
Integrative Analysis to Identify Genes Associated with Stemness and Immune Infiltration in Glioblastoma



Abstract
1. Introduction
2. Materials and Methods
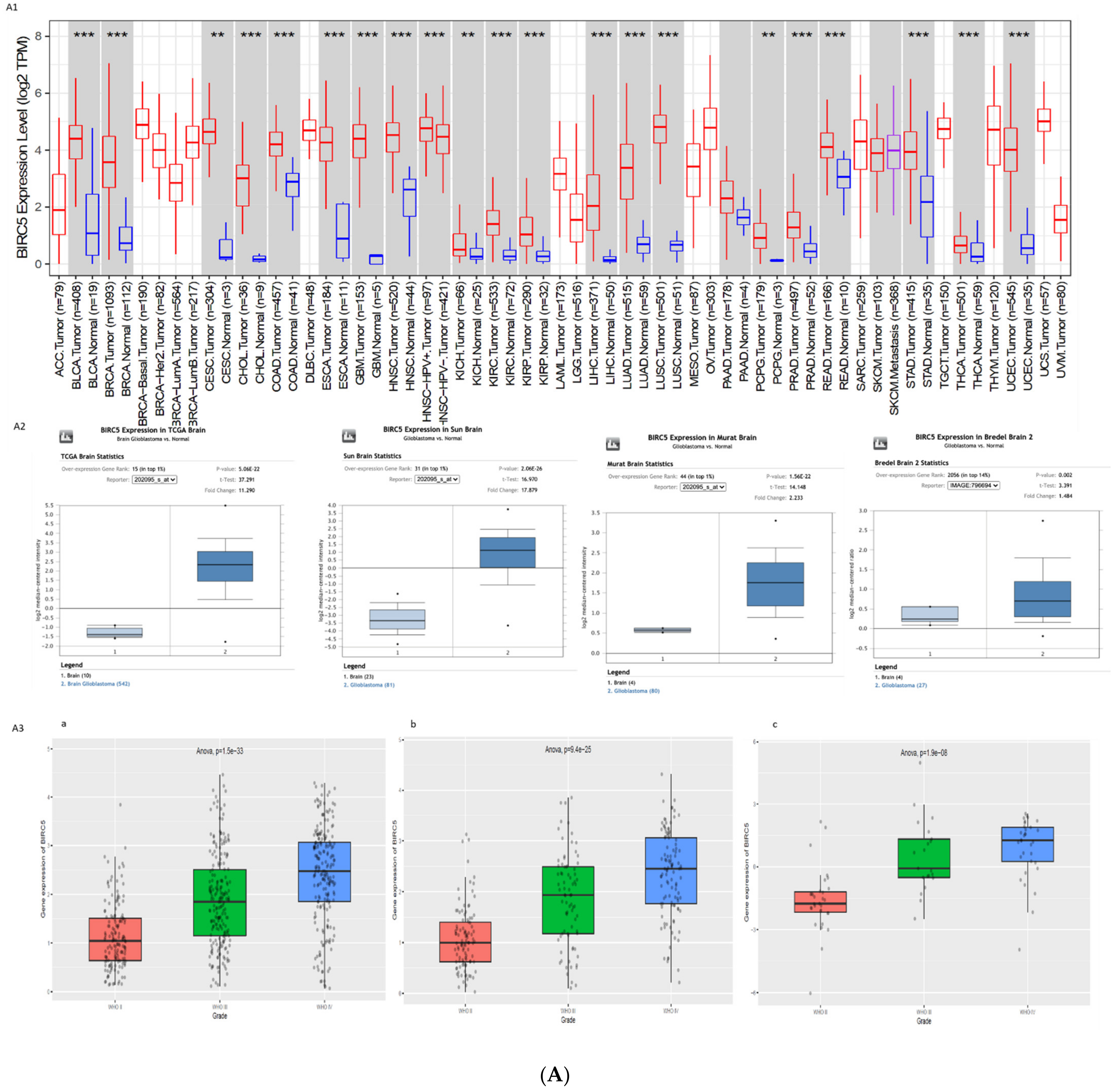
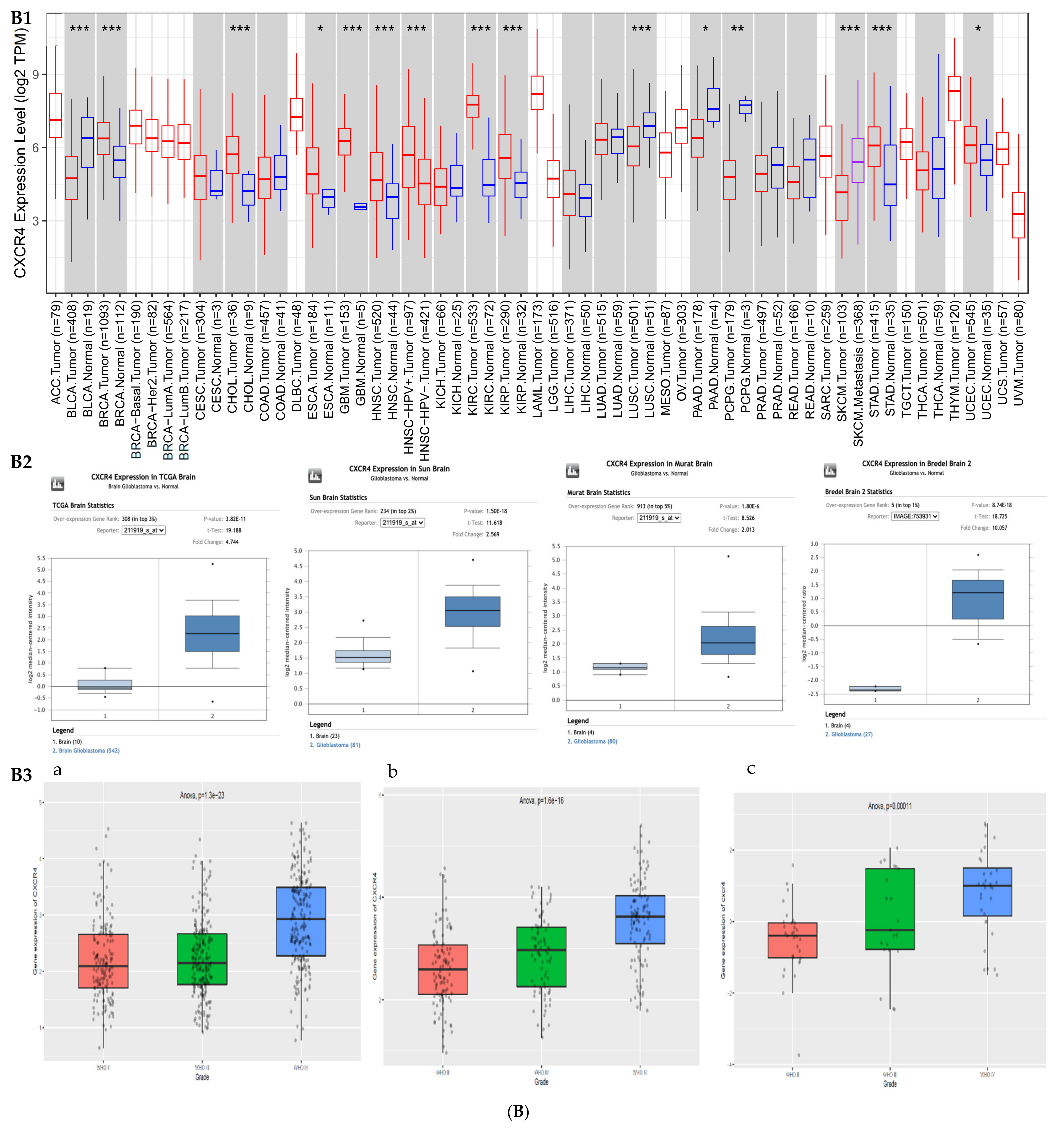
2.1. Expression Profiles of BIRC5 and CXCR4
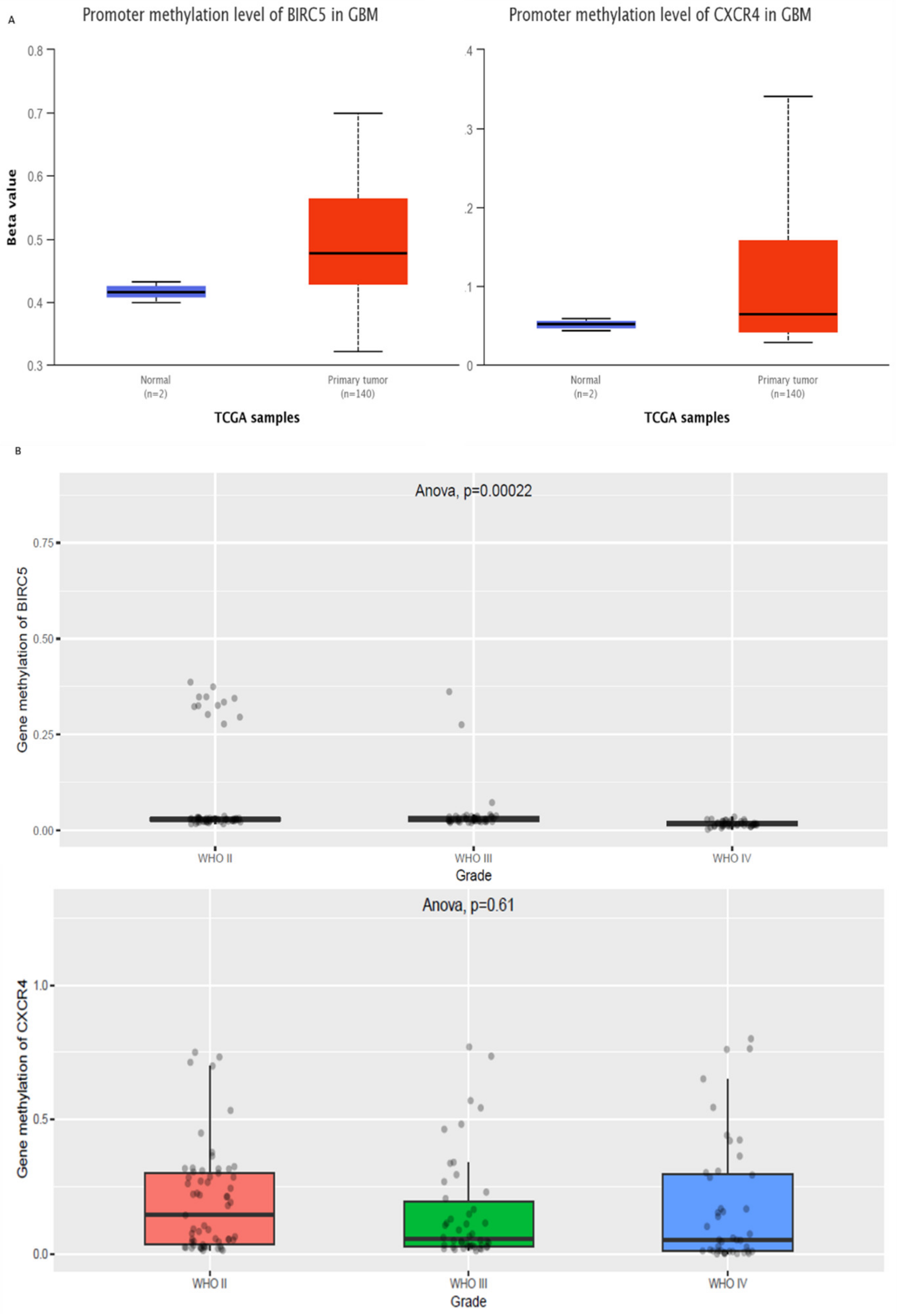
2.2. Role of Methylation in the Regulation of BIRC5 and CXCR4
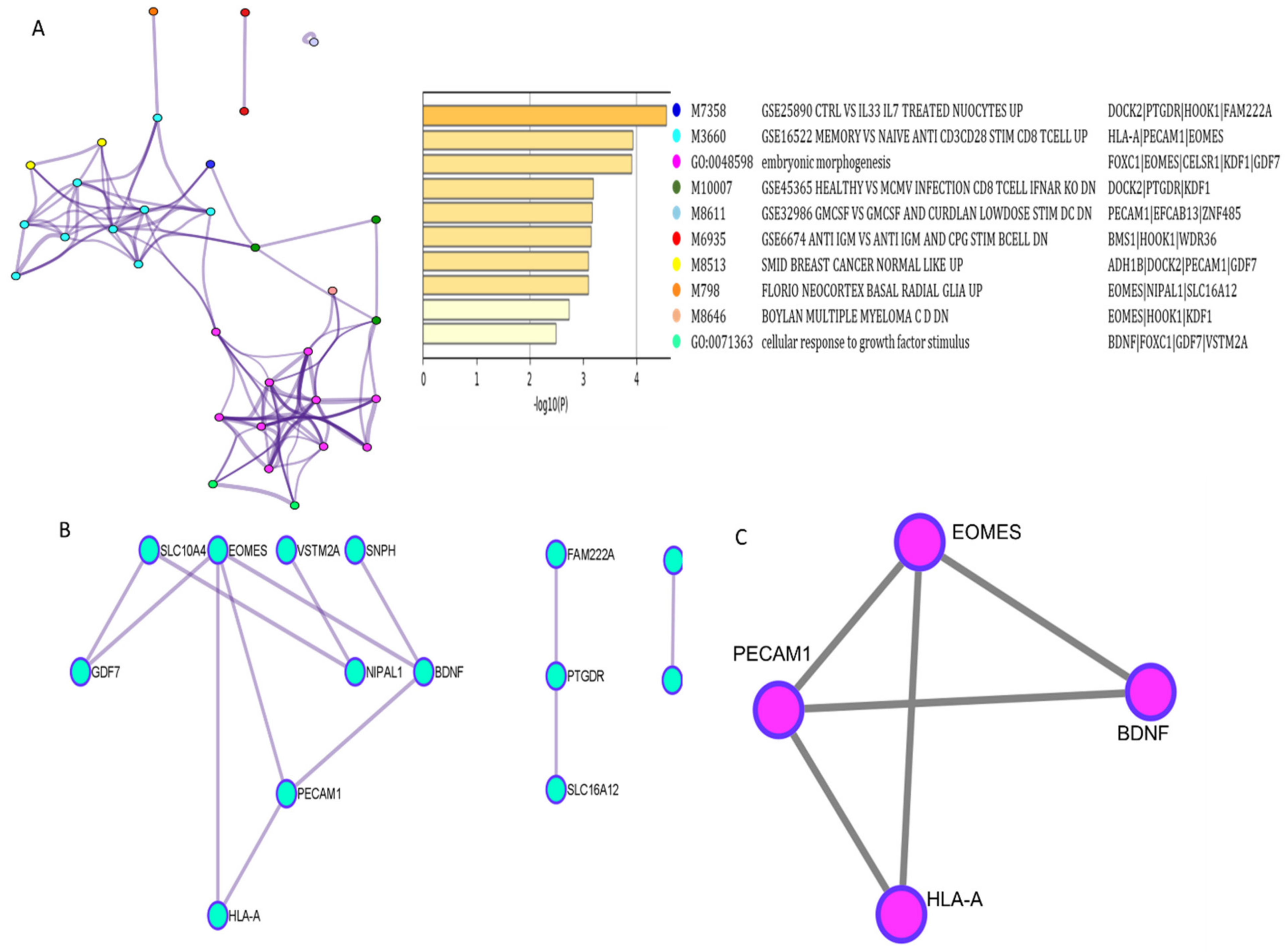
2.3. Functional Enrichment Analysis
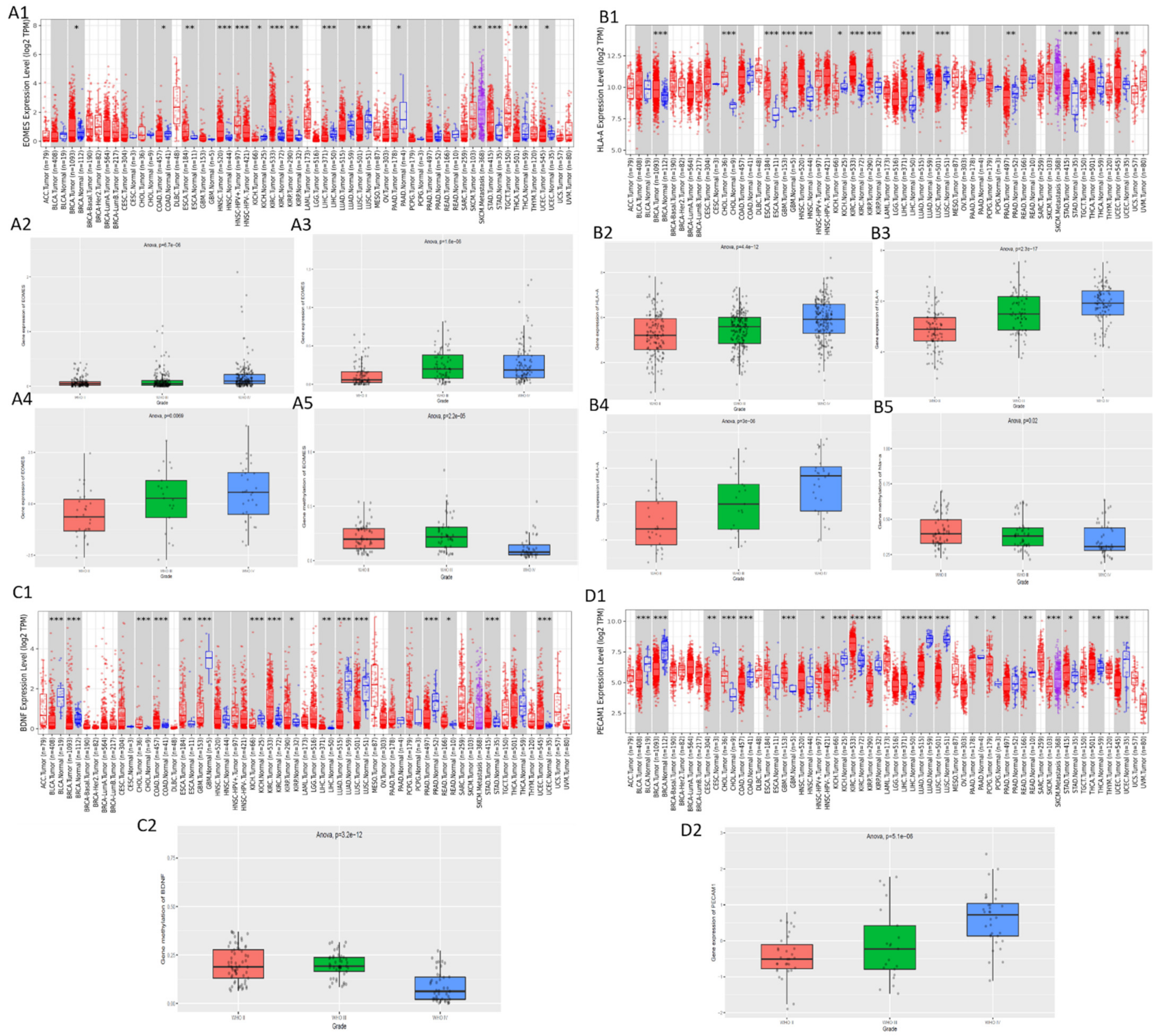
2.4. Validation of Expression and Methylation of EOMES, BDNF, PECAM1, and HLA-A
2.5. Role of Genes in Stemness
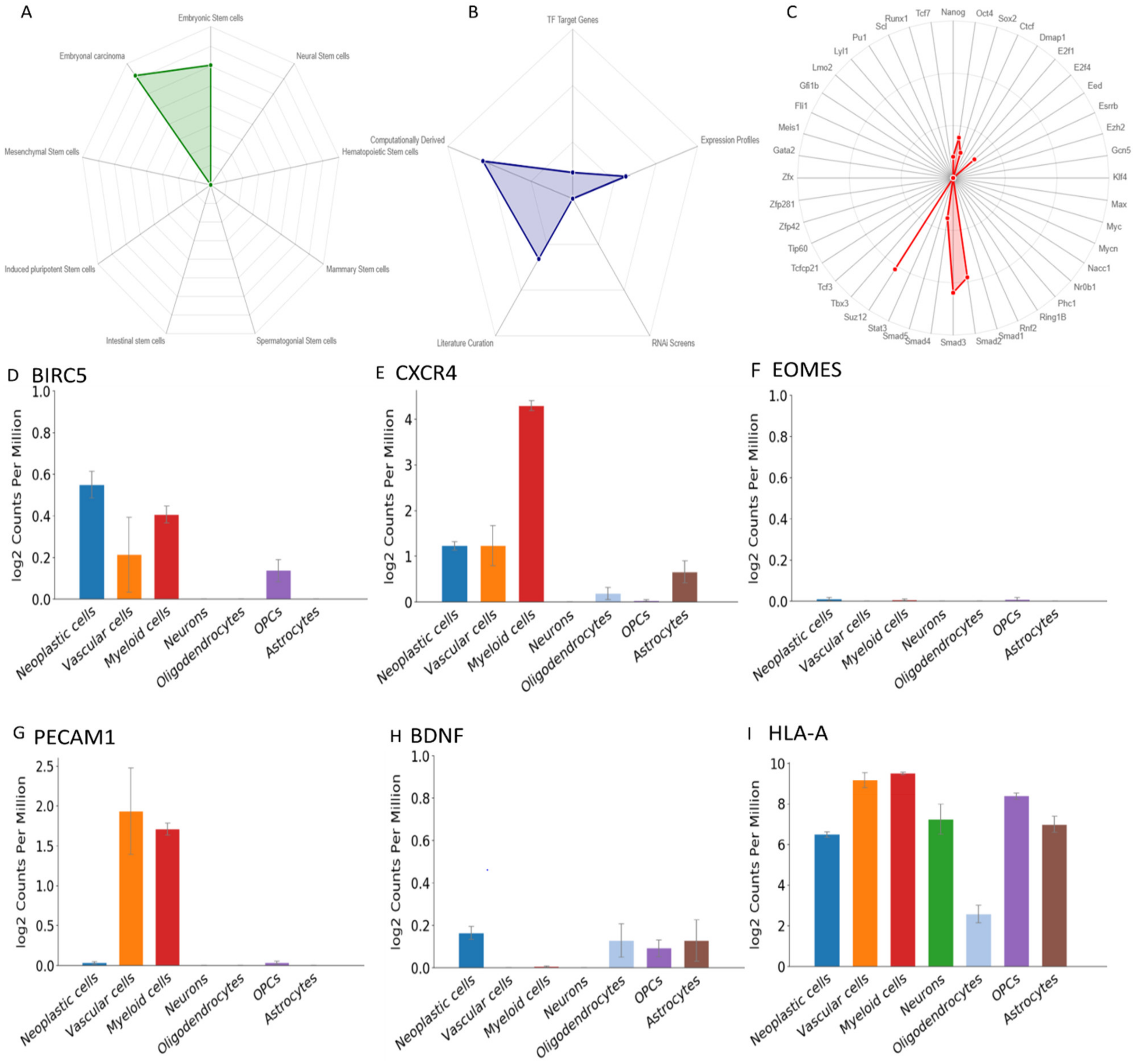
2.6. Single Cell Expression of the Genes
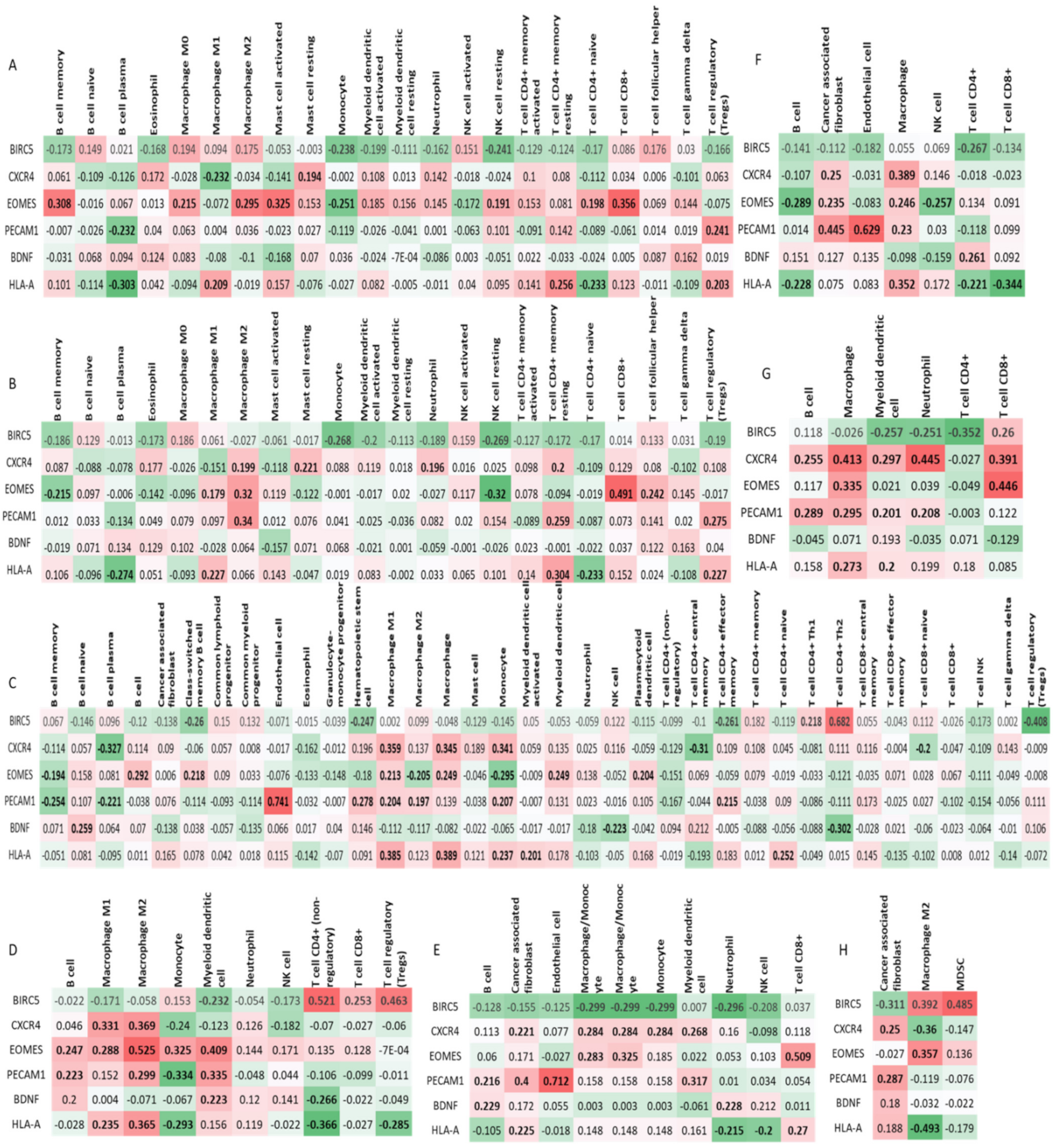
2.7. Immune Infiltration Analysis
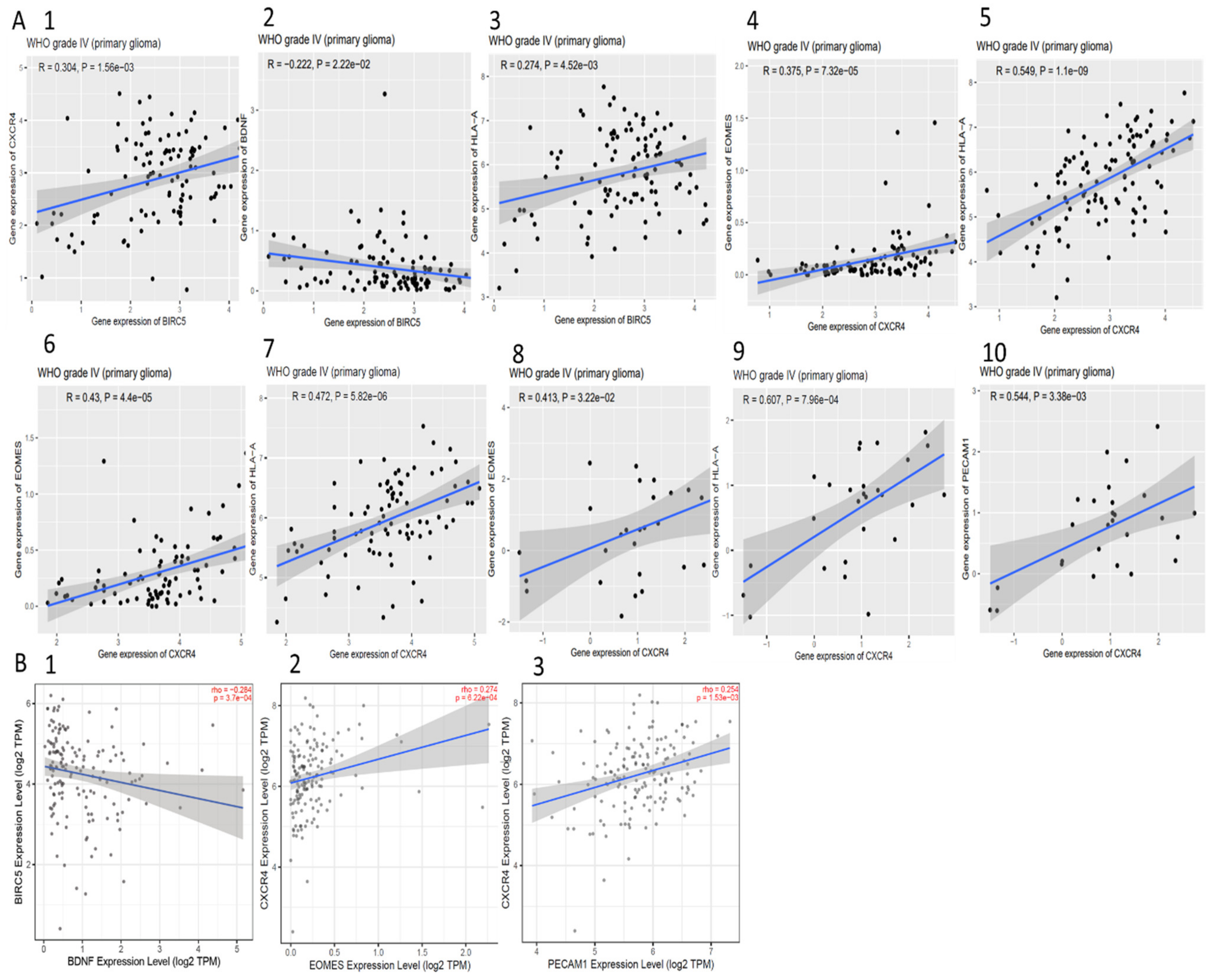
2.8. Correlation between Genes
2.9. Statistical Analysis
3. Results
3.1. Gene Expression Profile of BIRC5 and CXCR4
3.2. Epigenetic Regulation of BIRC5 and CXCR4
3.3. Epigenetic Regulation of Genes in Normal vs. GBM
3.4. Functional and Network Analysis of Methylated Genes
3.5. Validation of the Expression and Methylation Profiles of EOMES, BDNF, PECAM1, and HLA-A
3.6. The Role of the Genes in Stemness
3.7. ScRNA Expression Profile
3.8. Immune Infiltration
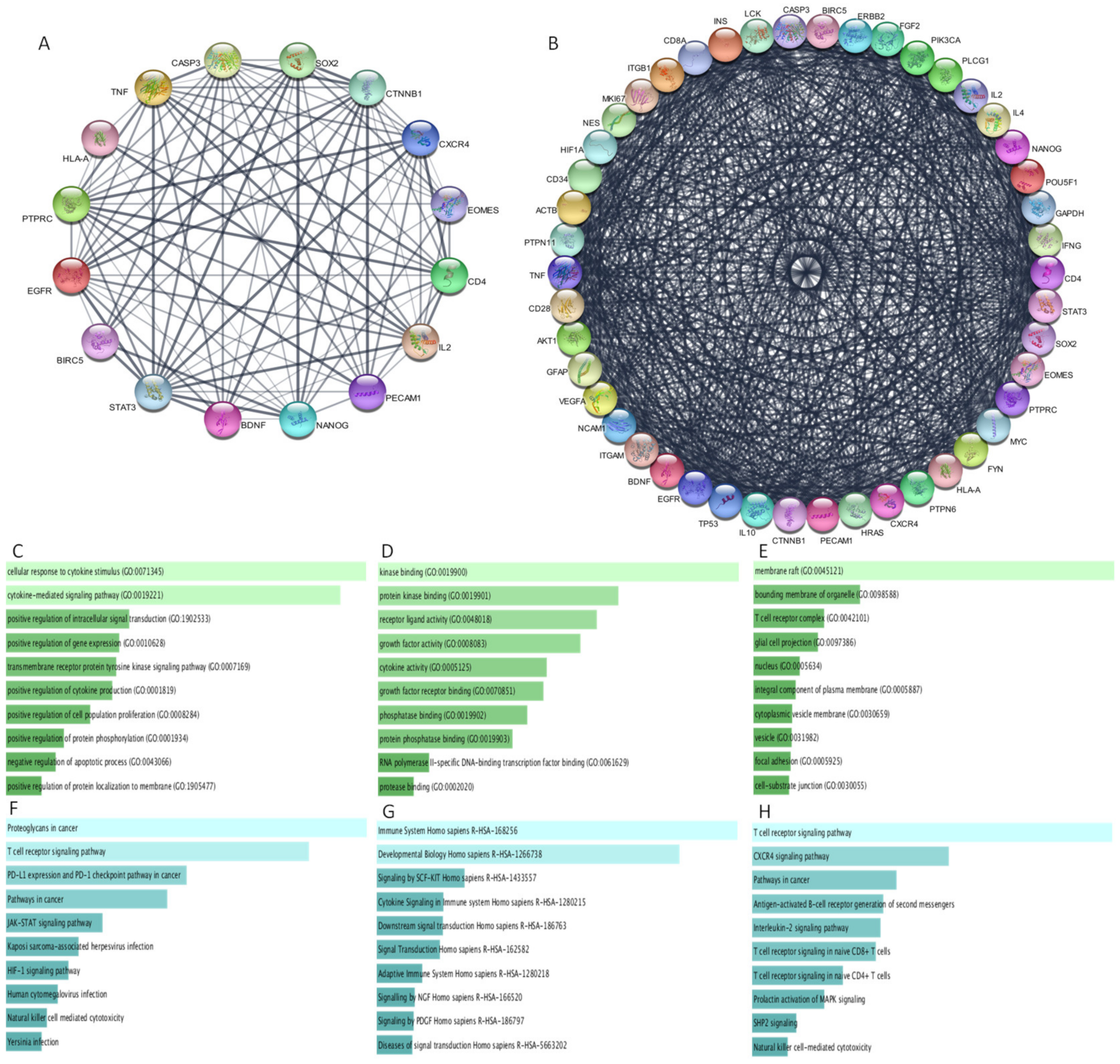
3.9. Protein–Protein Interaction
3.10. Functional Analysis of Protein–Protein Interaction Network
3.11. Correlation between BIRC5, CXCR4, and Methylated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BIRC5 | Baculoviral IAP Repeat Containing 5 |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| EOMES | Eomesodermin |
| BDNF | Brain derived Neurotrophic Factor |
| HLA-A | Human Leukocyte Antigen A |
| DMRs | Differentially methylated regions |
| PECAM1 | Platelet Endothelial Cell Adhesion Molecule 1 |
| GBM | Glioblastoma Multiform |
