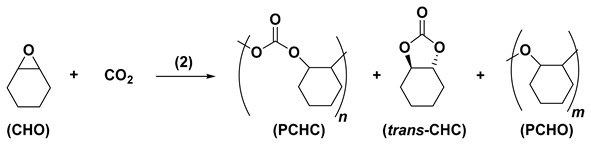
Abstract
The zinc complex Zn(C6F5)2(toluene) (1) behaves as a very active and selective catalyst in cyclohexene oxide (CHO) polymerization to produce poly(cyclohexene oxide) (PCHO) by the trans-ring-opening of CHO with remarkable TOF values at room temperature. The ring-opening copolymerization (ROCOP) of CO2 with CHO catalysed by 1 yields poly(cyclohexene carbonate) (PCHC) when using benzyl alcohol (BnOH) as an initiator at 120 °C. The 1H NMR monitoring of the in situ reaction of 1 with BnOH highlighted the formation of the dinuclear species [(C6F5)2Zn2(BnO)2 (2) that was isolated and found an active catalyst in the ROCOP of CO2 with CHO in the absence of initiators. Interestingly, PCHCs by 2 in solventless conditions show polydispersity index (Mw/Mn) values close to 2, corresponding to those expected for a single-site catalyst; on the contrary, a broader polydispersity index of the polymer products was found in toluene solution, suggesting the formation of new zinc catalysts during the polymerization reaction.
1. Introduction
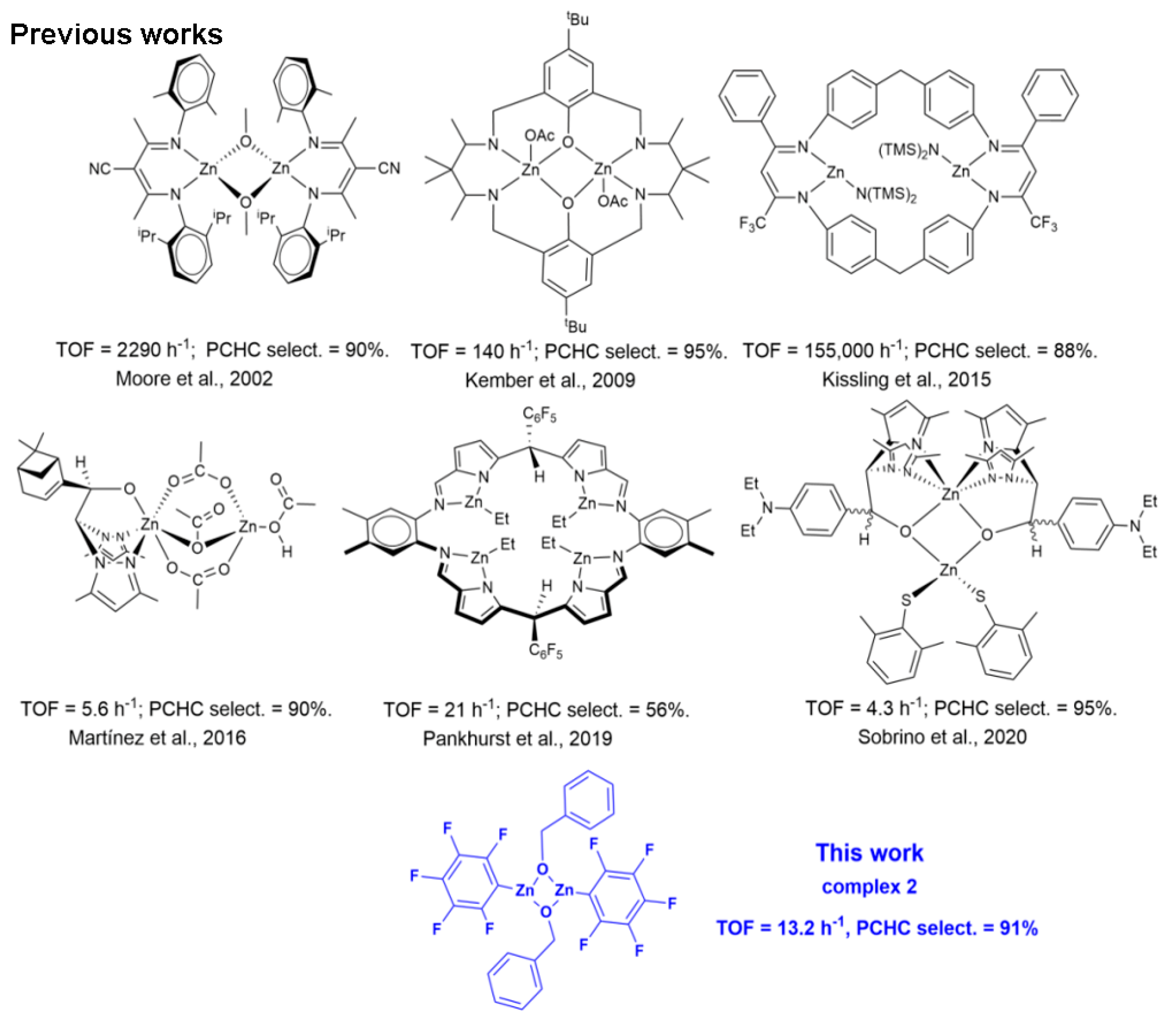
The last two decades witnessed a growing interest in the utilization of carbon dioxide (CO2) as an abundant, non-toxic and cheap C1 feedstock for the synthesis of fine chemicals and polymers [1,2]. In this field, one of the most studied reactions is the coupling of CO2 with epoxides that yields cyclic carbonates (COCs) [3,4] or aliphatic polycarbonates (APCs), depending on the reaction conditions and the catalytic system [5]. The most active catalysts for these reactions are based on binary systems consisting of a metal complex, acting as a Lewis acid activating the epoxide ring, and an onium salt (quaternary ammonium or phosphonium salts) delivering the nucleophile that allows the ring-opening of the oxirane ring. Parallel to the growing interest in the use of CO2, another important trend in green and sustainable chemistry is the use of Earth-crust-abundant, more environmentally benign metals in catalysis [6]. In this scenario, zinc complexes are ideal candidates in the CO2/epoxide reaction for the abundance, low cost and low toxicity of this metal; some successful examples of mono- and dinuclear zinc catalysts active in the CO2/epoxide coupling are pictured in Scheme 1 [7]. In particular, Zn β-diiminate [Zn(BDI)] complexes developed by Coates and co-workers [8,9] are one of the first examples of metal catalysts suitable for the synthesis of APCs by the Ring-Opening Alternating Copolymerization (ROCOP) of CO2 with epoxides that sparked the interest in dinuclear [10,11,12,13,14,15,16] and multinuclear zinc catalysts [17,18]. Based on kinetic and mechanistic studies, it was soon evident that good activity and high selectivity in the formation of APCs vs. COCs depend on the dinuclear metal complex architecture that allows the ring-opening of the monomer and the polymer chain growth. A bimetallic mechanism has also been postulated for the catalysts developed by Rieger [10,19] and Williams [12,20] which are among the most active catalysts in the ROCOP of cyclohexene oxide (CHO) with CO2. Intriguingly, dinuclear zinc complexes based on different ligand frameworks are less active, highlighting the fundamental role of the ligand structure in defining the optimal distance between the two metal centres active in catalysis. Of note, Zn-O-Zn structural motifs are also involved in the homopolymerization of oxiranes, giving the corresponding polyethers [21], with the possibility of controlling the stereochemistry of the polymeric product by introducing chiral ligands on the metal centre [22]. Herein we report on the application of Zn(C6F5)2(toluene) (1) as the active catalyst in the homopolymerization of CHO and the alternating copolymerization of CHO with CO2 using benzyl alcohol (BnOH) as the initiator. The investigation of the reaction of 1 with BnOH allowed the synthesis of the new dinuclear (C6F5)2Zn2(BnO)2 catalyst (2), which was found active in the ROCOP of CO2 with CHO in the absence of further initiators.
