
Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p



Abstract
:1. Introduction
2. Materials and Methods
2.1. Culturing and Stimulaton of Cells
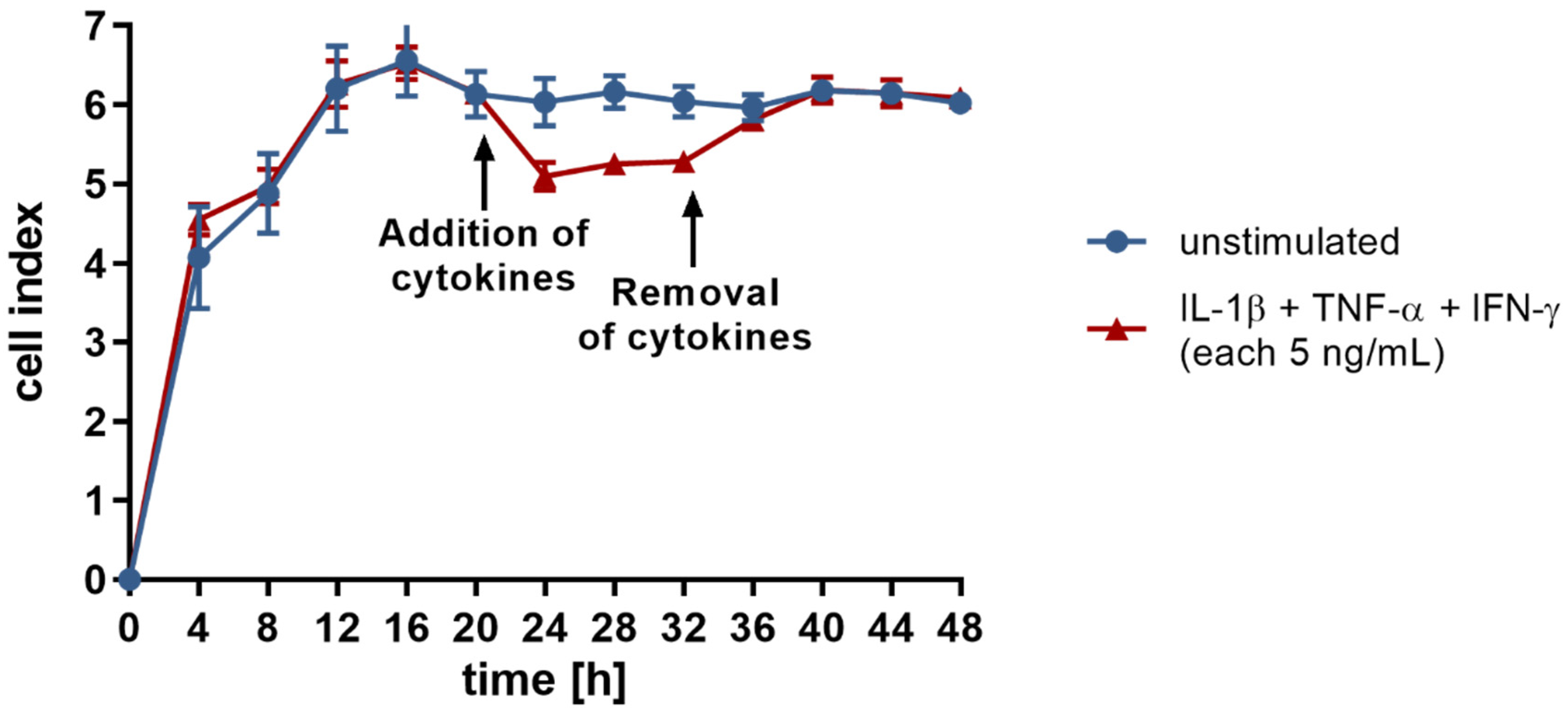
2.2. Impedance Analysis
2.3. RNA Isolation
2.4. MRNA Expression (Transcriptome) Screening
2.5. MiRNA Expression Screening
2.6. Droplet Digital PCR (ddPCR)
2.7. In Silico Analyses
3. Results
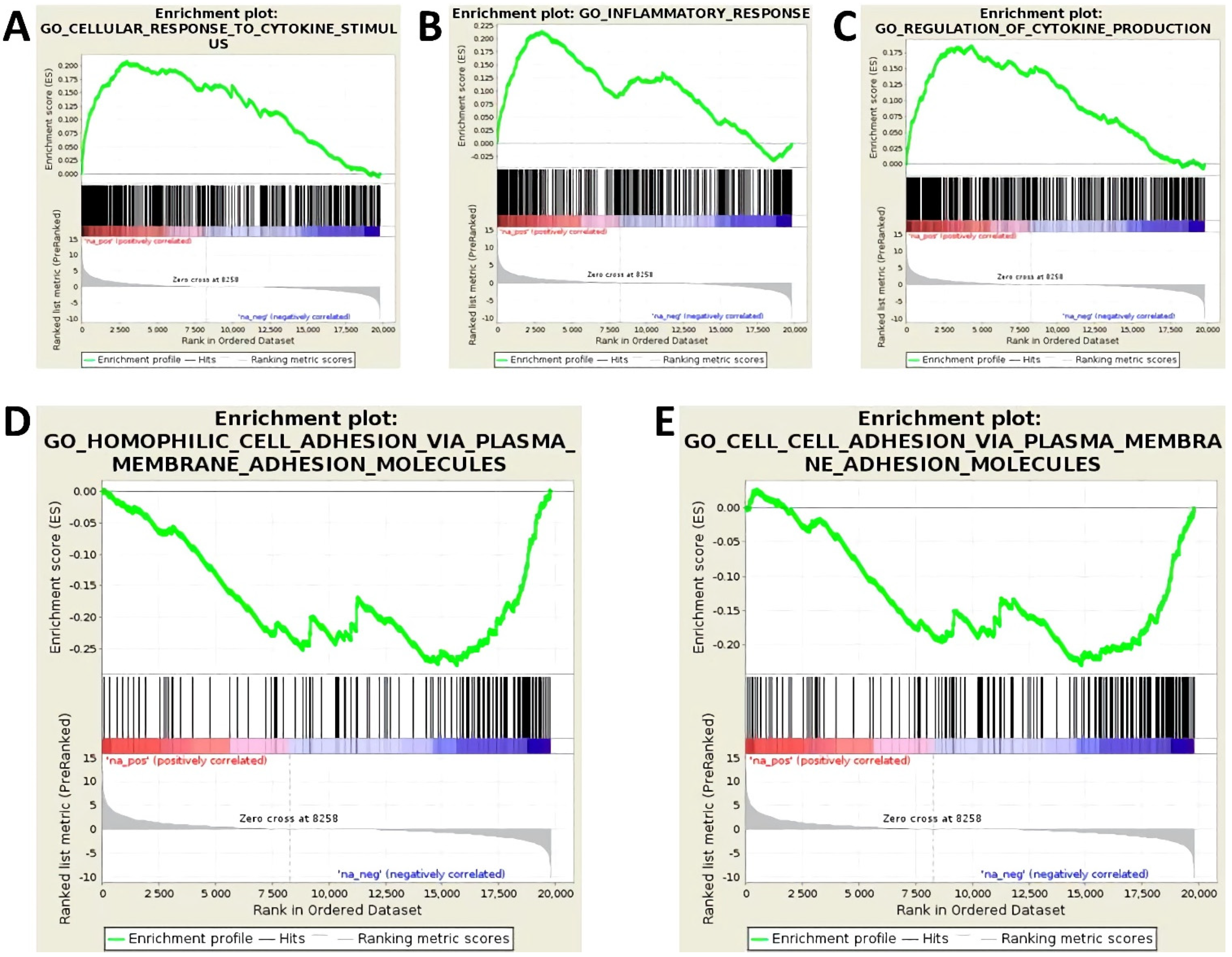
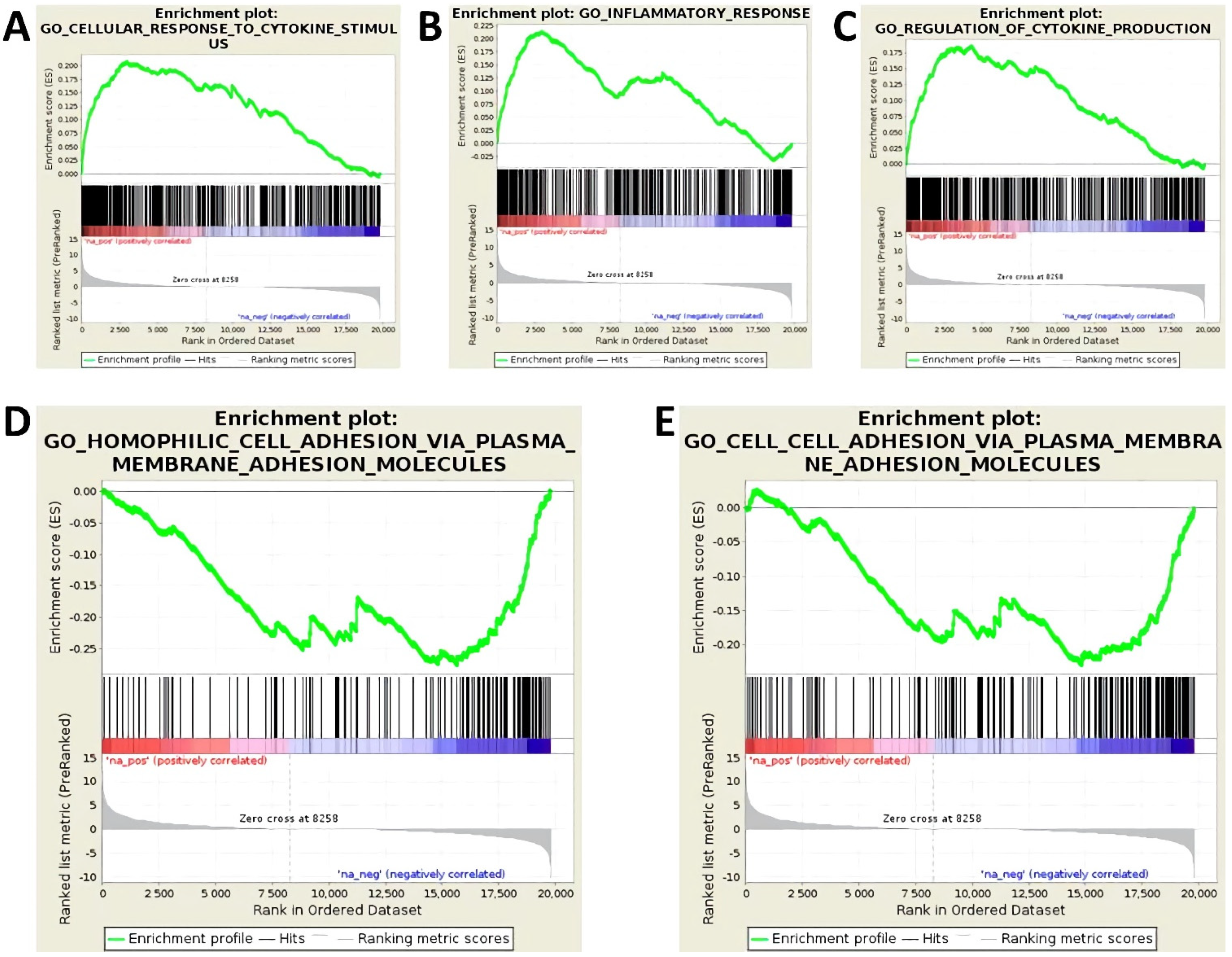
3.1. Treatment with Proinflammatory Cytokines Attenuates Endothelial Expression of Molecules of Homophilic Cell–Cell Adhesion and Results in Loss of Endothelial Cell Layer Integrity
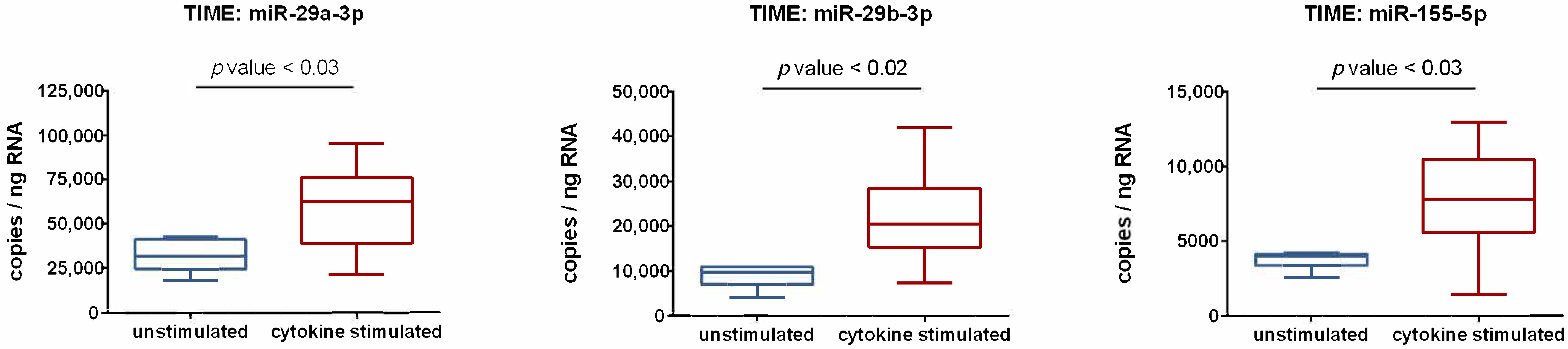
3.2. Activation of Endothelial Cells by Proinflammatory Cytokines Induces the Upregulation of miR-29a-3p, miR-29b-3p, and miR-155-5p
3.3. Expression of miR-29a-3p, miR-29b-3p, and miR-155-5p Is Subject to the Influence of Cytokine-Induced Signaling Cascades
3.4. miR-29a-3p, miR-29b-3p, and miR-155-5p Upregulated by Cytokine Treatment Target Key Mediators of Endothelial Barrier Function
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlegel, N.; Waschke, J. cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier—A signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef]
- Reglero-Real, N.; Colom, B.; Bodkin, J.V.; Nourshargh, S. Endothelial Cell Junctional Adhesion Molecules: Role and Regulation of Expression in Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, J.A.G.; Ridley, A.J. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J. Cell. Physiol. 2007, 213, 221–228. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Maury, E.; Lehoux, S.; Guidet, B.; Offenstadt, G. The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010, 36, 1286–1298. [Google Scholar] [CrossRef]
