
Fibroblast Memory in Development, Homeostasis and Disease


{kind=link}
{kind=link}
Abstract
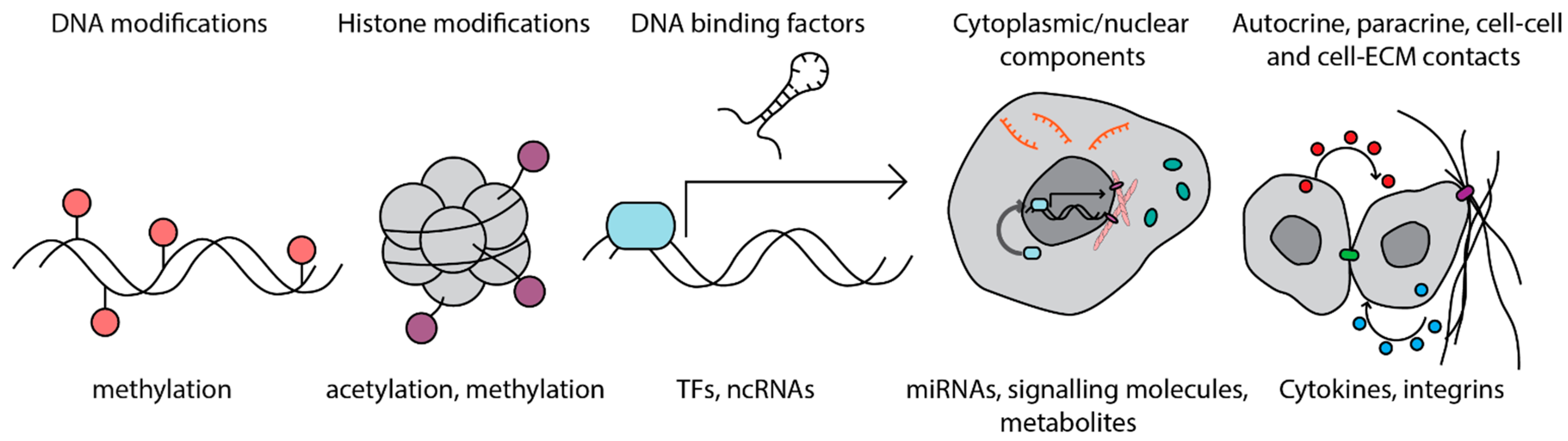
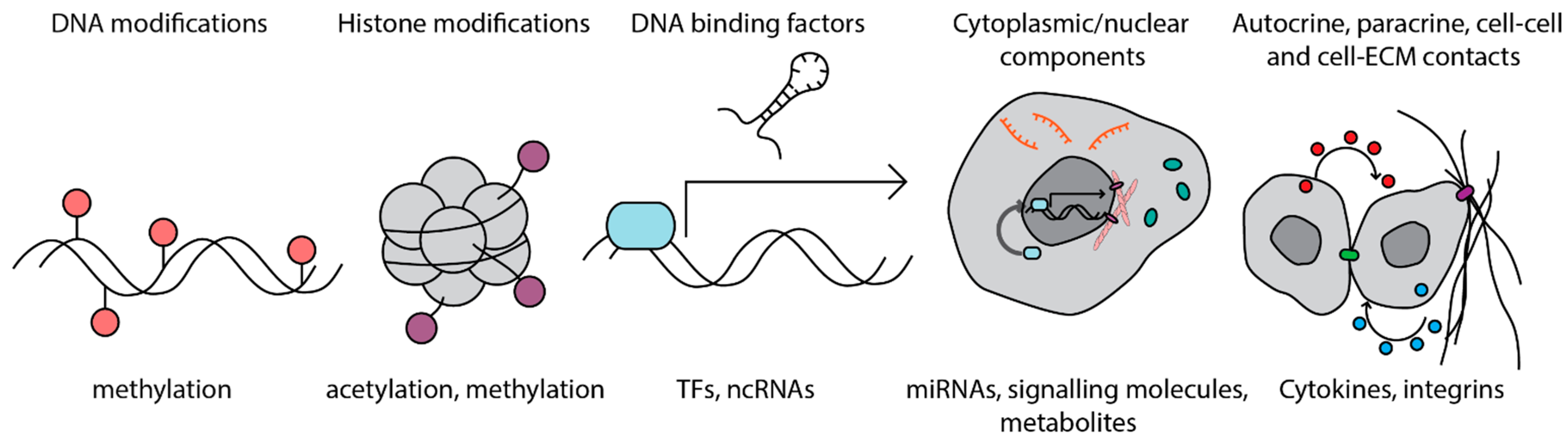
:1. Introduction
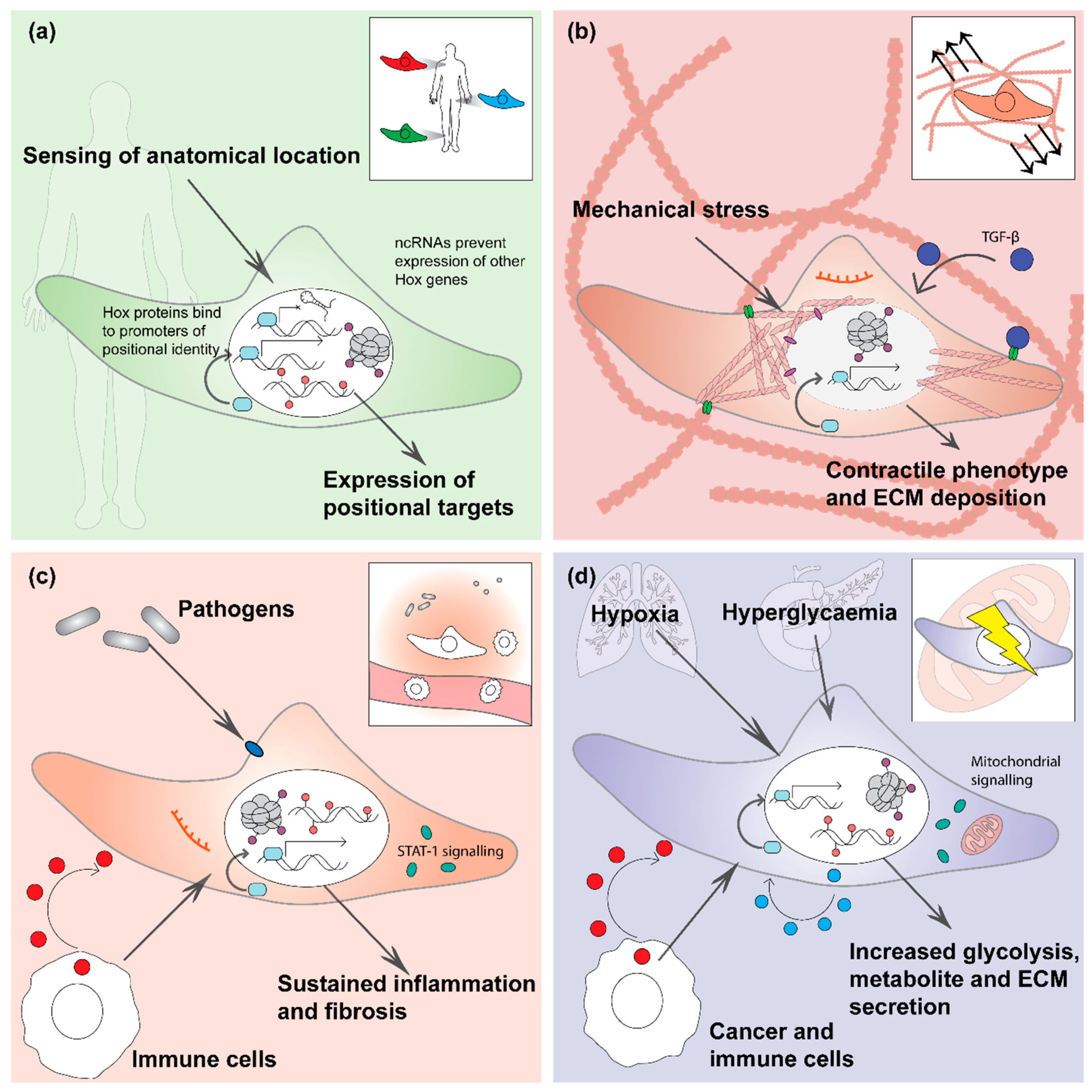
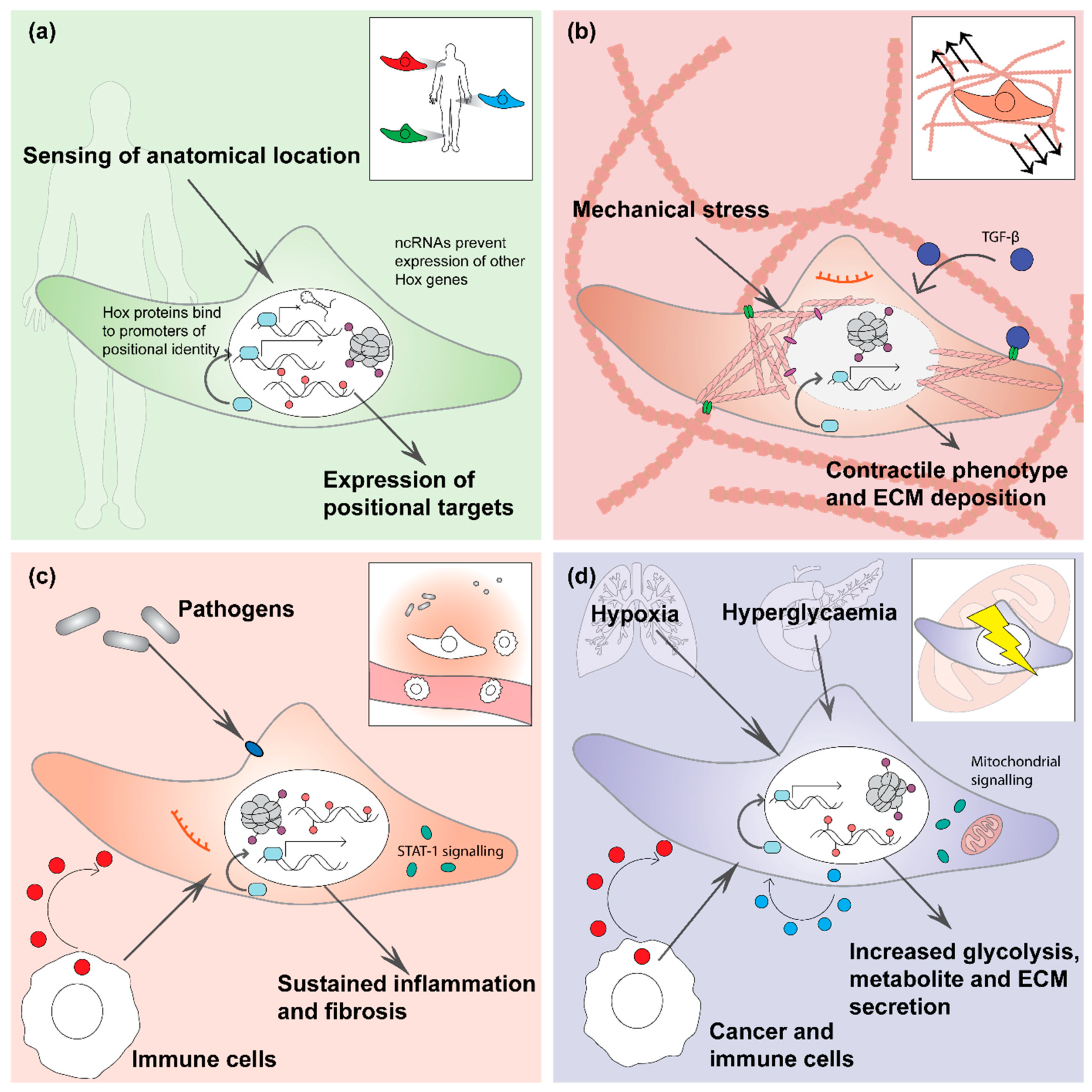
2. Positional Memory
3. Mechanical Memory
4. Inflammatory Memory
5. Metabolic Memory
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Netea, M.G.; Latz, E.; Mills, K.H.G.; O’Neill, L.A.J. Innate Immune Memory: A Paradigm Shift in Understanding Host Defense. Nat. Immunol. 2015, 16, 675–679. [Google Scholar] [CrossRef]
- Naik, S.; Larsen, S.B.; Gomez, N.C.; Alaverdyan, K.; Sendoel, A.; Yuan, S.; Polak, L.; Kulukian, A.; Chai, S.; Fuchs, E. Inflammatory Memory Sensitizes Skin Epithelial Stem Cells to Tissue Damage. Nature 2017, 550, 475–480. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Rognoni, E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr. Rheumatol. Rep. 2020, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-Tissue Organization of the Fibroblast Lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y. Anatomic Demarcation of Cells: Genes to Patterns. Science 2009, 326, 1206–1207. [Google Scholar] [CrossRef]
- Chang, H.Y.; Chi, J.-T.J.-T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, Topographic Differentiation, and Positional Memory in Human Fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Bondre, C.; Gladstone, H.B.; Brown, P.O.; Chang, H.Y. Anatomic Demarcation by Positional Variation in Fibroblast Gene Expression Programs. PLoS Genet. 2006, 2, e119. [Google Scholar] [CrossRef]
- Rinn, J.L.; Wang, J.K.; Allen, N.; Brugmann, S.A.; Mikels, A.J.; Liu, H.; Ridky, T.W.; Stadler, H.S.; Nusse, R.; Helms, J.A.; et al. A Dermal HOX Transcriptional Program Regulates Site-Specific Epidermal Fate. Genes Dev. 2008, 22, 303–307. [Google Scholar] [CrossRef] [Green Version]
- Frank-Bertoncelj, M.; Trenkmann, M.; Klein, K.; Karouzakis, E.; Rehrauer, H.; Bratus, A.; Kolling, C.; Armaka, M.; Filer, A.; Michel, B.A.; et al. Epigenetically-Driven Anatomical Diversity of Synovial Fibroblasts Guides Joint-Specific Fibroblast Functions. Nat. Commun. 2017, 8, 14852. [Google Scholar] [CrossRef] [Green Version]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue Specific Human Fibroblast Differential Expression Based on RNAsequencing Analysis. BMC Genom. 2019, 20, 308. [Google Scholar] [CrossRef]
- Leavitt, T.; Hu, M.S.; Borrelli, M.R.; Januszyk, M.; Garcia, J.T.; Ransom, R.C.; Mascharak, S.; DesJardins-Park, H.E.; Litzenburger, U.M.; Walmsley, G.G.; et al. Prrx1 Fibroblasts Represent a Pro-Fibrotic Lineage in the Mouse Ventral Dermis. Cell Rep. 2020, 33, 108356. [Google Scholar] [CrossRef]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Identification and Isolation of a Dermal Lineage with Intrinsic Fibrogenic Potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef] [Green Version]
- Terranova, R.; Agherbi, H.; Boned, A.; Meresse, S.; Djabali, M. Histone and DNA Methylation Defects at Hox Genes in Mice Expressing a SET Domain-Truncated Form of Mll. Proc. Natl. Acad. Sci. USA 2006, 103, 6629–6634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshnikova, N.; Duboule, D. Epigenetic Temporal Control of Mouse Hox Genes in Vivo. Science 2009, 324, 1320–1323. [Google Scholar] [CrossRef]