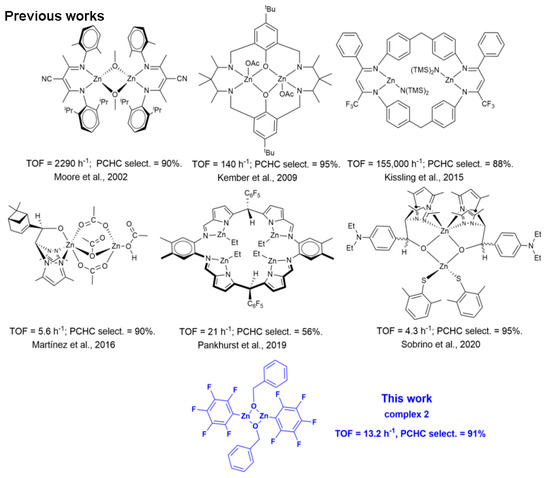
Scheme 1.
Dinuclear zinc catalysts for the alternating ROCOP of CO2 and CHO (Moore et al., 2002 [23], Kember et al., 2009 [12], Kissling et al., 2015 [10], Martínez et al., 2016 [13], Pankhurst et al., 2019 [24], Sobrino et al., 2020 [25]).
2. Results and Discussion
2.1. Ring-Opening Polymerization of Cyclohexene Oxide Promoted by the Complex 1
The toluene adduct of the bis(perfluoroaryl) zinc complex Zn(C6F5)2(toluene) (1) was synthesized and isolated as previously described in the literature [26]. The role of this strong Lewis acid as the catalyst in the ring-opening polymerization (ROP) of CHO was preliminarily screened in the absence of solvent under different experimental conditions (Table 1). An instantaneous and highly exothermic polymerization process was observed in 1 min, with a catalyst loading in the range of 0.02–0.1 mol%, corresponding to a CHO/1 molar ratio of 1000–5000 in the reaction feed (entries 1–4, Table 1). The monomer conversion was assessed in the crude reaction mixture by the integration of the 1H signals at 3.12 ppm for CHO and 3.37–3.53 ppm for poly(cyclohexene oxide) (PCHO) (Figure S8) yielding a value of 90 % of CHO conversion for entry 2 of Table 1. The full monomer conversion was never reached, likely because of the gelation of the solvent and the high viscosity of the polymerization medium in a short time. The turnover frequency (TOF) values increased as the monomer concentration increased, until reaching the remarkable value of 4200 min−1 at the catalyst loading of 0.02 mol%, (CHO/1 molar ratio in the feed of 5000; entry 4 of Table 1). The polydispersity index (Đ = Mw/Mn) of the PCHOs is in the range of 1.9–2.0, as expected for a single-site catalyst in a process where chain transfer reactions are active. The reduced polymerization control could result from the experimental conditions employed, e.g., neat monomer and poor control of temperature in a highly exothermic and rapid process. The polymerization of CHO in toluene ([CHO] = 1 M–5 M) was unfruitful at room temperature (entry 5, Table 1) whereas in dichloromethane solution ([CHO] = 5 M), only 35 % of the monomer conversion in 20 h was observed, and the Đ of the resulting PCHO is 1.7 (entry 6, Table 1).

Table 1.
Polymerization of neat CHO catalysed by complex 1.
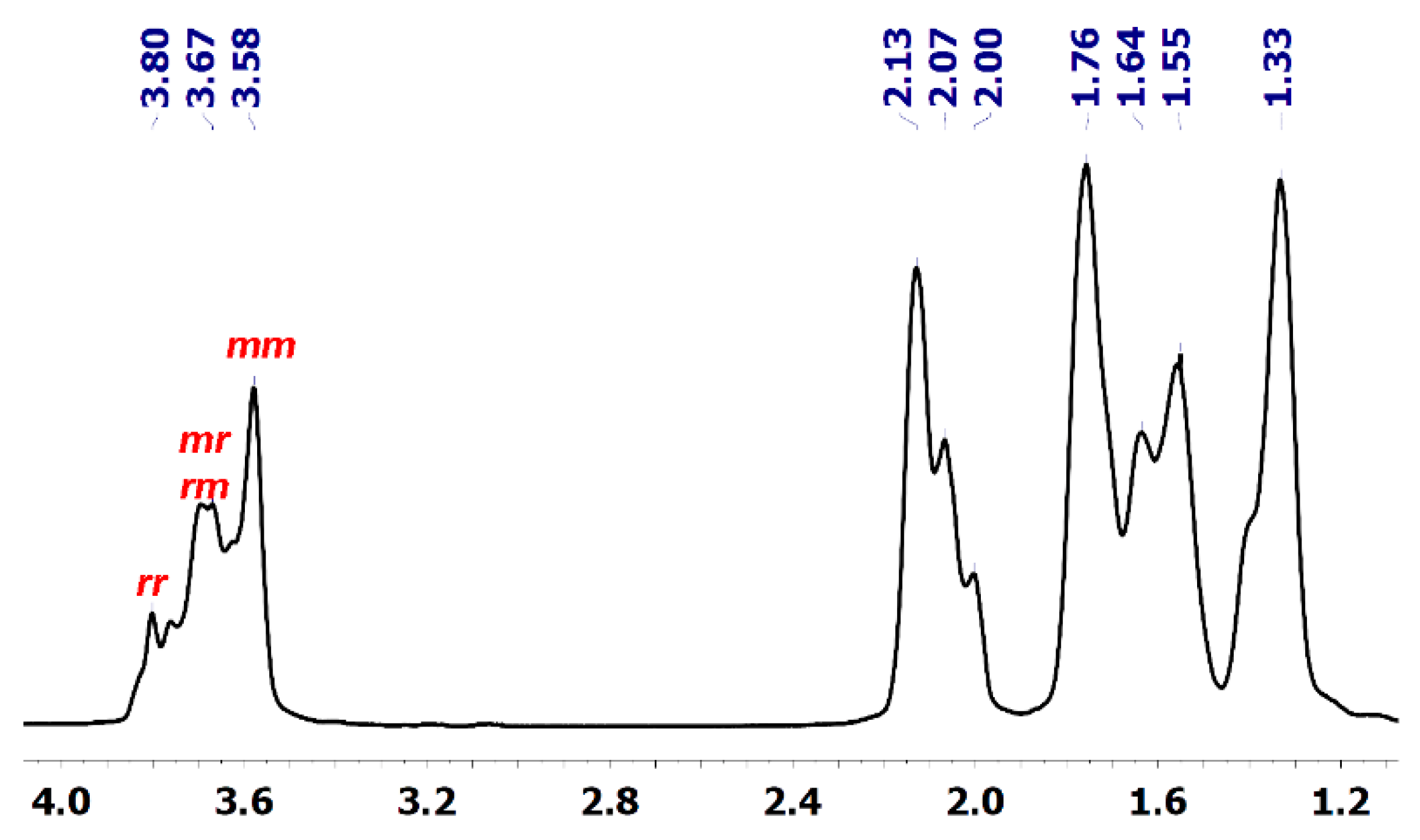
To gain information on the CHO polymerization mechanism catalysed by 1, the tacticity of the PCHOs was investigated by 1H and 13C NMR spectroscopy. The methine regions of the 1H NMR and 13C NMR spectra, respectively, at 3.58–3.80 ppm (Figure 1) and 76.71–78.64 ppm (Figures S9–S11), are diagnostic of the polymer stereochemistry. The threo-disyndiotactic triad (rr) resulting from the stereoselective anti opening of the oxirane ring produces broad 1H signals centred at about 3.8 ppm, whereas the corresponding heterotactic (mr) and threo-di isotactic triads (mm) are observed as a broad signal centred at 3.75 ppm and sharp signal at 3.55 ppm, respectively [27,28,29]. The integration of methine signals at 78.64, 78.11 and 76.7–77.7 ppm in the 13C NMR spectrum, which is better resolved than the corresponding 1H pattern, provides a ratio of about 1:1:1 for the threo-diisotactic, heterotactic and threo-disyndiotactic diads, respectively (Figure S11; Entry 5, Table 1; see additionally COSY and HSQC spectrum of Figures S12–S14, respectively, for the corresponding