
The Synthesis, Crystal Structure, DFT Calculations and Optical Properties of Orcinolic Derivatives as OH− Indicators

 , ,
, , _CHAINOK.jpg)
Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Equipment
2.3. Methods
2.3.1. Crystallographic Methods
2.3.2. Computational Details
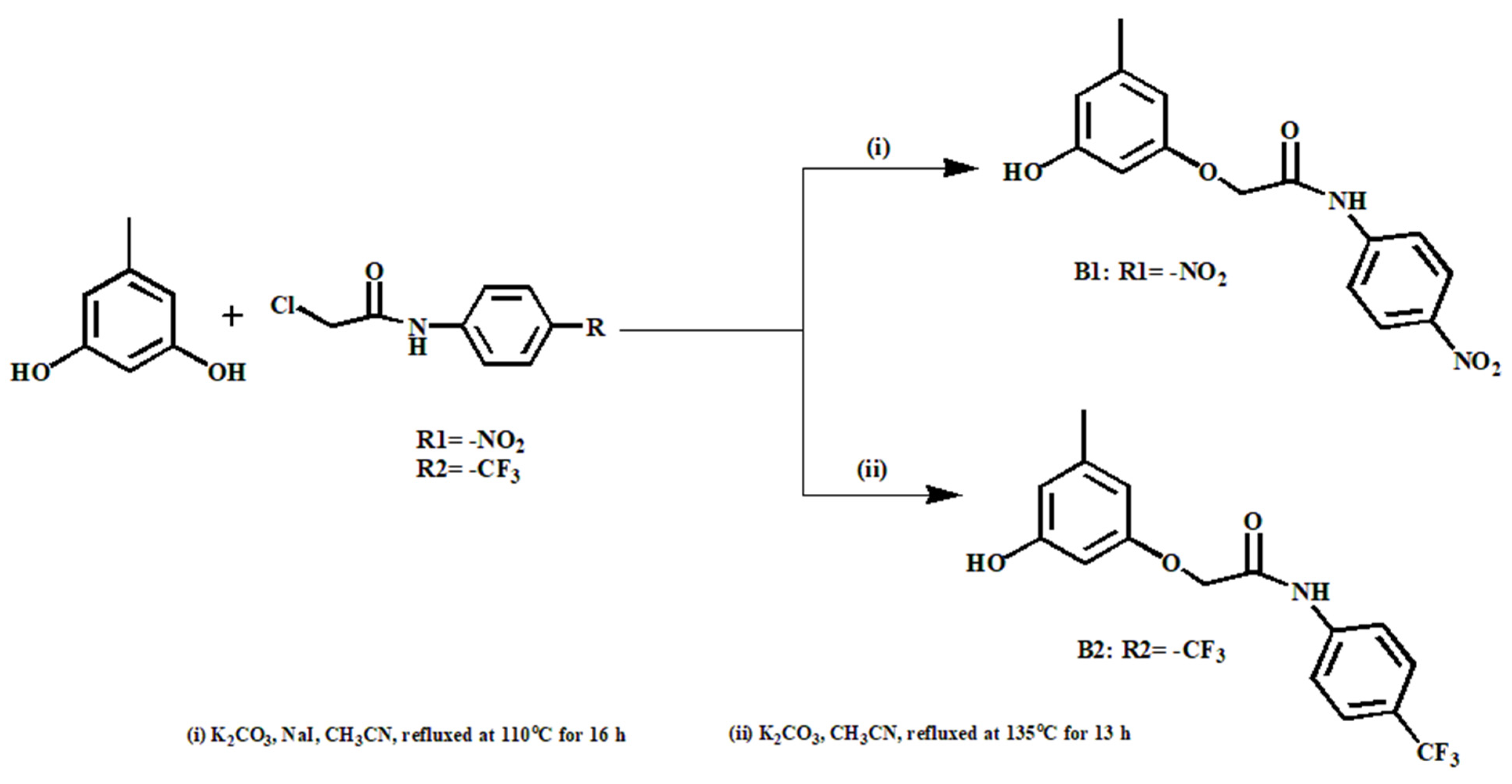
2.4. Synthesis of Compounds B1 and B2
3. Results and Discussion
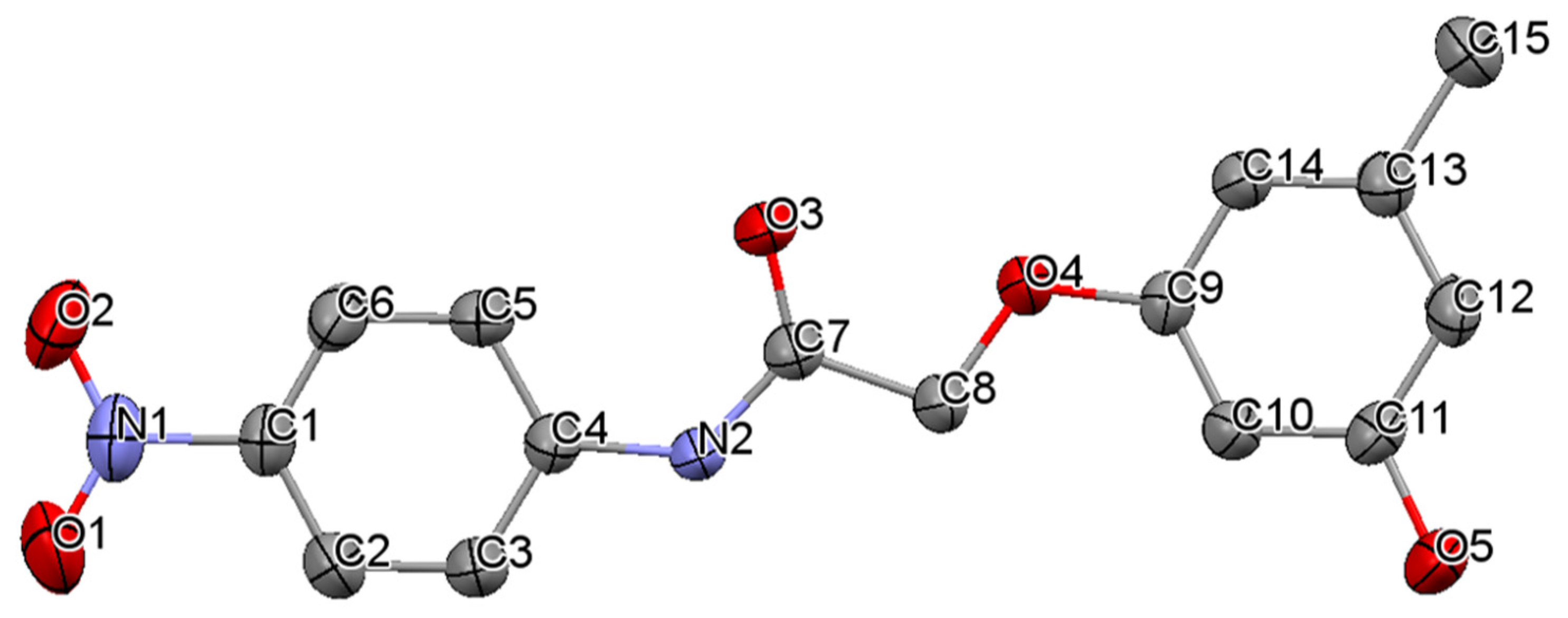
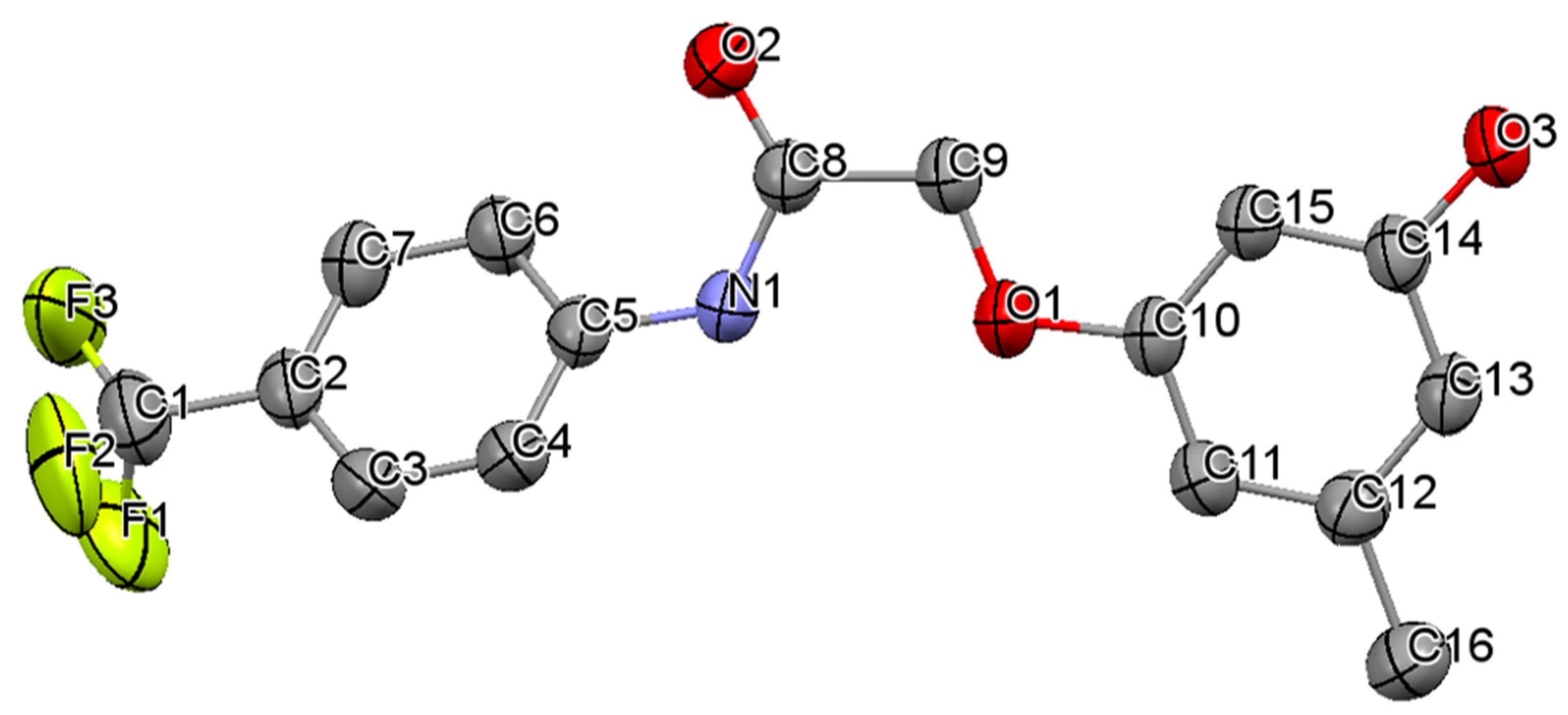
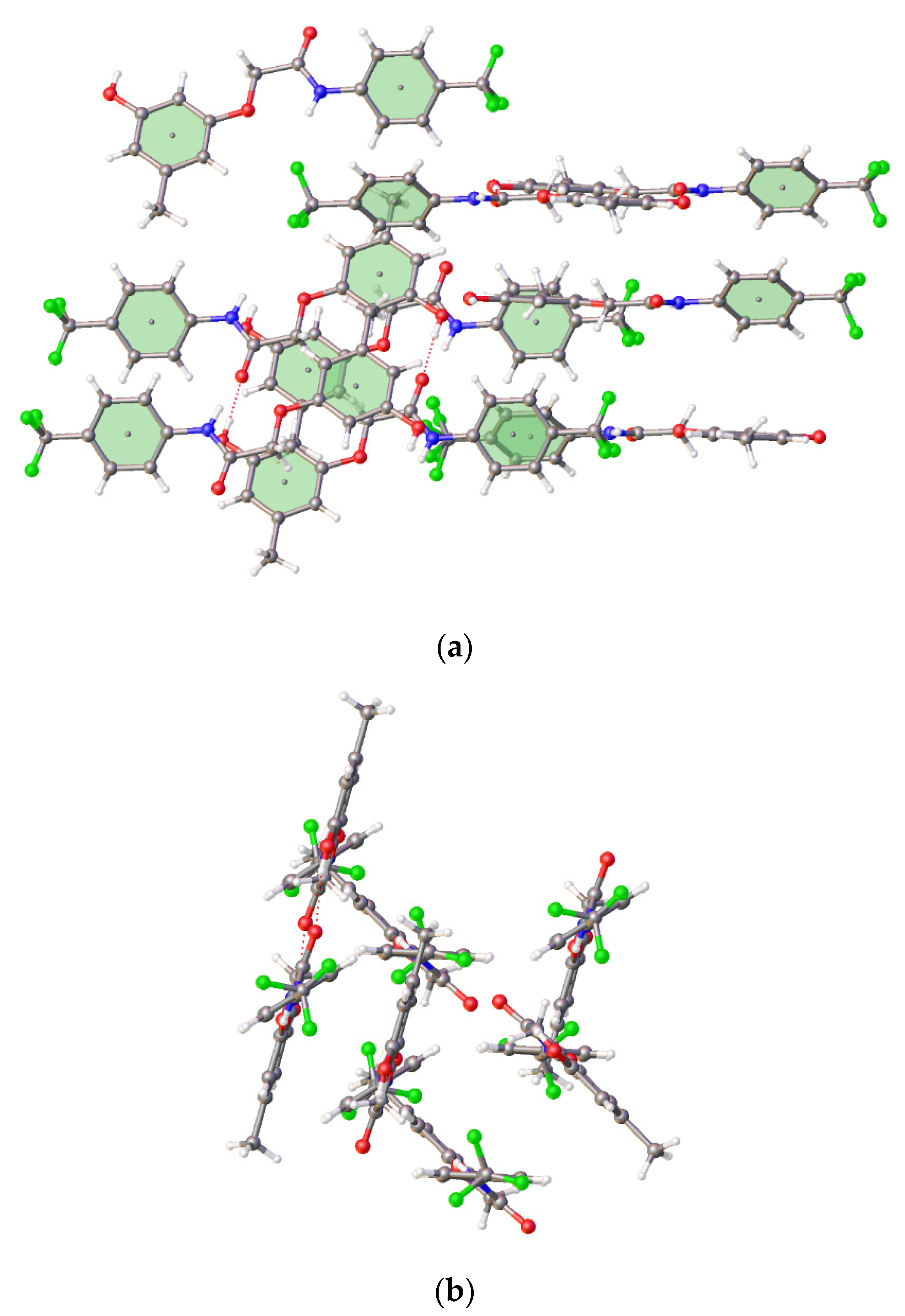
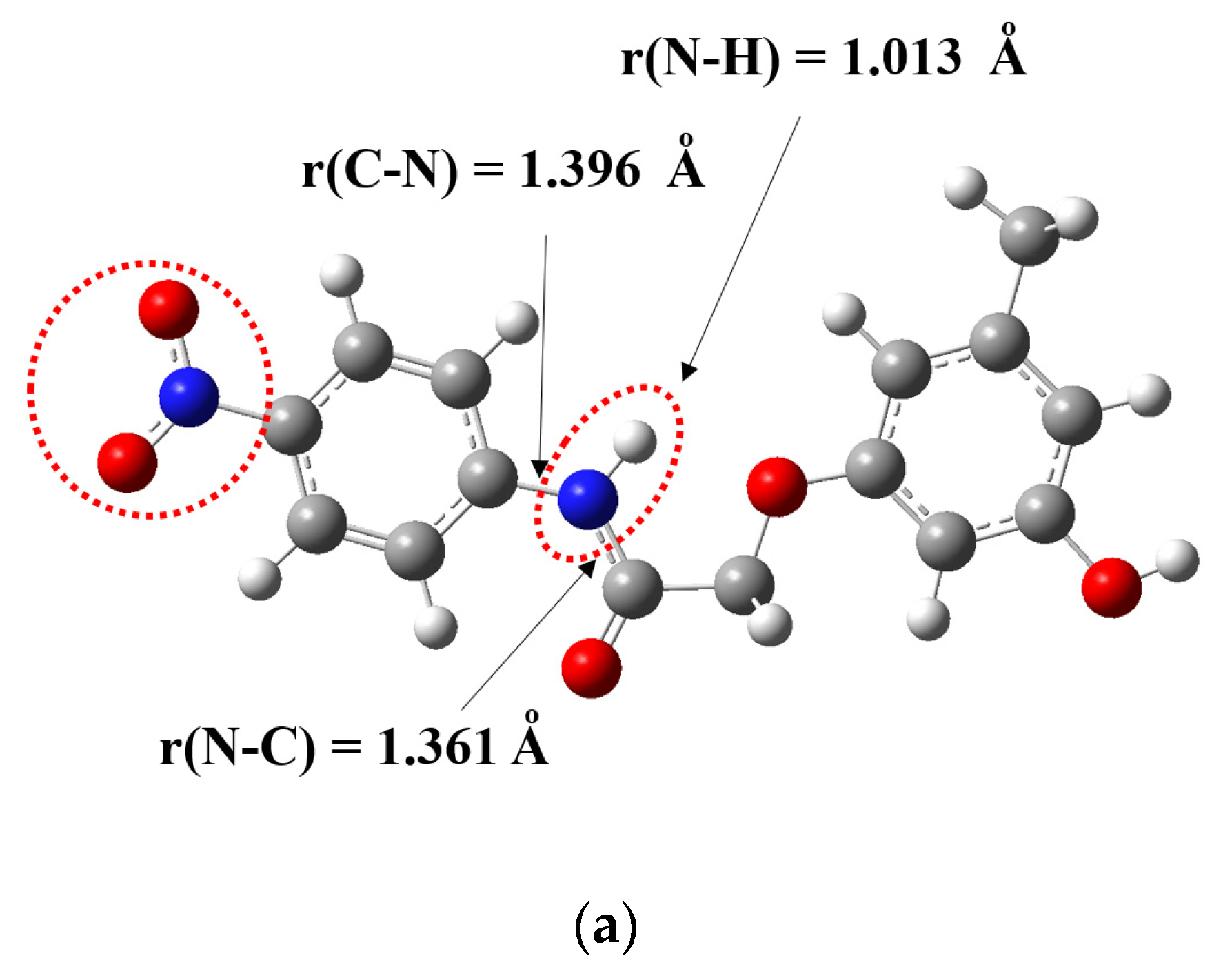
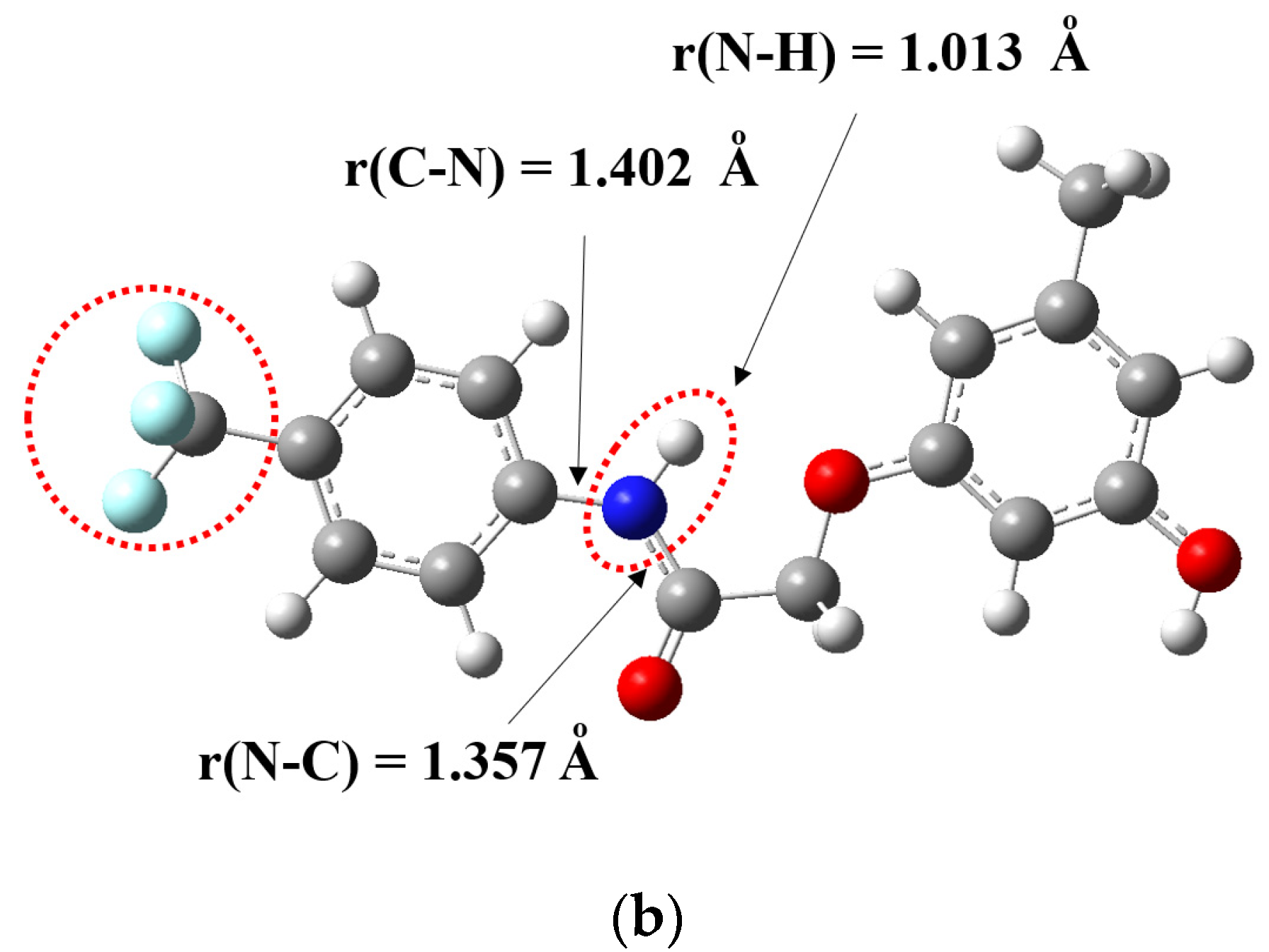
3.1. Crystal Structures of Compounds B1 and B2
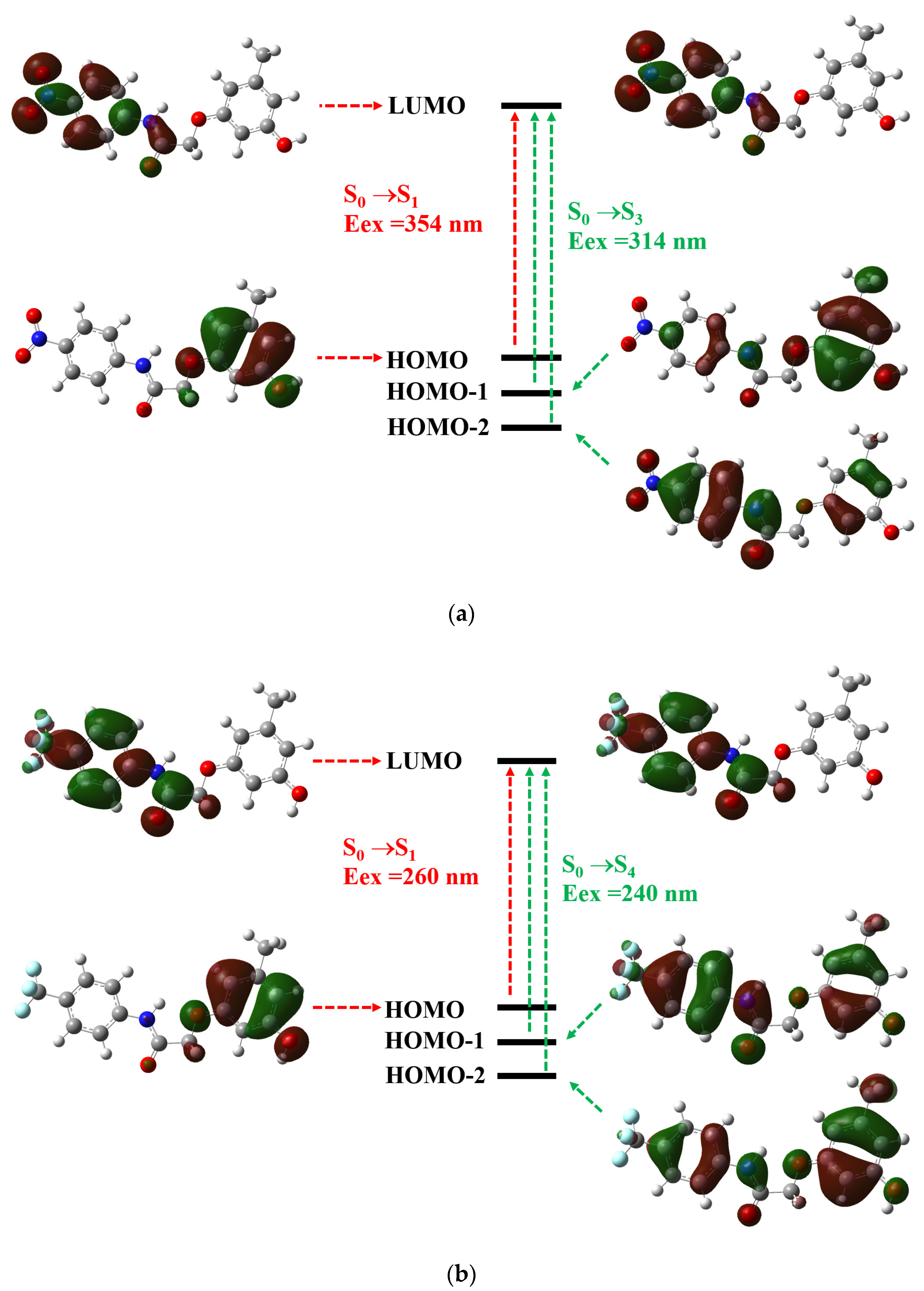
3.2. Theoretical Calculations
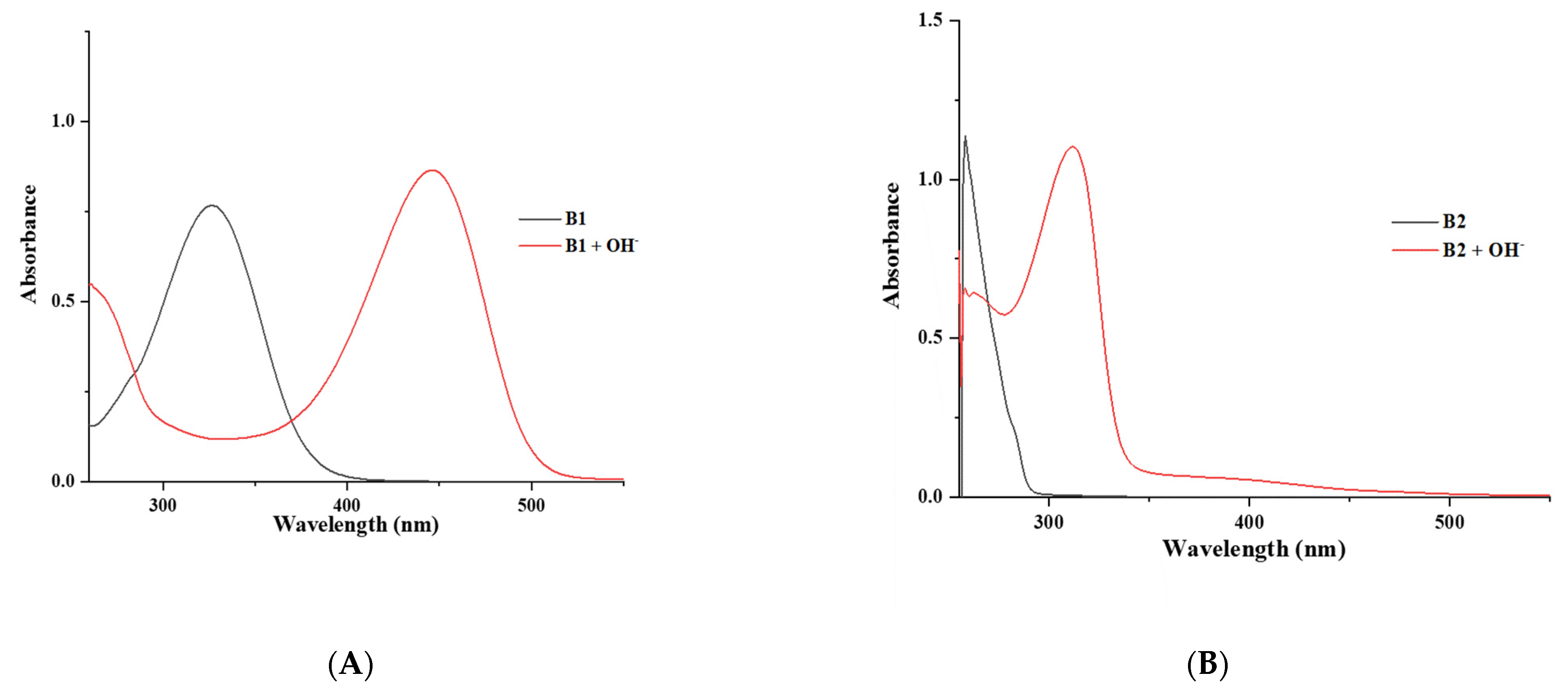
3.3. The Optical Properties of Compounds B1 and B2 by UV–Visible Spectroscopy
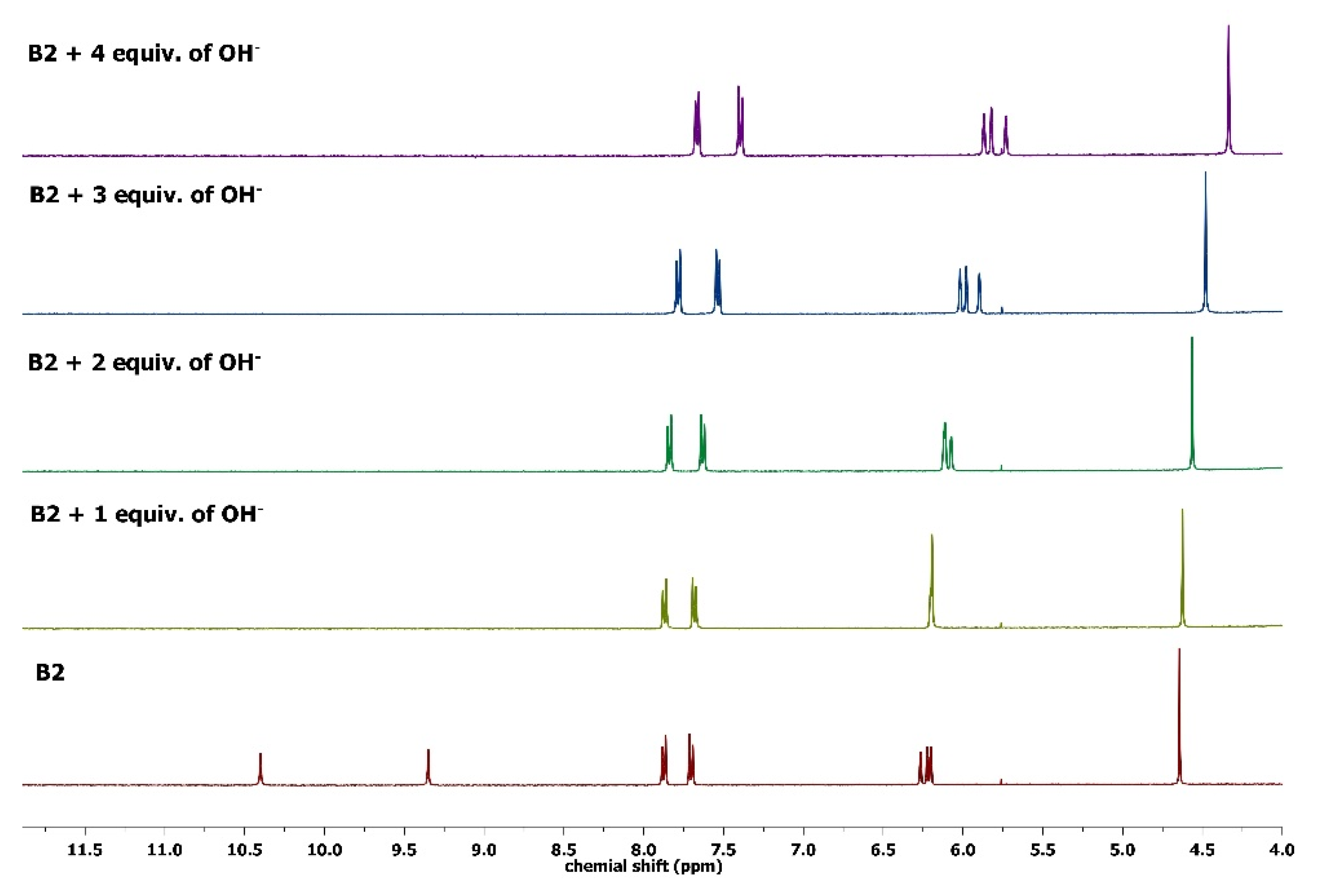
3.4. The Interactions between Compounds (B1 and B2) and OH− Ion by 1H NMR Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardullo, N.; Muccilli, V.; Pulvirenti, L.; Corrado Tringali, C. Natural isoflavones and semisynthetic derivatives as pancreatic lipase inhibitors. J. Nat. Prod. 2021, 84, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Taborga, L.; Díaz, K.; Olea, A.F.; Reyes-Bravo, P.; Flores, M.E.; Peña-Cortés, H.; Espinoza, L. Effect of polymer micelles on antifungal activity of geranylorcinol compounds against botrytis cinerea. J. Agric. Food Chem. 2015, 63, 6890–6896. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Zhanga, D.B.; Wang, J.Y.; Lua, R.M.; Zhenga, C.H.; Pua, S.Z. A novel diarylethene-hydrazinopyridine-based probe for fluorescent detection of aluminum ion and naked-eye detection of hydroxide ion. Sens. Actuators B 2017, 245, 263–272. [Google Scholar] [CrossRef]
- Mariappan, K.; Shrestha, P.K.; Hussain, A.; Sykes, A.G. A chemodosimeter for the detection of hydroxide using an anthraquinone-based receptor: Photophysical properties and X-ray crystallography. J. Mol. Struct. 2022, 1267, 133585. [Google Scholar] [CrossRef]
- Kuo, E.A.; Hambleton, P.T.; Kay, D.P.; Evans, P.L.; Matharu, S.S.; Little, E.; McDowall, N.; Jones, C.B.; Hedgecock, C.J.R.; Yea, C.M.; et al. Synthesis, Structure−Activity Relationships, and Pharmacokinetic Properties of Dihydroorotate Dehydrogenase Inhibitors: 2-Cyano-3-cyclopropyl-3-hydroxy- N-[3‘-methyl-4‘-(trifluoromethyl)phenyl]propenamide and Related Compounds. J. Med. Chem. 1996, 39, 4608–4621. [Google Scholar] [CrossRef] [PubMed]
- Aro-Heinilä, A.; Lepistö, A.; Äärelä, A.; Lönnberg, T.A.; Virta, P. 2 Trifluoromethyl-6-mercurianiline Nucleotide, a Sensitive 19F NMR Probe for Hg(II)-mediated Base Pairing. J. Org. Chem. 2022, 87, 137–146. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, W.; Hao, X.; Redshaw, C.; Chen, L.; Sun, W.-H. 6-Benzhydryl-4-methyl-2-(1H-benzoimidazol-2-yl)phenol ligands and their zinc complexes: Syntheses, characterization and photoluminescence behavior. Inorg. Chim. Acta 2012, 392, 345–353. [Google Scholar] [CrossRef]