- Lu, L.; Zhu, G.; Zhang, C.; Deng, Q.; Katsaros, D.; Mayne, S.T.; Risch, H.A.; Mu, L.; Canuto, E.M.; Gregori, G.; et al. Association of Large Noncoding RNA HOTAIR Expression and Its Downstream Intergenic CpG Island Methylation with Survival in Breast Cancer. Breast Cancer Res. Treat. 2012, 136, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto Attolini, C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [Green Version]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.-P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-Cell Transcriptomes of the Human Skin Reveal Age-Related Loss of Fibroblast Priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast Proliferation and Heterogeneity Are Supported by Macrophages during Skin Repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.A.; Kretzschmar, K.; Watt, F.M. Reprogramming Adult Dermis to a Neonatal State through Epidermal Activation of β-Catenin. Development 2011, 138, 5189–5199. [Google Scholar] [CrossRef] [Green Version]
- Philippeos, C.; Telerman, S.B.; Oulès, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Investig. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Korosec, A.; Frech, S.; Gesslbauer, B.; Vierhapper, M.; Radtke, C.; Petzelbauer, P.; Lichtenberger, B.M. Lineage Identity and Location within the Dermis Determine the Function of Papillary and Reticular Fibroblasts in Human Skin. J. Investig. Dermatol. 2019, 139, 342–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, A.; Iwabuchi, T.; Kishimoto, J.; Hamazaki, T.S.; Okochi, H. Long-Term Culture of Mouse Vibrissal Dermal Papilla Cells and De Novo Hair Follicle Induction. Tissue Eng. 2007, 13, 975–982. [Google Scholar] [CrossRef]
- Chen, H.; Wang, X.; Wang, J.; Shi, X.; Li, X.; Wang, J.; Li, D.; Zhu, Y.; Tan, W.; Tan, Z. In Vitro Adipogenesis and Long-Term Adipocyte Culture in Adipose Tissue-Derived Cell Banks. Biofabrication 2021, 13, 035052. [Google Scholar] [CrossRef]
- Rognoni, E.; Watt, F.M. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Goss, G.E.; Rognoni, E.; Salameti, V.; Watt, F.M. Distinct Fibroblast Lineages Give Rise to NG2+ Pericyte Populations in Mouse Skin Development and Repair. Front. Cell Dev. Biol. 2021, 9, 675080. [Google Scholar] [CrossRef]
- Abbasi, S.; Sinha, S.; Labit, E.; Rosin, N.L.; Yoon, G.; Rahmani, W.; Jaffer, A.; Sharma, N.; Hagner, A.; Shah, P.; et al. Distinct Regulatory Programs Control the Latent Regenerative Potential of Dermal Fibroblasts during Wound Healing. Cell Stem Cell 2020, 27, 396–412.e6. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.S.; Rognoni, E.; Lichtenberger, B.M.; Driskell, R.R.; Kretzschmar, K.; Hoste, E.; Watt, F.M. Fate of Prominin-1 Expressing Dermal Papilla Cells during Homeostasis, Wound Healing and Wnt Activation. J. Investig. Dermatol. 2015, 135, 2926–2934. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in Murine Cutaneous Fibrosis Originate From Adiponectin-Positive Intradermal Progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Mano, O.; Rutenberg-Schoenberg, M.; Rudolph, M.C.; Zirak, B.; Rivera-Gonzalez, G.C.; López-Giráldez, F.; Zarini, S.; Rezza, A.; et al. Dermal Adipocyte Lipolysis and Myofibroblast Conversion Are Required for Efficient Skin Repair. Cell Stem Cell 2020, 26, 880–895.e6. [Google Scholar] [CrossRef]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.-C.; et al. Regeneration of Fat Cells from Myofibroblasts during Wound Healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrini, J.L.; Chaudhry, S.; Sarrazy, V.; Koehler, A.; Hinz, B. The Mechanical Memory of Lung Myofibroblasts. Integr. Biol. 2012, 4, 410. [Google Scholar] [CrossRef]