assignments). The noteworthy cationic polymerization of CHO initiated by strong Brønsted or Lewis acids such as ZnR2/H2O (R = Et, Ph) [(ZnEt2/H2O) [30,31,32] or metal catalysts such as yttrium alkoxides [33] and aluminium amine-phenolate catalysts [34] yielded PCHOs with the same NMR fingerprints. The polymer structure was in all cases defined as atactic, not considering that a truly atactic pattern would correspond to the intensity ratio of 1:2:1 for the threo-diisotactic, heterotactic and threo-disyndiotactic diads. Independently of the proposed polymerization mechanism, namely, coordination/insertion (with metal alkoxides), active chain end mechanism (ACE) (ionic polymerization) or activated monomer mechanism (AMM), the resulting PCHOs show the same stereoregularity possibly resulting from a chain end stereocontrol.
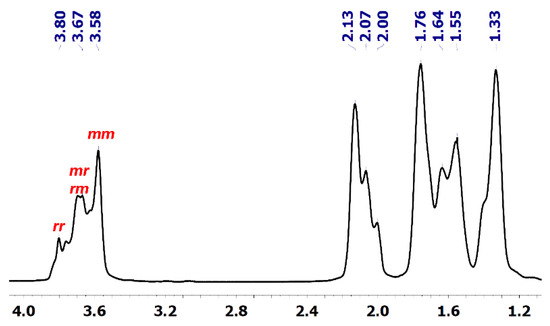
Figure 1.
1H NMR spectrum of PCHO (600 MHz, C6D6, 25 °C) (entry 5 of Table 1) with the signals for the isotactic (mm), heterotactic (mr and rm) and syndiotactic (rr) triads labelled.
Coming to the catalytic performances of 1 in CHO polymerization we observed that the addition of THF during the polymerization run terminates the polymerization process; moreover, CHO polymerization is inhibited at low temperature, and the TOF values dramatically decrease in solution. All these features suggest an AMM active in the CHO polymerization process catalysed by 1. Furthermore, the number of polymer chains can be roughly calculated from the mass of the PCHO recovered at the end of the polymerization and the Mn values determined by GPC analysis (Table 1); these values were found in the range of 2.5–19 polymer chains per zinc atom, which can be ascribed to the presence of trace amounts of water in the reaction mixture, acting both as initiator and terminating agent, to produce hydroxyl-terminated polymer chains [35].
2.2. ROCOP of CHO with CO2 Promoted by 1 and BnOH as Initiator
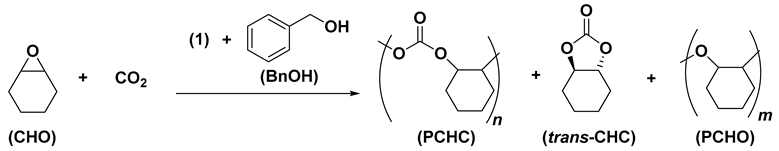
Based on the above-discussed results, we were prompted to investigate the coupling of CO2 with CHO catalysed by 1 in toluene, introducing benzyl alcohol (BnOH) as an initiator in variable amounts (Table 2). Interestingly, the most abundant product of this reaction was poly(cyclohexene carbonate) (PCHC), resulting from the alternating copolymerization of CO2 with CHO. The loading of 1 was fixed to 0.2 mol% (corresponding to a CHO/(1) molar ratio of 500), the CO2 pressure was set at 2.0 MPa and the reaction temperature at 120 °C. The CHO conversion is weakly affected by the BnOH/1 molar ratio in the range from 1 to 10; the highest TOF of 8 molCHO mol(1)−1 h−1 was observed when using 5 equiv. of BnOH, whereas the conversion decreases at a molar ratio of 10 (entries 1–4, Table 2). A strong excess of benzyl alcohol (BnOH/1 = 10) at 120 °C probably induces the partial decomposition of the active zinc catalyst and leads to lower CHO conversion and higher molecular weight because of the higher CHO/zinc catalyst molar ratio (entry 4, Table 2). The polymerization temperature strongly influences the activity of the 1-BnOH catalyst: actually, the TOF value progressively decreased as the polymerization temperature was decreased from 120 °C to 60 °C (compare entries 5–7 with entry 2, Table 2). The increase in CO2 pressure in the range of 0.5–2.0 MPa produces an increase in the TOFs (compare entries 8–9 with entry 3, Table 2).
Table 2.