- Lee, W.L.; Liles, W.C. Endothelial activation, dysfunction and permeability during severe infections. Curr. Opin. Hematol. 2011, 18, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Simard, M.J.; Huot, J. Endothelial microRNAs regulating the NF-kappa B pathway and cell adhesion molecules during inflammation. FASEB J. 2018, 32, 4070–4084. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Kim, S.; Patterson, N.; Rooney, K.; Searles, C.D. MiRNA-155 targets myosin light chain kinase and modulates actin cytoskeleton organization in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1192–H1203. [Google Scholar] [CrossRef] [Green Version]
- Kingsley, S.M.K.; Bhat, B.V. Role of microRNAs in sepsis. Inflamm. Res. 2017, 66, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Pena-Philippides, J.C.; Gardiner, A.S.; Caballero-Garrido, E.; Pan, R.; Zhu, Y.; Roitbak, T. Inhibition of MicroRNA-155 Supports Endothelial Tight Junction Integrity Following Oxygen-Glucose Deprivation. J. Am. Heart Assoc. 2018, 7, e009244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Ma, C.; Zheng, X. MicroRNA-155 in the Pathogenesis of Atherosclerosis: A Conflicting Role? Heart Lung Circ. 2013, 22, 811–818. [Google Scholar] [CrossRef]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Peng, H.; Mastej, V.; Chen, W. MicroRNA Regulation of Endothelial Junction Proteins and Clinical Consequence. Mediat. Inflamm. 2016, 2016, 5078627. [Google Scholar] [CrossRef]