- Rognoni, E.; Pisco, A.O.; Hiratsuka, T.; Sipilä, K.H.; Belmonte, J.M.; Mobasseri, S.A.; Philippeos, C.; Dilão, R.; Watt, F.M. Fibroblast State Switching Orchestrates Dermal Maturation and Wound Healing. Mol. Syst. Biol. 2018, 14, 1–20. [Google Scholar] [CrossRef]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The Inflammatory Speech of Fibroblasts. Immunol. Rev. 2021, 302, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The Myofibroblast. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Wipff, P.-J.; Rifkin, D.B.; Meister, J.-J.; Hinz, B. Myofibroblast Contraction Activates Latent TGF-Β1 from the Extracellular Matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.-Y.; Lee, K.-W.; Shin, D.; An, S.; Cho, K.-H.; Kim, S.-H. A Positive Feedback Loop Bi-Stably Activates Fibroblasts. Nat. Commun. 2018, 9, 3016. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaauboer, M.E.; Smit, T.H.; Hanemaaijer, R.; Stoop, R.; Everts, V. Cyclic Mechanical Stretch Reduces Myofibroblast Differentiation of Primary Lung Fibroblasts. Biochem. Biophys. Res. Commun. 2011, 404, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achterberg, V.F.; Buscemi, L.; Diekmann, H.; Smith-Clerc, J.; Schwengler, H.; Meister, J.-J.; Wenck, H.; Gallinat, S.; Hinz, B. The Nano-Scale Mechanical Properties of the Extracellular Matrix Regulate Dermal Fibroblast Function. J. Investig. Dermatol. 2014, 134, 1862–1872. [Google Scholar] [CrossRef] [Green Version]
- Childers, R.C.; Lucchesi, P.A.; Gooch, K.J. Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts. Int. J. Mol. Sci. 2021, 22, 6231. [Google Scholar] [CrossRef]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical Regulation of Cardiac Fibroblast Profibrotic Phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic Lineage Tracing Defines Myofibroblast Origin and Function in the Injured Heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized Fibroblast Differentiated States Underlie Scar Formation in the Infarcted Mouse Heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Sun, J.; Liu, Q.; Dodlapati, S.; Ming, H.; Wang, L.; Li, Y.; Li, R.; Jiang, Z.; Francis, J.; et al. The Landscape of Accessible Chromatin in Quiescent Cardiac Fibroblasts and Cardiac Fibroblasts Activated after Myocardial Infarction. Epigenetics 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and Fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janmey, P.A.; Wells, R.G.; Assoian, R.K.; McCulloch, C.A. From Tissue Mechanics to Transcription Factors. Differentiation 2013, 86, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Nava, M.M.; Wickström, S.A. Emerging Roles of Mechanical Forces in Chromatin Regulation. J. Cell Sci. 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ Drives Fibroblast Activation and Fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsharkawy, A.M.; Mann, D.A. Nuclear Factor-ΚB and the Hepatic Inflammation-Fibrosis-Cancer Axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef]
- Tajik, A.; Zhang, Y.; Wei, F.; Sun, J.; Jia, Q.; Zhou, W.; Singh, R.; Khanna, N.; Belmont, A.S.; Wang, N. Transcription Upregulation via Force-Induced Direct Stretching of Chromatin. Nat. Mater. 2016, 15, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.-J.; Han, W.M.; Szczesny, S.E.; Cosgrove, B.D.; Elliott, D.M.; Lee, D.A.; Duncan, R.L.; Mauck, R.L. Mechanically Induced Chromatin Condensation Requires Cellular Contractility in Mesenchymal Stem Cells. Biophys. J. 2016, 111, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Le, H.Q.; Ghatak, S.; Yeung, C.-Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical Regulation of Transcription Controls Polycomb-Mediated Gene Silencing during Lineage Commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tibbitt, M.W.; Basta, L.; Anseth, K.S. Mechanical Memory and Dosing Influence Stem Cell Fate. Nat. Mater. 