Dynamic localization of tripartite motif-containing 22 in nuclear and nucleolar bodies. Exp. Cell Res. 2009, 315, 1521–1532. [Google Scholar] [CrossRef]
- Lee, W.L.; Yen, M.S.; Chao, K.C.; Yuan, C.C.; Ng, H.T.; Chao, H.T.; Lee, F.K.; Wang, P.H. Hormone therapy for patients with advanced or recurrent endometrial cancer. J. Chin. Med. Assoc. 2014, 77, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zhao, S.; Xu, L.; Chen, A.; Dai, S. Expression of Efp, VEGF and bFGF in normal, hyperplastic and malignant endometrial tissue. Oncol. Rep. 2010, 23, 795–799. [Google Scholar] [PubMed]
- Guidi, A.J.; Abu-Jawdeh, G.; Tognazzi, K.; Dvorak, H.F.; Brown, L.F. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in endometrial carcinoma. Cancer 1996, 78, 454–460. [Google Scholar] [CrossRef]
- Wallace, A.E.; Gibson, D.A.; Saunders, P.T.; Jabbour, H.N. Inflammatory events in endometrial adenocarcinoma. J. Endocrinol. 2010, 206, 141–157. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Kato, T.; Enomoto, A.; Nakamura, N.; Shimono, Y.; Jijiwa, M.; Asai, N.; Murakumo, Y.; Shibata, K.; Kikkawa, F.; et al. Expression of Ret finger protein correlates with outcomes in endometrial cancer. Cancer Sci. 2009, 100, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.S.; Poncet-Montange, G.; Liu, G.; Petrocchi, A.; Reyna, N.; Subramanian, G.; Theroff, J.; Yau, A.; Kost-Alimova, M.; Bardenhagen, J.P.; et al. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J. Med. Chem. 2016, 59, 1440–1454. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauletto, E.; Eickhoff, N.; Padrão, N.A.; Blattner, C.; Zwart, W. TRIMming Down Hormone-Driven Cancers: The Biological Impact of TRIM Proteins on Tumor Development, Progression and Prognostication. Cells 2021, 10, 1517. https://doi.org/10.3390/cells10061517
Pauletto E, Eickhoff N, Padrão NA, Blattner C, Zwart W. TRIMming Down Hormone-Driven Cancers: The Biological Impact of TRIM Proteins on Tumor Development, Progression and Prognostication. Cells. 2021; 10(6):1517. https://doi.org/10.3390/cells10061517
Chicago/Turabian StylePauletto, Eleonora, Nils Eickhoff, Nuno A. Padrão, Christine Blattner, and Wilbert Zwart. 2021. "TRIMming Down Hormone-Driven Cancers: The Biological Impact of TRIM Proteins on Tumor Development, Progression and Prognostication" Cells 10, no. 6: 1517. https://doi.org/10.3390/cells10061517
APA StylePauletto, E., Eickhoff, N., Padrão, N. A., Blattner, C., & Zwart, W. (2021). TRIMming Down Hormone-Driven Cancers: The Biological Impact of TRIM Proteins on Tumor Development, Progression and Prognostication. Cells, 10(6), 1517. https://doi.org/10.3390/cells10061517