- Chatterjee, V.; Beard, R.S., Jr.; Reynolds, J.J.; Haines, R.; Guo, M.; Rubin, M.; Guido, J.; Wu, M.H.; Yuan, S.Y. MicroRNA-147b Regulates Vascular Endothelial Barrier Function by Targeting ADAM15 Expression. PLoS ONE 2014, 9, e110286. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Amado, T.; Schmolka, N.; Metwally, H.; Silva-Santos, B.; Gomes, A.Q. Cross-regulation between cytokine and microRNA pathways in Tcells. Eur. J. Immunol. 2015, 45, 1584–1595. [Google Scholar] [CrossRef] [Green Version]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]
- Hehlgans, T.; Pfeffer, K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: Players, rules and the games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta-Mol. Cell Res. 2002, 1592, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Daun, J.M.; Fenton, M.J. Interleukin-1/toll receptor family members: Receptor structure and signal transduction pathways. J. Interferon Cytokine Res. 2000, 20, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; O’Neill, L.A. The role of the interleukin-1/Toll-like receptor superfamily in inflammation and host defence. Microbes Infect. 2000, 2, 933–943. [Google Scholar] [CrossRef]
- Bhat, M.Y.; Solanki, H.S.; Advani, J.; Khan, A.A.; Prasad, T.S.K.; Gowda, H.; Thiyagarajan, S.; Chatterjee, A. Comprehensive network map of interferon gamma signaling. J. Cell Commun. Signal. 2018, 12, 745–751. [Google Scholar] [CrossRef]