| CSCs | Cancer Stem Cells |
| GSCs | Glioblastoma Stem Cells |
| TCGA | The Cancer Genome Atlas |
| CGGA | Chinese Glioma Genome Atlas |
| SDF-1 | Stromal Cell-Derived Factor |
| Sox-2 | SRY (Sex Determining Region Y)-Box 2 |
| Oct-4 | Octamer-Binding Transcription Factor 4 |
| HIF | Hypoxia Inducible Factor |
| JAK | Janus Kinase |
| Stat | Signal Transducer and Activator of Transcription |
| VEGF | Vascular Endothelial Growth Factor |
| PI3K | Phosphoinositide-3 Kinase |
| EGFR | Epidermal Growth Factor Receptor |
| IGF | Insulin-like Growth Factor |
| mTOR | Mammalian Target of Rapamycin |
| DOCK2 | Dedicator Of Cytokinesis 2 |
| CELSR1 | Cadherin EGF LAG Seven-Pass G-Type Receptor 1 |
| KDF-1 | Keratinocyte Differentiation Factor 1 |
| PTGDR | Prostaglandin D₂ receptor |
| GDF7 | Growth Differentiation Factor 7 |
| NF-ĸB | Nuclear Factor Kap-pa-Light-Chain-Enhancer of Activated B Cells |
| TGF-β | transforming growth factor-β |
| L1CAM | L1 Cell Adhesion Molecule |
| SALL4 | Sal-like protein 4 |
| OLIG2 | Oligodendrocyte Transcription Factor 2 |
| GFAP | Glial fibrillary acidic protein |
| KLF4 | Kruppel-like factor 4 |
| IL | Interleukin |
| ALDH | Aldehyde dehydrogenase |
| IFN | Interferon |
| ITG | Integrin |
| PTPN | Protein Tyrosine Phosphatase Non-Receptor |
| CTNNB1 | Catenin beta-1 |
| PTPRC | Protein Tyrosine Phosphatase Receptor Type C |
| PD-L1 | Programmed death-ligand 1 |
| TNF | Tumour necrosis factor |
| DC | Dendritic Cells |
| NK cells | Natural Killer cells |
| TH1 | Type 1 T helper |
References
- Oliver, L.; Lalier, L.; Salaud, C.; Heymann, D.; Cartron, P.F.; Vallette, F.M. Drug resistance in glioblastoma: Are persisters the key to therapy? Cancer Drug Resist. 2020, 3, 287–301. [Google Scholar] [CrossRef]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastomas. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Ou, A.; Alfred Yung, W.K.; Majd, N. Molecular mechanisms of treatment resistance in glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Ferreira, W.A.S.; Pinheiro, D.D.R.; Da Costa Junior, C.A.; Rodrigues-Antunes, S.; Araújo, M.D.; Leão Barros, M.B.; Teixeira, A.C.D.S.; Faro, T.A.S.; Burbano, R.R.; De Oliveira, E.H.C.; et al. An update on the epigenetics of glioblastomas. Epigenomics 2016, 8, 1289–1305. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic targeting of glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef]
- López-Gil, J.C.; Martin-Hijano, L.; Hermann, P.C.; Sainz, B. The CXCL12 crossroads in cancer stem cells and their niche. Cancers 2021, 13, 469. [Google Scholar] [CrossRef]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage Migration Inhibitory Factor–CXCR4 Is the Dominant Chemotactic Axis in Human Mesenchymal Stem Cell Recruitment to Tumors. J. Immunol. 2015, 194, 3463–3474. [Google Scholar] [CrossRef]
- Singh, P.; Mohammad, K.S.; Pelus, L.M. CXCR4 expression in the bone marrow microenvironment is required for hematopoietic stem and progenitor cell maintenance and early hematopoietic regeneration after myeloablation. Stem Cells 2020, 38, 849–859. [Google Scholar] [CrossRef]
- Oh, S.; Jeong, H.; Park, H.; Choi, K.A.; Hwang, I.; Lee, J.; Cho, J.; Hong, S. Activation of CXCL12-CXCR4 signalling induces conversion of immortalised embryonic kidney cells into cancer stem-like cells. Artif. Cells Nanomed. Biotechnol. 2020, 48, 1303–1313. [Google Scholar] [CrossRef]
- Yi, L.; Tong, L.; Li, T.; Hai, L.; Abeysekera, I.R.; Tao, Z.; Ma, H.; Liu, P.; Xie, Y.; Li, J.; et al. Bioinformatic analyses reveal the key pathways and genes in the CXCR4 mediated mesenchymal subtype of glioblastoma. Mol. Med. Rep. 2018, 18, 741–748. [Google Scholar] [CrossRef]