2014, 13, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Venkatachalapathy, S.; Ratna, P.; Wang, Y.; Jokhun, D.S.; Nagarajan, M.; Shivashankar, G.v. Laterally Confined Growth of Cells Induces Nuclear Reprogramming in the Absence of Exogenous Biochemical Factors. Proc. Natl. Acad. Sci. USA 2018, 115, E4741–E4750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Yuan, L.; Lee, Y.; Bharti, A.; Mitra, A.; Shivashankar, G.V. Fibroblast Rejuvenation by Mechanical Reprogramming and Redifferentiation. Proc. Natl. Acad. Sci. USA 2020, 117, 10131–10141. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal Adhesion Kinase Links Mechanical Force to Skin Fibrosis via Inflammatory Signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognoni, E.; Walko, G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells 2019, 8, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in Tissue Fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo Pathway Regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-J.; Byun, M.R.; Furutani-Seiki, M.; Hong, J.-H.; Jung, H.-S. YAP and TAZ Regulate Skin Wound Healing. J. Investig. Dermatol. 2014, 134, 518–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walko, G.; Woodhouse, S.; Pisco, A.O.; Rognoni, E.; Liakath-Ali, K.; Lichtenberger, B.M.; Mishra, A.; Telerman, S.B.; Viswanathan, P.; Logtenberg, M.; et al. A Genome-Wide Screen Identifies YAP/WBP2 Interplay Conferring Growth Advantage on Human Epidermal Stem Cells. Nat. Commun. 2017, 8, 14744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.R.L.; Gerard-O’Riley, R.; Harrington, M.; Pavalko, F.M. Activation of NF-ΚB by Fluid Shear Stress, but Not TNF-α, Requires Focal Adhesion Kinase in Osteoblasts. Bone 2010, 47, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; DiMaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-Related Transcription Factor-A Controls Myofibroblast Activation and Fibrosis in Response to Myocardial Infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Crider, B.J.; Risinger, G.M.; Haaksma, C.J.; Howard, E.W.; Tomasek, J.J. Myocardin-Related Transcription Factors A and B Are Key Regulators of TGF-Β1-Induced Fibroblast to Myofibroblast Differentiation. J. Investig. Dermatol. 2011, 131, 2378–2385. [Google Scholar] [CrossRef] [Green Version]
- Willer, M.K.; Carroll, C.W. Substrate Stiffness-Dependent Regulation of SRF/Mkl1 Requires the Inner Nuclear Membrane Protein Emerin. J. Cell Sci. 2017, 130, 2111–2118. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the Nucleus and Cytoplasm: Role of the LINC Complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Talele, N.P.; Boo, S.; Koehler, A.; Knee-Walden, E.; Balestrini, J.L.; Speight, P.; Kapus, A.; Hinz, B. MicroRNA-21 Preserves the Fibrotic Mechanical Memory of Mesenchymal Stem Cells. Nat. Mater. 2017, 16, 379–389. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, H.; Liu, Z.; Wu, Q.; Zhu, R.; Liu, J. MiR-21 Promotes Cardiac Fibroblast-to-Myofibroblast Transformation and Myocardial Fibrosis by Targeting Jagged1. J. Cell. Mol. Med. 2018, 22, 3816–3824. [Google Scholar] [CrossRef]
- Killaars, A.R.; Grim, J.C.; Walker, C.J.; Hushka, E.A.; Brown, T.E.; Anseth, K.S. Extended Exposure to Stiff Microenvironments Leads to Persistent Chromatin Remodeling in Human Mesenchymal Stem Cells. Adv. Sci. 2019, 6, 1801483. [Google Scholar] [CrossRef] [Green Version]