ROCOP of CHO with CO2 promoted by complex 1 using BnOH as initiator.
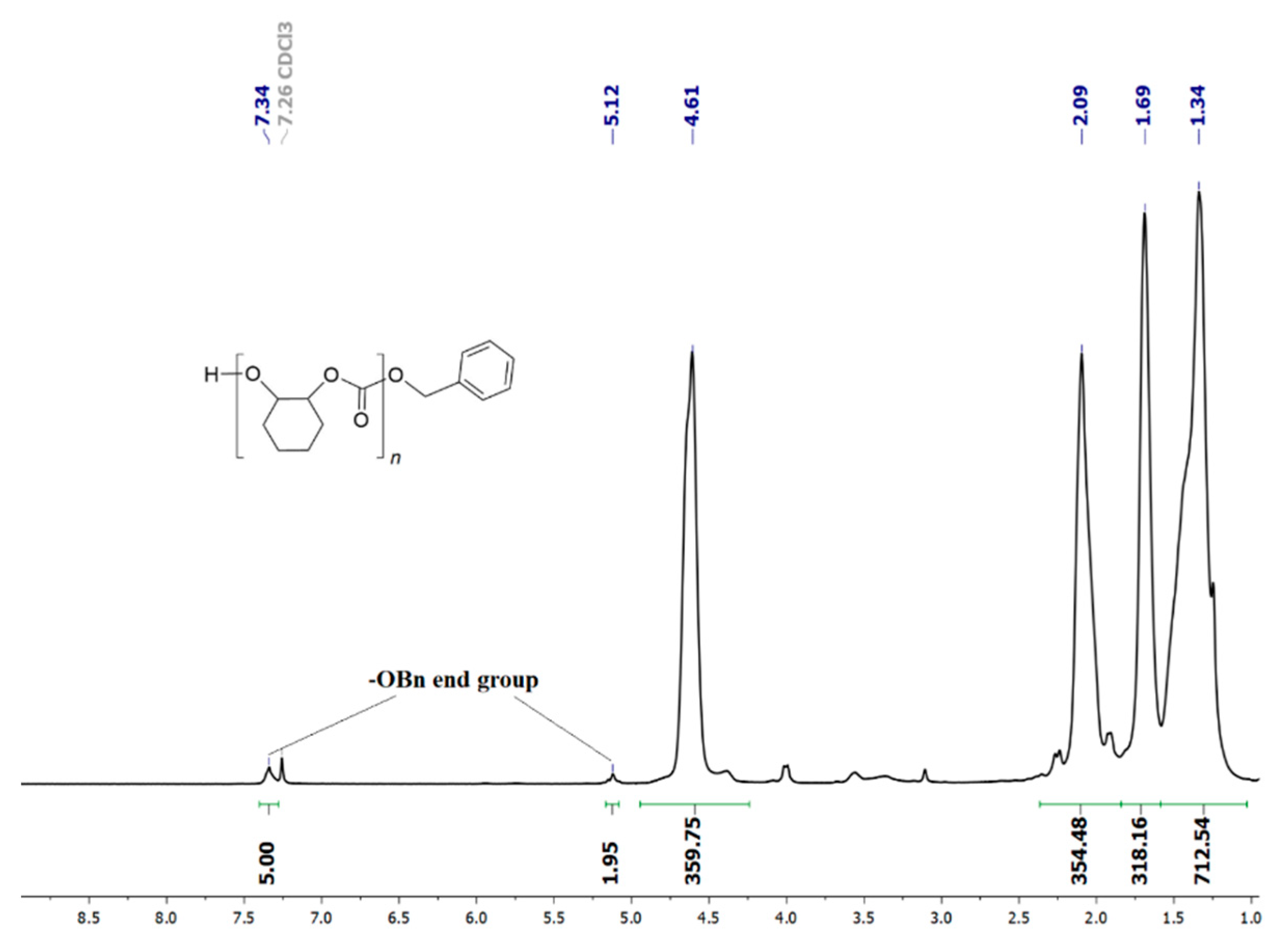
Selectivity in APCs vs. COCs formation and the block lengths of the polyether sequences (PCHO) vs. the polycarbonate sequences (PCHC) are the most important parameters in the ROCOP of CO2 with CHO [36]. The 1H NMR spectra of the crude reaction mixture resulting from the entries of Table 2 show the presence of small amounts of trans-cyclohexene carbonate (4–10 mol%), assessed by the presence of a methine 1H signal at 4.01–4.03 ppm (Figure S15) [37,38,39,40]. The selective formation of PCHC vs. PCHO was determined by the integration of 1H NMR signals with the methine protons of PCHC and PCHO at 4.6 and 3.3–3.6 ppm, respectively (see Figures S15 and S16 for the corresponding 13C NMR spectrum) [35]. The copolymer samples obtained at higher CO2 pressure (2.0 MPa) and temperature (80–120 °C) show good selectivity in favour of PCHC sequences (83–91 mol%; entries 1–6, Table 2); on the contrary, the samples obtained at low temperature (60 °C; entry 7, Table 2) or low PCO2 pressure (0.5 MPa; entry 9, Table 2) show 1H signals due to polyether sequences with higher intensity corresponding to PCHC selectivity in the range of 62–64 mol%. Higher reaction temperature and CO2 pressure improve the alternating incorporation of CO2 and CHO, affording a higher concentration of polycarbonate segments with respect to polyether segments (see Table 2). The 1H NMR spectra of the PCHCs by catalyst 1 show benzyloxy end groups (see e.g., Figure 2) to prove that the initiation reaction occurs through the CO2 insertion onto the Zn-OBn bond resulting from protonolysis of 1 with BnOH.
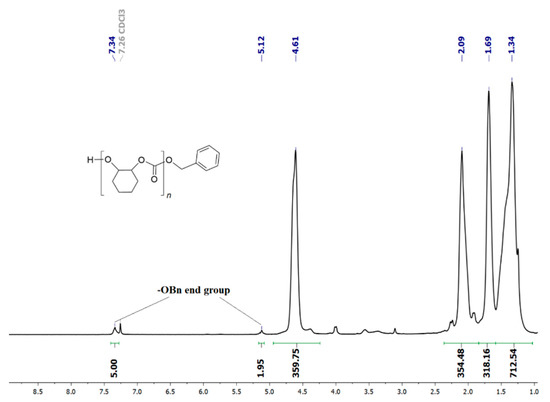
Figure 2.
1H NMR spectrum (400 MHz, CDCl3, 25 °C) of PCHC (from entry 2 of Table 2).