- Richardson, P.J. CXCR4 and Glioblastoma. Anticancer Agents Med. Chem. 2016, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, 132. [Google Scholar] [CrossRef]
- Warrier, N.M.; Agarwal, P.; Kumar, P. Emerging Importance of Survivin in Stem Cells and Cancer: The Development of New Cancer Therapeutics. Stem Cell Rev. Rep. 2020, 16, 828–852. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 24, 5. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Peng, S.; Dhruv, H.; Armstrong, B.; Salhia, B.; Legendre, C.; Kiefer, J.; Parks, J.; Virk, S.; Sloan, A.E.; Ostrom, Q.T.; et al. Integrated genomic analysis of survival outliers in glioblastoma. Neuro. Oncol. 2017, 19, 833–844. [Google Scholar] [CrossRef]
- Court, F.; Le Boiteux, E.; Fogli, A.; Müller-Barthélémy, M.; Vaurs-Barrière, C.; Chautard, E.; Pereira, B.; Biau, J.; Kemeny, J.L.; Khalil, T.; et al. Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms. Genome Res. 2019, 29, 1605–1621. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Pinto, J.P.; Kalathur, R.K.; Oliveira, D.V.; Barata, T.; Machado, R.S.R.; Machado, S.; Pacheco-Leyva, I.; Duarte, I.; Futschik, M.E. StemChecker: A web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015, 43, W72–W77. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Cho, S.; Lee, J.H.; Cho, S.B.; Yoon, K.W.; Park, S.Y.; Lee, W.S.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; et al. Epigenetic methylation and expression of caspase 8 and survivin in hepatocellular carcinoma. Pathol. Int. 2010, 60, 203–211. [Google Scholar] [CrossRef]
- Nitschkowski, D.; Marwitz, S.; Kotanidou, S.A.; Reck, M.; Kugler, C.; Rabe, K.F.; Ammerpohl, O.; Goldmann, T. Live and let die: Epigenetic modifications of Survivin and Regucalcin in non-small cell lung cancer tissues contribute to malignancy. Clin. Epigenet. 2019, 11, 157. [Google Scholar] [CrossRef]