- Ligresti, G.; Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Choi, K.M.; Haak, A.J.; Aravamudhan, A.; Roden, A.C.; Prakash, Y.S.; et al. CBX5/G9a/H3K9me-Mediated Gene Repression Is Essential to Fibroblast Activation during Lung Fibrosis. JCI Insight 2019, 4, e127111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct Fibroblast Subsets Drive Inflammation and Damage in Arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Strle, K.; Locchead, R.; Pianta, A.; Crowley, J.T.; Arvikar, S. Fibroblast-like Synoviocytes Shape and Perpetuate the Inflammatory Immune Responses Associated with Antibiotic-Refractory Lyme Arthritis.—ACR Meeting Abstracts. Arthritis Rheumatol. 2015, 67, 9–10. [Google Scholar]
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma Pathogenesis: A Pivotal Role for Fibroblasts as Effector Cells. Arthritis Res. Ther. 2013, 15, 215. [Google Scholar] [CrossRef] [Green Version]
- Berroth, A.; Kühnl, J.; Kurschat, N.; Schwarz, A.; Stäb, F.; Schwarz, T.; Wenck, H.; Fölster-Holst, R.; Neufang, G. Role of Fibroblasts in the Pathogenesis of Atopic Dermatitis. J. Allergy Clin. Immunol. 2013, 131, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2018, 66, 213–227. [Google Scholar] [CrossRef]
- Kyburz, D.; Brentano, F.; Schorr, O.; Michel, B.A.; Gay, R.E.; Gay, S. Toll-like Receptor Expression in Synovial Fibroblasts and Normal Skin Fibroblasts and Induction of Matrix Metallo-Proteinase Expression by Various Toll-like Receptor Ligands. Arthritis Res. Ther. 2005, 7, P119. [Google Scholar] [CrossRef]
- Klein, K.; Frank-Bertoncelj, M.; Karouzakis, E.; Gay, R.E.; Kolling, C.; Ciurea, A.; Bostanci, N.; Belibasakis, G.N.; Lin, L.-L.; Distler, O.; et al. The Epigenetic Architecture at Gene Promoters Determines Cell Type-Specific LPS Tolerance. J. Autoimmun. 2017, 83, 122–133. [Google Scholar] [CrossRef]
- Sohn, C.; Lee, A.; Qiao, Y.; Loupasakis, K.; Ivashkiv, L.B.; Kalliolias, G.D. Prolonged Tumor Necrosis Factor α Primes Fibroblast-like Synoviocytes in a Gene-Specific Manner by Altering Chromatin. Arthritis Rheumatol. 2015, 67, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Crowley, T.; O’Neil, J.D.; Adams, H.; Thomas, A.M.; Filer, A.; Buckley, C.D.; Clark, A.R. Priming in Response to Pro-Inflammatory Cytokines Is a Feature of Adult Synovial but Not Dermal Fibroblasts. Arthritis Res. Ther. 2017, 19, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangishetti, U.; Ramirez-Perez, S.; Jones, K.; Arif, A.; Drissi, H.; Bhattaram, P. Chronic Exposure to TNF Reprograms Cell Signaling Pathways in Fibroblast-like Synoviocytes by Establishing Long-Term Inflammatory Memory. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ghazarian, A.; Garner, W.L.; Ehrlich, H.P. Memory of Past Exposure to the Chemokine IL-8 Inhibits the Contraction of Fibroblast-Populated Collagen Lattices. Exp. Mol. Pathol. 2000, 69, 242–247. [Google Scholar] [CrossRef]
- Nayar, S.; Campos, J.; Smith, C.G.; Iannizzotto, V.; Gardner, D.H.; Mourcin, F.; Roulois, D.; Turner, J.; Sylvestre, M.; Asam, S.; et al. Immunofibroblasts Are Pivotal Drivers of Tertiary Lymphoid Structure Formation and Local Pathology. Proc. Natl. Acad. Sci. USA 2019, 116, 13490–13497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, S.; Ciechomska, M.; Fullard, N.; Przyborski, S.; Van Laar, J.M. IL-13 Mediates Collagen Deposition via STAT6 and MicroRNA-135b: A Role for Epigenetics. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Davidson-Moncada, J.; Papavasiliou, F.N.; Tam, W. MicroRNAs of the Immune System: Roles in Inflammation and Cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Saferding, V.; Puchner, A.; Goncalves-Alves, E.; Hofmann, M.; Bonelli, M.; Brunner, J.S.; Sahin, E.; Niederreiter, B.; Hayer, S.; Kiener, H.P.; et al. MicroRNA-146a Governs Fibroblast Activation and Joint Pathology in Arthritis. J. Autoimmun. 