- Gough, D.J.; Levy, D.E.; Johnstone, R.W.; Clarke, C.J. IFN-gamma signaling-Does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008, 19, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Trommer, S.; Leimert, A.; Bucher, M.; Schumann, J. Impact of Unsaturated Fatty Acids on Cytokine-Driven Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2017, 18, 2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trommer, S.; Leimert, A.; Bucher, M.; Schumann, J. Polyunsaturated Fatty Acids Induce ROS Synthesis in Microvascular Endothelial Cells. Adv. Exp. Med. Biol. 2018, 1072, 393–397. [Google Scholar] [CrossRef]
- Duan, Q.; Mao, X.; Xiao, Y.; Liu, Z.; Wang, Y.; Zhou, H.; Zhou, Z.; Cai, J.; Xia, K.; Zhu, Q.; et al. Super enhancers at the miR-146a and miR-155 genes contribute to self-regulation of inflammation. Biochim. Biophys. Acta-Gene Regul. Mech. 2016, 1859, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Shah, K.M.; Coles, M.C.; Sharp, T.V.; Lagos, D. MicroRNA-155 induction via TNF- and IFN-suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J. Biol. Chem. 2017, 292, 20683–20693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Ma, J.; Li, T.; Han, X. miR-29b aggravates lipopolysaccharide-induced endothelial cells inflammatory damage by regulation of NF-kappa B and JNK signaling pathways. Biomed. Pharmacother. 2018, 99, 451–461. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Pathway | miR-29a-3p | miR-29b-3p | miR-155-5p |
|---|---|---|---|
| NFκB pathway | RELA, RELB, NFKBIZ | NFKBIZ | RELB |