- Chauhan, P.; Yan, N. Novel nitroaniline-cellulose nanohybrids: Nitro radical photo-release and its antibacterial action. Carbohydr. Polym. 2017, 174, 1106–1113. [Google Scholar] [CrossRef]
- Mohan, B.; Choudhary, M.; Bharti, S.; Jana, A.; Das, N.; Muhammad, S.; Al-Sehemi, A.G.; Kumar, S. Syntheses, characterizations, crystal structures and efficient NLO applications of new organic compounds bearing 2-methoxy-4-nitrobenzeneamine moiety and copper (II) complex of (E)-N’-(3, 5-dichloro-2-hydroxybenzylidene) benzohydrazide. J. Mol. Struc. 2019, 1190, 54–67. [Google Scholar] [CrossRef]
- Prabukanthan, P.; Lakshmi, R.; Harichandran, G.; Kumar, C.S. Synthesis, structural, optical and thermal properties of N-methyl–N-aryl benzamide organic single crystals grown by a slow evaporation technique. J. Mol. Struc. 2018, 1156, 62–73. [Google Scholar] [CrossRef]
- Nakayama, H.; Nishida, J.-I.; Takada, N.; Hiroyasu Sato, H.; Yamashita, Y. Crystal Structures and Triboluminescence Based on Trifluoromethyl and Pentafluorosulfanyl Substituted Asymmetric N-Phenyl Imide Compounds. Chem. Mater. 2012, 24, 671–676. [Google Scholar] [CrossRef]
- Ahmad, I.; Rehman, Z.U.; Waseem, A.; Tariq, M.; Beth, C.M.; Bacsa, J.; Venkataraman, D.; Rajakumar, A.; Ullah, N.; Tabassum, S. Organotin (IV) derivatives of amide-based carboxylates: Synthesis, spectroscopic characterization, single crystal studies and antimicrobial, antioxidant, cytotoxic, anti-leishmanial, hemolytic, noncancerous, anticancer activities. Inorg. Chim. Acta 2020, 505, 119433. [Google Scholar] [CrossRef]