- Ma, X.; Shang, F.; Zhu, W.; Lin, Q. CXCR4 expression varies significantly among different subtypes of glioblastoma multiforme (GBM) and its low expression or hypermethylation might predict favorable overall survival. Expert Rev. Neurother. 2017, 17, 941–946. [Google Scholar] [CrossRef]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Allen, B.K.; Stathias, V.; Maloof, M.E.; Vidovic, D.; Winterbottom, E.F.; Capobianco, A.J.; Clarke, J.; Schurer, S.; Robbins, D.J.; Ayad, N.G. Epigenetic Pathways and Glioblastoma Treatment: Insights from Signaling Cascades. J. Cell. Biochem. 2015, 116, 351–363. [Google Scholar] [CrossRef]
- Polano, M.; Fabbiani, E.; Adreuzzi, E.; Di Cintio, F.; Bedon, L.; Gentilini, D.; Mongiat, M.; Ius, T.; Arcicasa, M.; Skrap, M.; et al. A New Epigenetic Model to Stratify Glioma Patients According to Their Immunosuppressive State. Cells 2021, 10, 576. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, P.; Bai, H.J.; Huang, J.; Yang, H.T. TATA box-binding protein-related factor 3 drives the mesendoderm specification of human embryonic stem cells by globally interacting with the TATA box of key mesendodermal genes. Stem Cell Res. Ther. 2020, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, G.; Liu, Y.; Feng, L.; Wang, M.; Liu, J.; Chen, Y.; Ouyang, L. Development of small-molecule tropomyosin receptor kinase (TRK) inhibitors for NTRK fusion cancers. Acta Pharm. Sin. B 2021, 11, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Patel, P. BDNF: An oncogene or tumor suppressor? Anticancer Res. 2017, 37, 3983–3990. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, S.; Liu, Y.; Feng, R.; Wang, C.; Jiang, S.; Zhang, X.; Lan, F.; Li, Y. Survivin Improves Reprogramming Efficiency of Human Neural Progenitors by Single Molecule OCT4. Stem Cells Int. 2016, 2016, 4729535. [Google Scholar] [CrossRef]