| MAPK pathway | JUN, JUNB, JUND, FOS, FOSL2, CEBPA, CEBPB, CEBPG, CREB1, ATF1, ATF2, ATF3, ATF4, BATF | JUND, FOS, FOSL2, CEBPA, CEBPB, CEBPG, ATF3, BATF | JUNB, JUND, CEBPA, CEBPB, ATF2, BATF |
| JAK-STAT pathway | STAT3, STAT5A, IRF2, IRF4, IRF9 | STAT3, IRF9 | STAT5A, IRF4 |
| Target Genes | miR-29a-3p | miR-29b-3p | miR-155-5p |
|---|---|---|---|
| Validated by previous experiments | F11R, CLDN1, CLDN5, CTNNB1, JUP, CTNND1, VCL, TJP1, LIMA1 | F11R, CLDN1, CTNNB1, CTNND1, VCL, LIMA1 | F11R, CLDN1, CTNNA1, CTNNB1, JUP, CTNND1, TJP1, TJP2, LIMA1 |
| Putative due to sequence analogies | OCLN, JAM3, CDH5, CTNNA1, TJP2 | OCLN, JAM3, CLDN5, CDH5, CTNNA1, JUP, TJP1, TJP2 | OCLN, JAM2, JAM3, CLDN5, VCL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maucher, D.; Schmidt, B.; Schumann, J. Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p. Cells 2021, 10, 2843. https://doi.org/10.3390/cells10112843
Maucher D, Schmidt B, Schumann J. Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p. Cells. 2021; 10(11):2843. https://doi.org/10.3390/cells10112843
Chicago/Turabian StyleMaucher, Daniel, Birte Schmidt, and Julia Schumann. 2021. "Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p" Cells 10, no. 11: 2843. https://doi.org/10.3390/cells10112843
APA StyleMaucher, D., Schmidt, B., & Schumann, J. (2021). Loss of Endothelial Barrier Function in the Inflammatory Setting: Indication for a Cytokine-Mediated Post-Transcriptional Mechanism by Virtue of Upregulation of miRNAs miR-29a-3p, miR-29b-3p, and miR-155-5p. Cells, 10(11), 2843. https://doi.org/10.3390/cells10112843