- Sawant, A.S.; Kamble, S.S.; Pisal, P.M.; Sawant, S.S.; Hese, S.V.; Bagul, K.T.; Pinjari, R.V.; Kamble, V.T.; Meshram, R.J.; Gacche, R.N. Synthesis and evaluation of N-(4-(substituted)-3-(trifluoromethyl) phenyl) isobutyramides and their N-ethyl analogous as anticancer, anti-angiogenic & antioxidant agents: In vitro and in silico analysis. Comput. Biol. Chem. 2021, 92, 107484. [Google Scholar] [PubMed]
- Tian, C.; Wang, Q.; Wang, X.; An, G.; Li, G. Visible-Light Mediated ortho-Trifluoromethylation of Aniline Derivatives. J. Org. Chem. 2019, 84, 14241–14247. [Google Scholar] [CrossRef] [PubMed]
- Nath, J.; Baruah, B.J. Self-Assemblies of Solvates, Ionic Cocrystals, and a Salt Based on 4 {[(4-Nitrophenyl)carbamoyl]amino} N (pyrimidin-2-yl)benzene-1- sulfonamide: Study in the Solid and Solution States. Cryst. Growth Des. 2021, 21, 5325–5341. [Google Scholar] [CrossRef]
- Lobatto, V.L.; Argüello, G.A.; Caira, M.R.; Bujan, E.I. Trifluralin and two of its photodegradation products: Crystal structures and phase solubility/UV studies with cyclodextrins. J. Phys. Org. Chem. 2019, 32, e4006. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.D.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Yeh, M.-Y.; Lin, H.