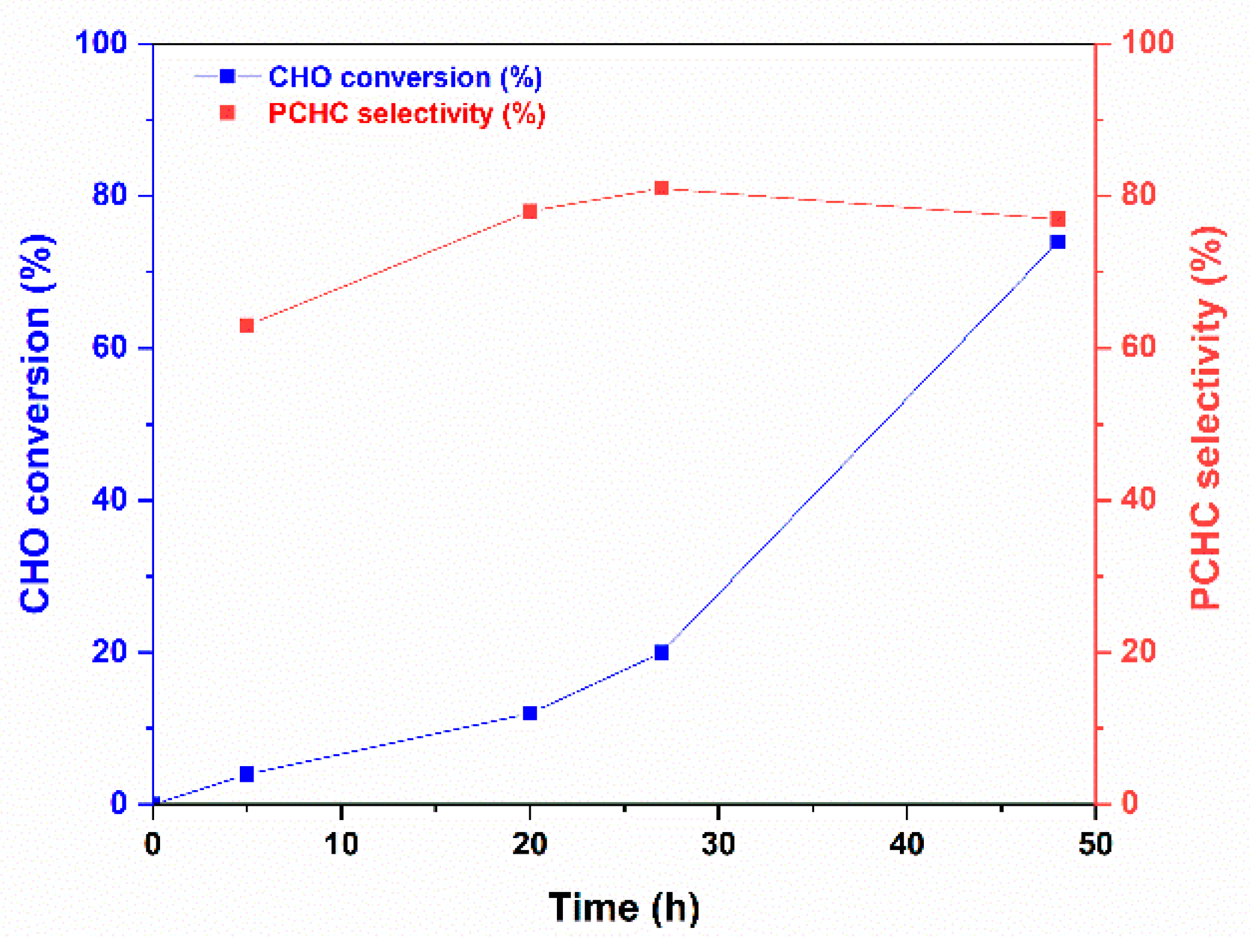
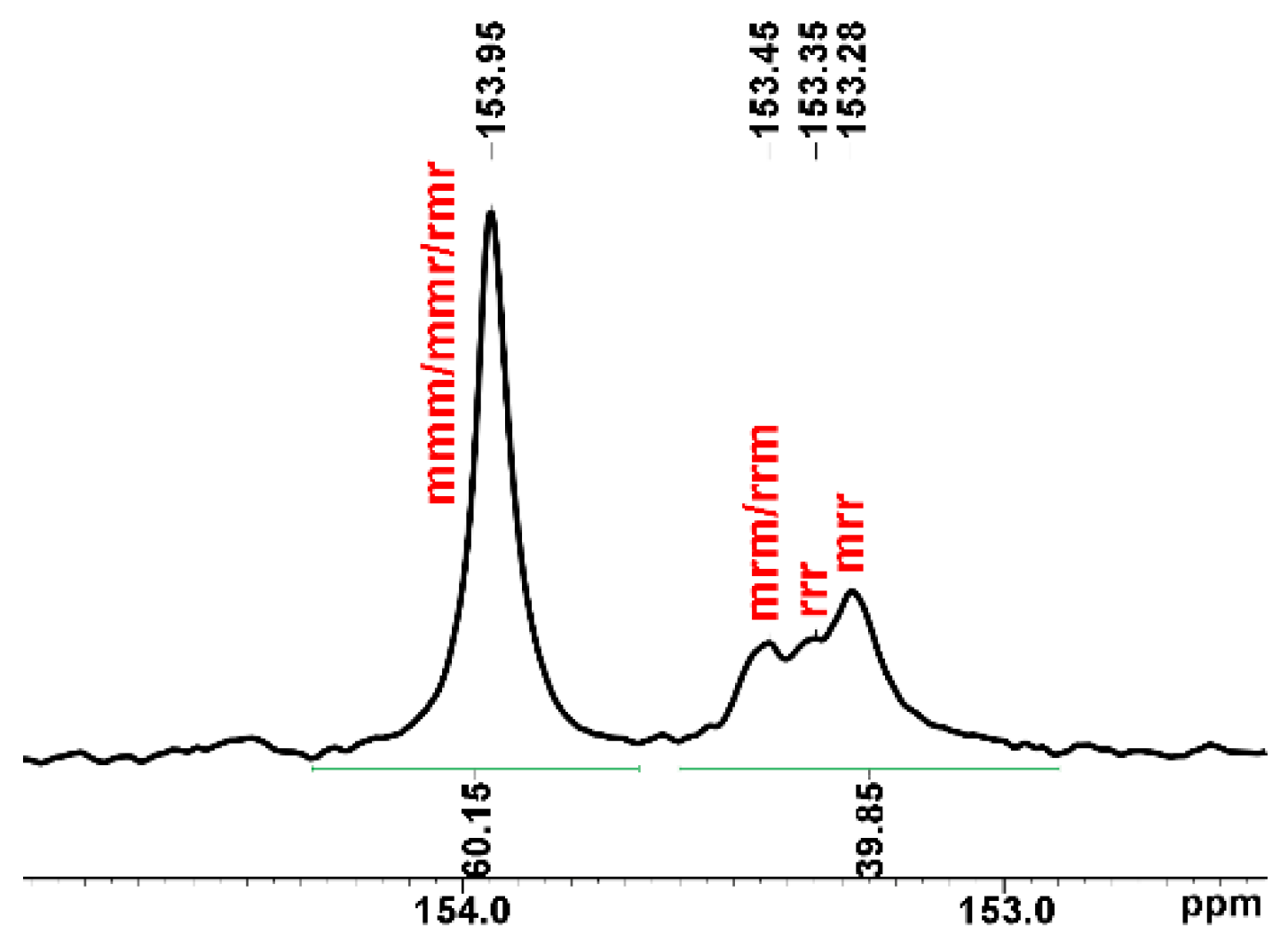
The ROCOP reaction profile in the presence of the 1 catalyst and BnOH initiator under the conditions of entries 10–13 of Table 2 is depicted in Figure 3. The polymerization run initially proceeds slowly and accelerates after 20 h. In parallel, the selectivity in APC is low at the beginning of the reaction and increases up to reach a plateau at the end of the reaction. Thus, after 20 h, additional catalytic species seem to appear in the polymerization solution that allow faster polymerization and higher selectivity in PCHC (see entry 5, Table 2 and Figure 3). These results prompted us to investigate the reaction of 1 with BnOH; the main results are discussed in the next paragraph. The polymer architecture of the PCHCs by 1/BnOH was investigated by 13C NMR spectroscopy, focusing the attention on the carbonyl spectral region that is diagnostic of the stereochemistry of the polycarbonate segments; a typical 13C NMR spectrum of PCHCs by 1 catalyst is shown in Figure 4 (see Figure S17 for the complete spectrum). The isotactic m-centred tetrads are expected to produce a sharp 13C signal at 154.0 ppm, whereas the syndiotactic (r)-centred tetrads produce broad signals in the range of 153.0–153.5 ppm; the integral values of the 13C NMR signals allow recognizing, for, e.g., the sample of entry 5 of Table 2, an isotactoid structure with a probability value of the meso tetrad Pm of 0.62. These results agree with those of most of the PCHCs obtained with zinc catalysts independently of the ligand environment.