- Llaó-Cid, L.; Roessner, P.M.; Chapaprieta, V.; Öztürk, S.; Roider, T.; Bordas, M.; Izcue, A.; Colomer, D.; Dietrich, S.; Stilgenbauer, S.; et al. EOMES is essential for antitumor activity of CD8+ T cells in chronic lymphocytic leukemia. Leukemia 2021. [Google Scholar] [CrossRef]
- Shimizu, K.; Sato, Y.; Kawamura, M.; Nakazato, H.; Watanabe, T.; Ohara, O.; Fujii, S.I. Eomes transcription factor is required for the development and differentiation of invariant NKT cells. Commun. Biol. 2019, 2, 1–13. [Google Scholar] [CrossRef]
- Sun, R.; Wu, Y.; Zhou, H.; Wu, Y.; Yang, Z.; Gu, Y.; Jiang, J.; Lu, B.; Zhu, Y. Eomes Impedes Durable Response to Tumor Immunotherapy by Inhibiting Stemness, Tissue Residency, and Promoting the Dysfunctional State of Intratumoral CD8+ T Cells. Front. Cell Dev. Biol. 2021, 9, 640224. [Google Scholar] [CrossRef]
- Lino, C.N.R.; Barros-Martins, J.; Oberdörfer, L.; Walzer, T.; Prinz, I. Eomes expression reports the progressive differentiation of IFN-γ-producing Th1-like γδ T cells. Eur. J. Immunol. 2017, 47, 970–981. [Google Scholar] [CrossRef]
- Eckert, F.; Schilbach, K.; Klumpp, L.; Bardoscia, L.; Sezgin, E.C.; Schwab, M.; Zips, D.; Huber, S.M. Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front. Immunol. 2018, 9, 3018. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Kong, L.Q.; Zhu, X.D.; Cai, H.; Wang, C.H.; Shi, W.K.; Cao, M.Q.; Li, X.L.; Li, K.S.; Zhang, S.Z.; et al. CD31 regulates metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma via the ITGB1-FAK-Akt signaling pathway. Cancer Lett. 2018, 429, 29–40. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Zong-Qing, Z.; Jin-Tao, C.; Ming-Cheng, Z.; Li-Juan, Y.; Jun-Ming, W.; Quan-Li, L.; Lu-Fei, C.; Zu-Cheng, Y.; Jin-Ming, L.; Zhi-Xiong, L. Nestin+/CD31+ Cells in the Hypoxic Perivascular Niche Regulate Glioblastoma Chemoresistance by Upregulating JAG1 and DLL4. Neuro. Oncol. 2021, 23, 905–919. [Google Scholar] [CrossRef]