-C. Theoretical analysis of the intermolecular interactions in naphthalene diimide and pyrene complexes. Phys. Chem. Chem. Phys. 2014, 16, 24216–24222. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gussion 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Jansukra, P.; Duangthongyou, T.; Suramitr, S.; Kittipong Chainok, K.; Wannalerse, B. Synthesis, Crystal Structure and Optical Properties of 2-(3-(Hexyloxy)-5-Methylphenoxy)-N-(4-Nitrophenyl)acetamide for Anion Detection. Crystals 2021, 11, 671. [Google Scholar] [CrossRef]
- Bhat, H.R.; Gupta, P.S.S.; Satyaranjan Biswal, S.; Rana, M.K. Anion Sensing by Novel Triarylboranes Containing Boraanthracene: DFT Functional Assessment, Selective Interactions, and Mechanism Demonstration. ACS Omega 2019, 4, 4505–4518. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Sarma, T.; Wang, F.; Yuan, N.; Duan, Z.; Sessler, J.L.; Zhang, Z. Air-Stable N,N′ Dihydroporphycene: A Quinoxaline-Fused Tetrapyrrolic Macrocycle That Detects Fluoride Anion via Deprotonation. Org. Lett. 2019, 21, 1849–1852. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | Compound B1 | Compound B2 |
|---|---|---|
| Empirical formula | C15H14N2O5 | C32H28F6N2O6 |
| Formula weight | 302.28 | 650.56 |
| Temperature/K | 273.15 | 296.15 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a/Å | 7.1943(19) | 10.3749(19) |
| b/Å | 14.499(4) | 7.2604(14) |
| c/Å | 13.550(3) | 20.582(4) |
| α/° | 90.00 | 90 |
| β/° | 94.159(9) | 97.360(7) |
| γ/° | 90.00 | 90 |