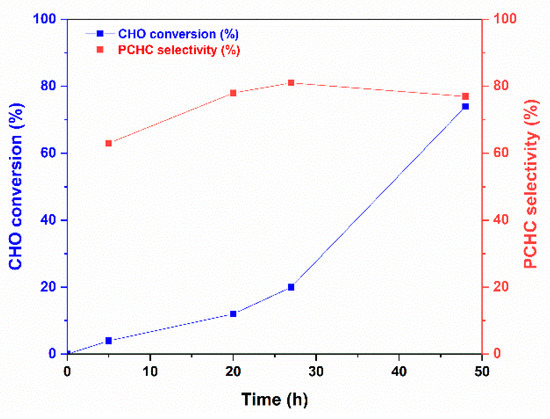
Figure 3.
PCHC selectivity in the ROCOP of CHO with CO2 catalysed by 1 using BnOH initiator (reaction conditions of entries 10–13 of Table 2).
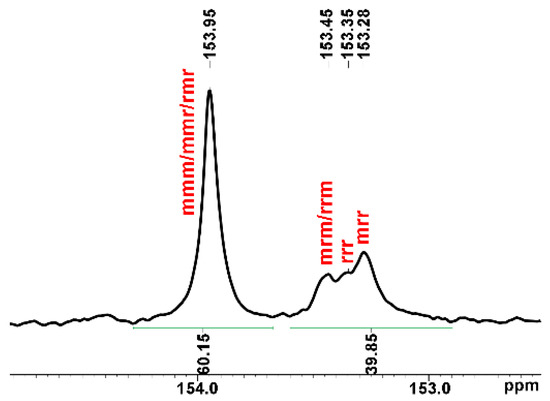
Figure 4.
Magnification of carbonyl signals in the 13C NMR spectrum (151 MHz, C6D6, 25 °C) of PCHC (entry 5, Table 2; full spectrum in Figure S17).
2.3. ROCOP of Cyclohexene Oxide and Carbon Dioxide Catalysed by Complexes 2
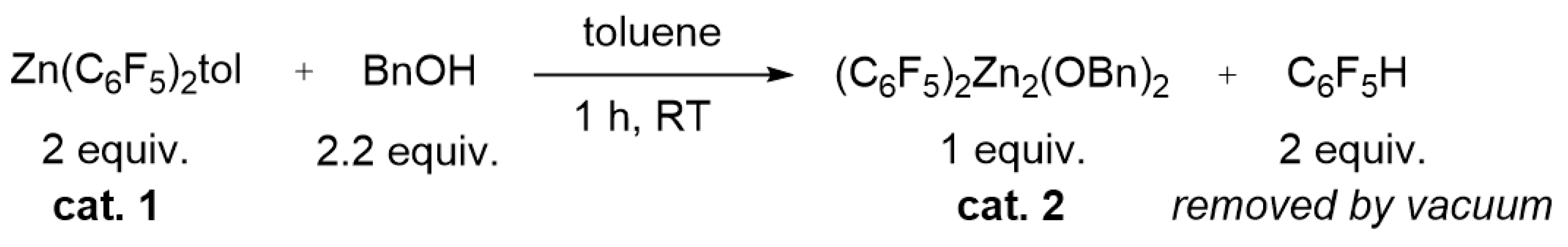
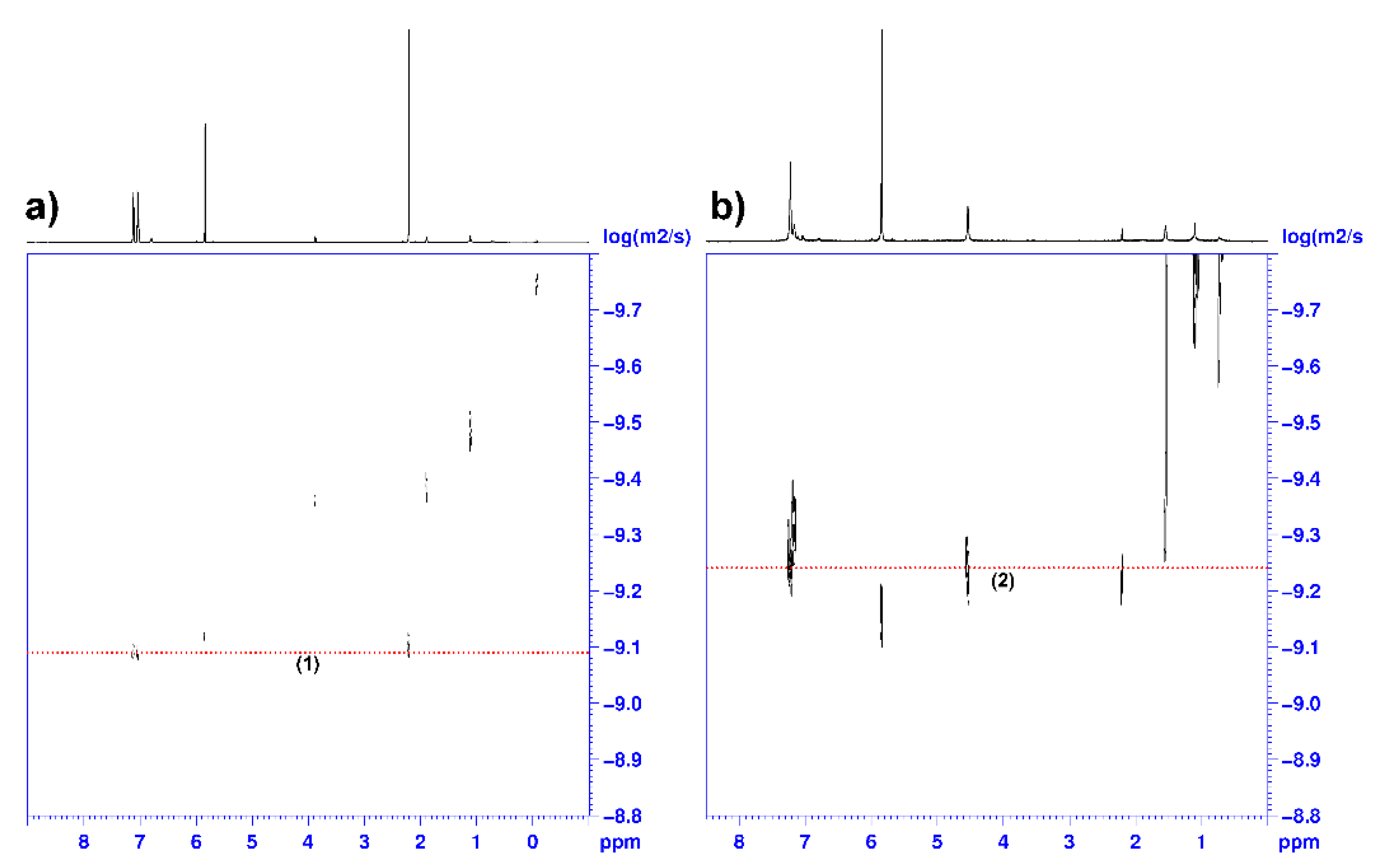
To understand the evolution of the catalyst mixture vs. time, we investigated the reaction of 1 and BnOH. Chisholm et al. showed that the reaction of 1 with an equimolar amount of the sterically demanding alcohol Pri2CHOH produced at room temperature the formation of the dinuclear species (C6F5)2Zn2(OCHPri2)2 which was structurally characterized by single crystal X-ray diffraction [41]. The reaction of 1 with 2 equiv. of Pri2CHOH in toluene at 85 °C for 20 h yielded the trinuclear complex (C6F5)2Zn3(OCHPri2)4 which is the kinetic product obtained in the presence of the excess of the alcohol, whereas the dinuclear zinc complex is the thermodynamic product [41]. (C6F5)2Zn2(OCHPri2)2 is a poorly active catalyst in the ROCOP of propylene oxide with CO2, producing traces of poly(propylene carbonate) at 60 °C in 20 h, whereas the trinuclear zinc complex is practically inactive under the same conditions [41]. Considering this information and these findings in the kinetic investigation of the ROCOP of CHO with CO2 in the presence of 1 and BnOH, we studied the initiation reaction between the latter compounds. When 1 was treated with 1 equiv. of BnOH in toluene solution at room temperature for 1 h (see Scheme 2), the high vacuum distillation of toluene and C6F5H yielded a white powder that was fully characterized by 1H, 13C (Figures S1 and S2), 19F (Figures S3 and S4), DOSY (Figure 5), NMR and mass spectroscopy (see Figure S6) and identified as (C6F5)2Zn2(OBn)2 (2). Of note, the 19F NMR analysis of 2 revealed an upfield shift