2017, 82, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, D.; Wikstrom, J.D.; Pivarcsi, A.; Sonkoly, E.; Ståhle, M.; Landén, N.X. MicroRNA-132 Promotes Fibroblast Migration via Regulating RAS P21 Protein Activator 1 in Skin Wound Healing. Sci. Rep. 2017, 7, 7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, M.G.; Artlett, C.M. The MicroRNA MiR-155 Is Essential in Fibrosis. Non-Coding RNA 2019, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, M.F.; Oliveira, P.G.; Garcia-Carbonell, R.; Croft, A.P.; Smith, J.M.; Serrano, R.L.; Sanchez-Lopez, E.; Liu, X.; Kisseleva, T.; Hay, N.; et al. Hexokinase 2 as a Novel Selective Metabolic Target for Rheumatoid Arthritis. Ann. Rheum. Dis. 2018, 77, 1636–1643. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Zhong, Q.; Kanwar, M. Metabolic Memory and Diabetic Retinopathy: Role of Inflammatory Mediators in Retinal Pericytes. Exp. Eye Res. 2010, 90, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, L.M.; Reddy, M.A.; Lanting, L.L.; Wang, M.; Meng, L.; Natarajan, R. Epigenetic Histone H3 Lysine 9 Methylation in Metabolic Memory and Inflammatory Phenotype of Vascular Smooth Muscle Cells in Diabetes. Proc. Natl. Acad. Sci. USA 2008, 105, 9047–9052. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Psarianos, P.; Ghoraie, L.S.; Yip, K.; Goldstein, D.; Gilbert, R.; Witterick, I.; Pang, H.; Hussain, A.; Lee, J.H.; et al. Metabolic Regulation of Dermal Fibroblasts Contributes to Skin Extracellular Matrix Homeostasis and Fibrosis. Nat. Metab. 2019, 1, 147–157. [Google Scholar] [CrossRef]
- Bernard, K.; Logsdon, N.J.; Ravi, S.; Xie, N.; Persons, B.P.; Rangarajan, S.; Zmijewski, J.W.; Mitra, K.; Liu, G.; Darley-Usmar, V.M.; et al. Metabolic Reprogramming Is Required for Myofibroblast Contractility and Differentiation. J. Biol. Chem. 2015, 290, 25427–25438. [Google Scholar] [CrossRef] [Green Version]
- Sang, L.; Coller, H.A.; Roberts, J.M. Control of the Reversibility of Cellular Quiescence by the Transcriptional Repressor HES1. Science 2008, 321, 1095–1100. [Google Scholar] [CrossRef] [Green Version]
- Coller, H.A.; Sang, L.; Roberts, J.M. A New Description of Cellular Quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemons, J.M.S.; Feng, X.-J.; Bennett, B.D.; Legesse-Miller, A.; Johnson, E.L.; Raitman, I.; Pollina, E.A.; Rabitz, H.A.; Rabinowitz, J.D.; Coller, H.A. Quiescent Fibroblasts Exhibit High Metabolic Activity. PLoS Biol. 2010, 8, e1000514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, E.J.; Remillard, M.Y.; Legesse-Miller, A.; Johnson, E.L.; Lemons, J.M.; Chapman, T.R.; Forman, J.J.; Kojima, M.; Silberman, E.S.; Coller, H.A. A MicroRNA Network Regulates Proliferative Timing and Extracellular Matrix Synthesis during Cellular Quiescence in Fibroblasts. Genome Biol. 2012, 13, R121. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Riddle, S.; Zhang, H.; D’Alessandro, A.; Flockton, A.; Serkova, N.J.; Hansen, K.C.; Moldvan, R.; McKeon, B.A.; Frid, M.; et al. Metabolic Reprogramming Regulates the Proliferative and Inflammatory Phenotype of Adventitial Fibroblasts in Pulmonary Hypertension Through the Transcriptional Corepressor C-Terminal Binding Protein-1. Circulation 2016, 134, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer-Associated Fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Tauber, J.; Li, M.; Zhang, H.; Flockton, A.R.; Pullamsetti, S.S.; Chelladurai, P.; D’Alessandro, A.; el Kasmi, K.C.; Ježek, P.; et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-Induced Epigenetic Modifications Are Associated with Cardiac Tissue Fibrosis and the Development of a Myofibroblast-like Phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terranova, A. The Effects of Diabetes Mellitus on Wound Healing. Plast. Surg.Nurs. Off. J. Am. Soc. Plast. Reconstr. Surg. Nurses 1991, 11, 20–25. [Google Scholar] [CrossRef]