| Volume/Å3 | 1409.7(6) | 1537.6(5) |
| Z | 4 | 2 |
| ρcalc g/cm3 | 1.424 | 1.405 |
| μ/mm−1 | 0.109 | 0.121 |
| F(000) | 632.0 | 672.0 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 6.02 to 50.12 | 5.956 to 54.068 |
| Index ranges | −8 ≤ h ≤ 8, −16 ≤ k ≤ 17, −16 ≤ l ≤ 16 | −13 ≤ h ≤ 13, −9 ≤ k ≤ 9, −26 ≤ l ≤ 26 |
| Reflections collected | 8786 | 48,473 |
| Independent reflections | 2500 [Rint = 0.1084, Rsigma = 0.0919] | 3361 [Rint = 0.0515, Rsigma = 0.0185] |
| Data/restraints/parameters | 2500/0/201 | 3361/105/239 |
| Goodness-of-fit on F2 | 0.975 | 1.029 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0556, wR2 = 0.1113 | R1 = 0.0428, wR2 = 0.1064 |
| Final R indexes [all data] | R1 = 0.1537, wR2 = 0.1435 | R1 = 0.0591, wR2 = 0.1228 |
| Largest diff. peak/hole/e Å−3 | 0.18/−0.22 | 0.20/−0.18 |
| Compound B1 | Compound B2 | ||||||
|---|---|---|---|---|---|---|---|
| Selected bond lengths | Selected bond lengths | ||||||
| N1–O1 | 1.221(4) | C7–C8 | 1.501(4) | C1–F1 | 1.336(4) | C8–C9 | 1.510(2) |
| N1–O2 | 1.226(4) | C8–O4 | 1.409(3) | C1–F2 | 1.244(6) | C9–O1 | 1.412(2) |
| C4–N2 | 1.408(4) | O4–C9 | 1.377(3) | C5–N1 | 1.419(2) | O1–C10 | 1.378(2) |
| N2–C7 | 1.352(4) | C13–C15 | 1.504(4) | N1–C8 | 1.338(2) | C12–C16 | 1.507(3) |
| C7–O3 | 1.211(4) | C11–O5 | 1.371(4) | C8–O2 | 1.222(2) | C14–O3 | 1.367(2) |
| Selected bond angles | Selected bond angles | ||||||
| C4–N2–C7 | 129.0(3) | C5–N1–C8 | 125.7(1) | ||||