of the orto-fluorine atoms from −118.9 ppm in 1 to −137.9 ppm in 2; moreover, the intensity ratio of the 13C signals of the aryloxo moieties are in a 1:1 ratio with the perfluoroaryl groups (Figures S3 and S4). The complex 2 still coordinates a toluene molecule, as demonstrated by the 1H singlet at 2.2 ppm in the 1H NMR spectrum (see Figures S1a and S2b) and the DOSY NMR analysis (Figure 5b) in which the 1H signals of the toluene and benzyloxy ligand show the same diffusivity.
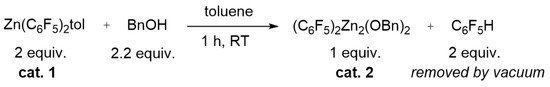
Scheme 2.
Reaction scheme for the synthesis of the catalyst 2.
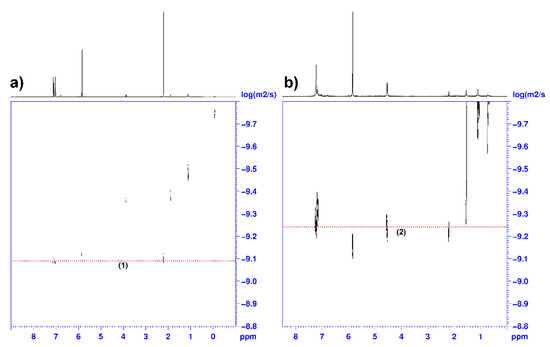
Figure 5.
DOSY 1H NMR spectra (600 MHz, 1,1,2,2-tetrachloroethane-d2, 25 °C) of complex 1 (a) and complex 2 (b).
The ROCOP of CHO with CO2 catalysed by 2 in the absence of an alcohol initiator was carried out at the variance of the polymerization temperature and PCO2, keeping constant the catalyst loading (0.2 mol% w.r.t. CHO; CHO/(2) molar ratio of 500) and polymerization time of 20 h; the main results are summarized in Table 3. The CHO conversion is near 50 mol% (39–54 mol%) in all runs and is scarcely affected by temperature (100–120 °C) and PCO2 (0.5–2.0 MPa). Interestingly the average molecular weights are higher, and the polydispersity index is lower than those reported for 1. Notably, one polymer chain for complex 2 was calculated from the weight of PCHC and the Mn determined by GPC. These results agree with the report by Nozaki et al., dealing with the alternating copolymerization of CHO with CO2 catalysed by the heterochiral dinuclear species Et2Zn2[diphenyl(pyrrolidine-2-yl)methoxy)2] in the presence of 1 equiv. of ethanol, where the zinc complex retains the dinuclear structure during polymerization and produces one polymer chain per Zn complex [42]. However, the formation of multinuclear zinc complexes, in particular of the stable heterocubane structures [43], cannot be ruled out and is hypothesized as active catalyst species in the polymerization process.
Table 3.
ROCOP between CO2 and CHO catalysed by complex 2.