- Caramori, M.L.; Kim, Y.; Natarajan, R.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Kuriyama, R.; Kirkpatrick, D.; Mauer, M. Differential Response to High Glucose in Skin Fibroblasts of Monozygotic Twins Discordant for Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E883–E889. [Google Scholar] [CrossRef] [Green Version]
- Park, L.K.; Maione, A.G.; Smith, A.; Gerami-Naini, B.; Iyer, L.K.; Mooney, D.J.; Veves, A.; Garlick, J.A. Genome-Wide DNA Methylation Analysis Identifies a Metabolic Memory Profile in Patient-Derived Diabetic Foot Ulcer Fibroblasts. Epigenetics 2014, 9, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- Al-Rikabi, A.H.A.; Tobin, D.J.; Riches-Suman, K.; Thornton, M.J. Dermal Fibroblasts Cultured from Donors with Type 2 Diabetes Mellitus Retain an Epigenetic Memory Associated with Poor Wound Healing Responses. Sci. Rep. 2021, 11, 1474. [Google Scholar] [CrossRef]
- Li, Q.; Qin, Z.; Nie, F.; Bi, H.; Zhao, R.; Pan, B.; Ma, J.; Xie, X. Metabolic Reprogramming in Keloid Fibroblasts: Aerobic Glycolysis and a Novel Therapeutic Strategy. Biochem. Biophys. Res. Commun. 2018, 496, 641–647. [Google Scholar] [CrossRef]
- Salmond, R.J. MTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Ma, G.; Samad, I.; Motz, K.; Yin, L.X.; Duvvuri, M.V.; Ding, D.; Namba, D.R.; Elisseeff, J.H.; Horton, M.R.; Hillel, A.T. Metabolic Variations in Normal and Fibrotic Human Laryngotracheal-Derived Fibroblasts: A Warburg-like Effect. Laryngoscope 2017, 127, E107–E113. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic Reprogramming of Cancer-Associated Fibroblasts by TGF-β Drives Tumor Growth: Connecting TGF-β Signaling with “Warburg-like” Cancer Metabolism and L-Lactate Production. Cell Cycle (Georget. Tex.) 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Kvacskay, P.; Yao, N.; Schnotz, J.-H.; Scarpone, R.; de Albuquerque Carvalho, R.; Klika, K.D.; Merkt, W.; Tretter, T.; Lorenz, H.-M.; Tykocinski, L.-O. Increase of Aerobic Glycolysis Mediated by Activated T Helper Cells Drives Synovial Fibroblasts towards an Inflammatory Phenotype: New Targets for Therapy? Arthritis Res. Ther. 2021, 23, 56. [Google Scholar] [CrossRef]
- Dabiri, G.; Tumbarello, D.A.; Turner, C.E.; van de Water, L. Hic-5 Promotes the Hypertrophic Scar Myofibroblast Phenotype by Regulating the TGF-Beta1 Autocrine Loop. J. Investig. Dermatol. 2008, 128, 2518–2525. [Google Scholar] [CrossRef] [Green Version]
- Mathur, J.; Shenoy, V.B.; Pathak, A. Mechanical Memory in Cells Emerges from Mechanotransduction with Transcriptional Feedback and Epigenetic Plasticity. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hahn, J.M.; McFarland, K.L.; Combs, K.A.; Anness, M.C.; Supp, D.M. Analysis of HOX Gene Expression and the Effects of HOXA9 Overexpression in Fibroblasts Derived from Keloid Lesions and Normal Skin. Wound Repair Regen. Off. Publ. Wound Health Soc. Eur. Tissue Repair Soc. 2021, 29, 777–791. [Google Scholar] [CrossRef]