| N2–C7–O3 | 123.7(3) | N1–C8–O2 | 124.7(1) | ||||
| N2–C7–C8 | 113.6(2) | N1–C8–C9 | 116.3(1) | ||||
| O3–C7–C8 | 122.7(3) | O2–C8–C9 | 119.0(1) | ||||
| C7–C8–O4 | 106.5(2) | C8–C9–O1 | 110.2(1) | ||||
| C8–O4–C9 | 118.3(2) | C9–O1–C10 | 117.8(1) | ||||
| C12–C13–C15 | 120.0(3) | C13–C12–C16 | 120.3(1) | ||||
| O5–C11–C12 | 122.7(3) | O3–C14–C13 | 117.8(1) | ||||
| Selected torsion angles | Selected torsion angles | ||||||
| O2–N1–C1–C6 | −177.0(3) | F3–C1–C2–C7 | −10.1(4) | ||||
| C7–N2–C4–C3 | −178.6(3) | C8–N1–C5–C4 | −142.3(2) | ||||
| C7–N2–C4–C5 | 1.8(5) | C8–N1–C5–C6 | 38.2(2) | ||||
| C4–N2–C7–O3 | 0.9(5) | C5–N1–C8–O2 | 0.7(2) | ||||
| C4–N2–C7–C8 | 179.1(3) | C5–N1–C8–C9 | −179.6(1) | ||||
| N2–C7–C8–O4 | 172.7(2) | N1–C8–C9–O1 | 1.7(2) | ||||
| C7–C8–O4–C9 | 174.2(2) | C8–C9–O1–C10 | 172.8(1) | ||||
| C8–O4–C9–C10 | 5.4(4) | C9–O1–C10–C11 | −177.1(1) | ||||
| C8–O4–C9–C14 | −174.3(2) | C9–O1–C10–C15 | 3.8(2) | ||||
| Compound B1 | ||||
| States | Eex (eV) | Eex (nm) | f | Electronic Transition |
| S0→S1 | 4.02 | 354 | 0.0048 | H→L(99%) |
| S0→S2 | 4.3 | 315 | 0.0006 | H-5→L(95%) |
| S0→S3 | 4.61 | 314 | 0.3465 | H-1→L(66%)+ H-2→L(33%) |
| S0→S4 | 4.78 | 279 | 0.0131 | H-2→L(65%)+ H-1→L(34%) |
| Compound B2 | ||||
| States | Eex (eV) | Eex (nm) | f | Electronic Transition |
| S0→S1 | 5.21 | 260 | 0.0062 | H→L(98%) |
| S0→S2 | 5.22 | 243 | 0.0397 | H-1→L+1(33%)+ H-2→L+1(28%) |
| S0→S3 | 5.33 | 242 | 0.1511 | H→L+2(70%)+ H-1→L+3(10%) |