- Wang, R.; Kang, Y.; Löhr, C.V.; Fischer, K.A.; Bradford, C.S.; Johnson, G.; Dashwood, W.M.; Williams, D.E.; Ho, E.; Dashwood, R.H. Reciprocal regulation of BMF and BIRC5 (Survivin) linked to Eomes overexpression in colorectal cancer. Cancer Lett. 2016, 381, 341–348. [Google Scholar] [CrossRef]
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, Y.; Akiyama, H.; Okada, K.; Ishida, S.; Nogami, A.; Oshikawa, G.; Kurosu, T.; Miura, O. Molecular mechanisms for enhancement of stromal cell-derived factor 1-induced chemotaxis by platelet endothelial cell adhesion molecule 1 (PECAM-1). J. Biol. Chem. 2017, 292, 19639–19655. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe Name | Nearest Gene Symbol | p Value | Adjusted p Value | Beta Difference | UCSC_Ref_Gene_Group | Relation to UCSC_CpG_Island |
|---|---|---|---|---|---|---|
| cg00236919 | NIPAL1 | 0.0266 | 0.0266 | −1.6098 | TSS1500 | N_Shore |
| cg02742775 | C12orf34 | 0.0062 | 0.0062 | 1.48821 | 5′UTR | Island |
| cg05454446 | BMS1 | 0.0044 | 0.0044 | −1.4252 | TSS200 | N_Shore |
| cg08085267 | C17orf57 | 0.0031 | 0.0031 | −1.9814 | 5′UTR | S_Shore |
| cg08209133 | SLC10A4 | 0.0031 | 0.0031 | −1.464 | 1stExon | Island |
| cg12297409 | ZNF485 | 0.0003 | 0.0003 | −1.5062 | TSS200 | N_Shore |
| cg18624900 | SLC16A12 | 0.0205 | 0.0205 | −1.6339 | TSS1500 | Island |
| cg19868631 | VSTM2A | 0.0343 | 0.0343 | 1.45488 | TSS1500 | Island |
| cg24368912 | ADH1B | 0.0031 | 0.0031 | 1.48875 | TSS1500 | |
| cg01912921 | FOXC1 | 0.0085 | 0.0085 | −1.6797 | 1stExon | Island |
| cg08699174 | PECAM1 | 0.0205 | 0.0205 | 2.181 | TSS1500 | |
| cg11585022 | WDR36 | 0.0205 | 0.0205 | 1.44761 | TSS1500 | N_Shore |
| cg11718030 | BDNF | 0.0117 | 0.0117 | −1.3965 | TSS1500 | Island |
| cg11792516 | HOOK1 | 0.0014 | 0.0014 | −1.5621 | TSS200 | N_Shore |