- Barallobre-Barreiro, J.; Woods, E.; Bell, R.E.; Easton, J.A.; Hobbs, C.; Eager, M.; Baig, F.; Ross, A.M.; Mallipeddi, R.; Powell, B.; et al. Cartilage-like Composition of Keloid Scar Extracellular Matrix Suggests Fibroblast Mis-Differentiation in Disease. Matrix Biol. Plus 2019, 4, 100016. [Google Scholar] [CrossRef] [PubMed]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-Associated Fibroblast Heterogeneity in Axillary Lymph Nodes Drives Metastases in Breast Cancer through Complementary Mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald O’Connor, E.J.; Badshah, I.I.; Addae, L.Y.; Kundasamy, P.; Thanabalasingam, S.; Abioye, D.; Soldin, M.; Shaw, T.J. Histone Deacetylase 2 Is Upregulated in Normal and Keloid Scars. J. Investig. Dermatol. 2012, 132, 1293–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Hirahashi, J.; Kawachi, S.; Fujita, T. Histone Deacetylase Modulates the Proinflammatory and -Fibrotic Changes in Tubulointerstitial Injury. Am. J. Physiol.-Ren. Physiol. 2010, 298, F133–F141. [Google Scholar] [CrossRef]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of Histone Deacetylase Activity Attenuates Renal Fibroblast Activation and Interstitial Fibrosis in Obstructive Nephropathy. Am. J. Physiol.-Ren. Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Fenning, A.; Lim, J.; Le, G.T.; Reid, R.C.; Halili, M.A.; Fairlie, D.P.; Brown, L. Antifibrotic Activity of an Inhibitor of Histone Deacetylases in DOCA-Salt Hypertensive Rats. Br. J. Pharmacol. 2010, 159, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the Class I Histone Deacetylase Ameliorates Renal Fibrosis and Inhibits Renal Fibroblast Activation via Modulating TGF-Beta and EGFR Signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, D.; Li, M.; Plecitá-Hlavatá, L.; D’Alessandro, A.; Tauber, J.; Riddle, S.; Kumar, S.; Flockton, A.; McKeon, B.A.; et al. Metabolic and Proliferative State of Vascular Adventitial Fibroblasts in Pulmonary Hypertension Is Regulated Through a MicroRNA-124/PTBP1 (Polypyrimidine Tract Binding Protein 1)/Pyruvate Kinase Muscle Axis. Circulation 2017, 136, 2468–2485. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M. HDAC Inhibitors as Anti-Inflammatory Agents. Br. J. Pharmacol. 2007, 150, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Korchynskyi, O.; Tak, P.P.; Reedquist, K.A. Histone Deacetylase Inhibitors Suppress Rheumatoid Arthritis Fibroblast-like Synoviocyte and Macrophage IL-6 Production by Accelerating MRNA Decay. Ann. Rheum. Dis. 2012, 71, 424–431. [Google Scholar] [CrossRef]
- Mascharak, S.; DesJardins-Park, H.E.; Davitt, M.F.; Griffin, M.; Borrelli, M.R.; Moore, A.L.; Chen, K.; Duoto, B.; Chinta, M.; Foster, D.S.; et al. Preventing Engrailed-1 Activation in Fibroblasts Yields Wound Regeneration without Scarring. Science 2021, 372, eaba2374. [Google Scholar] [CrossRef]
- Chen, K.; Kwon, S.H.; Henn, D.; Kuehlmann, B.A.; Tevlin, R.; Bonham, C.A.; Griffin, M.; Trotsyuk, A.A.; Borrelli, M.R.; Noishiki, C.; et al. Disrupting Biological Sensors of Force Promotes Tissue Regeneration in Large Organisms. Nat. Commun. 2021, 12, 5256. [Google Scholar] [CrossRef]
- Sacco, A.M.; Belviso, I.; Romano, V.; Carfora, A.; Schonauer, F.; Nurzynska, D.; Montagnani, S.; di Meglio, F.; Castaldo, C. Diversity of Dermal Fibroblasts as Major Determinant of Variability in Cell Reprogramming. J. Cell. Mol. Med. 2019, 23, 4256–4268. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirk, T.; Ahmed, A.; Rognoni, E. Fibroblast Memory in Development, Homeostasis and Disease. Cells 2021, 10, 2840. https://doi.org/10.3390/cells10112840
Kirk T, Ahmed A, Rognoni E. Fibroblast Memory in Development, Homeostasis and Disease. Cells. 2021; 10(11):2840. https://doi.org/10.3390/cells10112840
Chicago/Turabian StyleKirk, Thomas, Abubkr Ahmed, and Emanuel Rognoni. 2021. "Fibroblast Memory in Development, Homeostasis and Disease" Cells 10, no. 11: 2840. https://doi.org/10.3390/cells10112840
APA StyleKirk, T., Ahmed, A., & Rognoni, E. (2021). Fibroblast Memory in Development, Homeostasis and Disease. Cells, 10(11), 2840. https://doi.org/10.3390/cells10112840