| S0→S4 | 5.52 | 240 | 0.5093 | H-1→L(70%)+ H-2→L(18%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wannalerse, B.; Kwanmuang, P.; Jansukra, P.; Pinchaipat, B.; Duangthongyou, T.; Hasin, P.; Songsasen, A.; Chainok, K.; Suramitr, S. The Synthesis, Crystal Structure, DFT Calculations and Optical Properties of Orcinolic Derivatives as OH− Indicators. Crystals 2022, 12, 1252. https://doi.org/10.3390/cryst12091252
Wannalerse B, Kwanmuang P, Jansukra P, Pinchaipat B, Duangthongyou T, Hasin P, Songsasen A, Chainok K, Suramitr S. The Synthesis, Crystal Structure, DFT Calculations and Optical Properties of Orcinolic Derivatives as OH− Indicators. Crystals. 2022; 12(9):1252. https://doi.org/10.3390/cryst12091252
Chicago/Turabian StyleWannalerse, Boontana, Paradee Kwanmuang, Piangkwan Jansukra, Bussaba Pinchaipat, Tanwawan Duangthongyou, Panitat Hasin, Apisit Songsasen, Kittipong Chainok, and Songwut Suramitr. 2022. "The Synthesis, Crystal Structure, DFT Calculations and Optical Properties of Orcinolic Derivatives as OH− Indicators" Crystals 12, no. 9: 1252. https://doi.org/10.3390/cryst12091252
APA StyleWannalerse, B., Kwanmuang, P., Jansukra, P., Pinchaipat, B., Duangthongyou, T., Hasin, P., Songsasen, A., Chainok, K., & Suramitr, S. (2022). The Synthesis, Crystal Structure, DFT Calculations and Optical Properties of Orcinolic Derivatives as OH− Indicators. Crystals, 12(9), 1252. https://doi.org/10.3390/cryst12091252