| cg15308664 | EOMES | 0.0117 | 0.0117 | −1.7284 | TSS200 | Island |
| cg17341969 | SNPH | 0.0266 | 0.0266 | 1.62061 | 3’UTR | S_Shore |
| cg18552861 | GDF7 | 0.0205 | 0.0205 | −1.4586 | TSS1500 | Island |
| cg19585676 | HLA-A | 0.0434 | 0.0434 | 1.53707 | 3’UTR | S_Shore |
| cg21070172 | CELSR1 | 0.0343 | 0.0343 | −1.4559 | 1stExon | Island |
| cg23022053 | PTGDR | 0.0266 | 0.0266 | 1.63417 | TSS1500 | N_Shore |
| cg26503653 | DOCK2 | 0.0009 | 0.0009 | −1.496 | TSS1500 | N_Shore |
| cg27049594 | OR8A1 | 0.0205 | 0.0205 | 2.28489 | TSS1500 | |
| cg08231348 | HOOK1 | 0.0266 | 0.0266 | −1.586 | TSS200 | N_Shore |
| cg10706454 | C1orf172 | 0.0205 | 0.0205 | −1.5193 | TSS200 | Island |
| cg15533046 | C12orf34 | 0.0117 | 0.0117 | 1.4762 | 5’UTR | N_Shore |
| cg25340688 | MIR886 | 0.0266 | 0.0266 | −1.5062 | TSS200 | Island |
| cg25502144 | NCRNA00028 | 0.0044 | 0.0044 | −1.5589 | TSS200 | S_Shore |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warrier, N.M.; Agarwal, P.; Kumar, P. Integrative Analysis to Identify Genes Associated with Stemness and Immune Infiltration in Glioblastoma. Cells 2021, 10, 2765. https://doi.org/10.3390/cells10102765
Warrier NM, Agarwal P, Kumar P. Integrative Analysis to Identify Genes Associated with Stemness and Immune Infiltration in Glioblastoma. Cells. 2021; 10(10):2765. https://doi.org/10.3390/cells10102765
Chicago/Turabian StyleWarrier, Neerada Meenakshi, Prasoon Agarwal, and Praveen Kumar. 2021. "Integrative Analysis to Identify Genes Associated with Stemness and Immune Infiltration in Glioblastoma" Cells 10, no. 10: 2765. https://doi.org/10.3390/cells10102765
APA StyleWarrier, N. M., Agarwal, P., & Kumar, P. (2021). Integrative Analysis to Identify Genes Associated with Stemness and Immune Infiltration in Glioblastoma. Cells, 10(10), 2765. https://doi.org/10.3390/cells10102765