Interestingly, a lower polydispersity index of Đ = 2.1 (entry 3, Table 3) was observed when the ROCOP was performed in solventless conditions indicating that a single-site catalyst is active in such conditions. The stereoregularity of the PCHCs by 2 is the same as the ones from 1 (compare Figure 4 and Figures S19–S21), indicating that the stereocontrol degree is independent of the ligand environment, as generally observed with zinc catalysts; the difference in Mn values in most of the catalytic runs catalysed by 1 could thus result from the stoichiometric excess of BnOH that acts as a chain transfer agent.
3. Materials and Methods
3.1. General Procedures and Materials
The manipulation of air/moisture-sensitive compounds was performed under an inert N2 atmosphere using Schlenk techniques or glovebox. Toluene (99.5%, Merck, NJ, USA) was used as received or pre-dried with calcium chloride, refluxed for 48 h over sodium and distilled before use in moisture- and oxygen-sensitive reactions. Cyclohexene oxide (98%, TCI chemicals) was dried over calcium hydride and distilled in vacuo prior to use. Carbon dioxide (4.8 purity grade, Rivoira, Italy), chloroform-d (99.8%, Merck), tetrahydrofuran (THF, chromatography grade, Merck), calcium hydride (98%, Merck), tris(pentafluorophenyl)borane (97%, TCI) and diethyl zinc (1.0 mol/L solution in toluene) were purchased from TCI and used as received. Toluene-d8 (99%, Merck), acetonitrile-d3 (99.4%, Merck), benzene-d6 (99.6%, Merck), benzyl alcohol (99%, Merck), pentane (99%, Merck) and anhydrous solvents were stored over molecular sieves (4 Å, Merck), which were preactivated under high vacuum at 200 °C for 48 h. Zn(C6F5)2toluene (Complex 1) was synthesized according to the procedure described in the literature [26].
3.2. Instrumentation and Analytical Methods
Nuclear magnetic resonance (NMR) spectra were acquired with 300, 400, 500 or 600 MHz AVANCE spectrometers from Bruker (Billerica, MA, USA). The chemical shifts were referenced to the residual proton signal of the deuterated solvent. Gel permeation chromatography (GPC) analyses were carried out using a Shimadzu chromatograph equipped with a RID10A refraction index detector by using THF (HPLC grade) as an eluent for the polymers at room temperature at a flow rate of 1.0 mL min−1. The instrumental calibration was performed with polystyrene standards with molecular weight in the range of 106 to 102 Da.
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) spectra were acquired with a Bruker SolariX XR Fourier transform ion cyclotron resonance mass spectrometer equipped with a 7 T refrigerated, actively-shielded superconducting magnet from Bruker Biospin. The samples were ionized in positive ion mode using 2,5-dihydroxybenzoic acid (DHB) as the matrix for MALDI with a laser power of 15% and 15 laser shots for each scan. The MS spectra were externally calibrated using a standard sample and internally recalibrated with DHB. The samples were prepared by mixing 10 μL of dichloromethane polymer solutions (1 mg·mL−1) with 10 μL saturated solution of DHB (30 mg·mL−1). The mixed solution was deposited onto the MALDI target and left to dry before the measurement. The ESI-mass spectra were performed using a Bruker solariX equipped with a Fourier transform mass spectrometer. The system contains a 7 T superconducting magnet refrigerated shield (Bruker Biospin). Initially, the samples undergo a negative ion mode ionization performed by an ESI ion source (Bruker Daltonik GmbH) in a flow of 120 μL/h. NaTFA was employed for the linear calibration. The mass range detection was performed between 150–3000 m/z. The voltage employed was −3.9 kV with a N2 flow rate of 4 L/min at 200 °C.
3.3. Synthesis of (C6F5)2Zn2(OBn)2 (Complex 2)
A 10 mL Schlenk tube equipped with a magnetic stir bar was sequentially charged with Zn(C6F5)2 (toluene) (1) (150 mg, 0.305 mmol), dissolved in 3 mL of toluene and benzyl alcohol (35 µL, 0.335 mmol, 1.1 equiv. w.r.t. 1). The system was kept under stirring at room temperature for 2 h. The solvent and HC6F5 were removed under high vacuum to yield (C6F5)2Zn2(OBn)2 (2) as a white solid (201 mg; yield of 97%). 1H NMR (1,1,2,2-tetrachloroethane-d2, 600 MHz): δ 7.38 (m, 4 H), 7.32 (t, 1 H), 4.69 (s, 1 H). 13C{1H, 19F} NMR (1,1,2,2-tetrachloroethane-d2, 151 MHz): δ 146.26 (d, J = 249.15 Hz), 141.82 (d, J = 261.23 Hz), 137.66 (d, J = 250.66 Hz), 128.84, 127.96, 127.25, 65.47. 19F{1H, 19F} NMR (1,1,2,2-tetrachloroethane-d2, 565 MHz): δ −137.87 (m, 4 F, o-F), −152.97 (t, 2 F, JFF = 20.5 Hz, p-F), −161.28 (m, 4 F, m-F).
3.4. Procedure for the Ring-Opening Polymerization of Cyclohexene Oxide Promoted by Complex 1 (Table 1)
A typical procedure is herein described as a representative example. Briefly, 0.32 mL of a toluene solution of 1 (20.3 mmol; 64.3 mM) was transferred into a 10 mL Schlenk tube, and the solvent was distilled off in vacuo. Cyclohexene oxide (0.82 mL, 8.14 mmol, 400 equiv.) was then injected at room temperature for initiating the polymerization. The run was terminated after 1 min by the addition of 3 mL of chloroform-d, and the solution was readily analysed by means of 1H NMR spectroscopy. The polymer was precipitated in cold methanol, recovered by filtration and dried under vacuum.
3.5. Typical Procedure for Ring-Opening Copolymerization (ROCOP) between Cyclohexene Oxide and CO2 Promoted by Complex 1 in Combination with Benzyl Alcohol (Table 2, Entry 4)
The following typical procedure is reported as a representative example. Initially, 0.32 mL of a toluene solution (64.3 mM) of 1 (20.3 µmol) was transferred into a Schlenk tube and the solvent was distilled off under high vacuum. Toluene-d8 (2 mL) and benzyl alcohol (10.6 mL, 102 mmol, 5 equiv. w.r.t. complex 1) were sequentially added in the order and, finally, cyclohexene oxide (1.0 mL, 10 mmol, 500 equiv.) was injected in at room temperature. The resulting mixture was immediately transferred into a 60 mL stainless steel pressure reactor. The pressure of CO2 was set at 2 MPa. The reactor was sealed and transferred to an oil bath equilibrated at the desired temperature. The reaction mixture after 48 h was cooled at room temperature, and 3 mL of chloroform-d was added; a sample of the solution was transferred into an NMR tube and readily analysed by 1H NMR for determining the composition. Poly(cyclohexene carbonate) was recovered from the reaction solution after coagulation in plenty of hexane; the polymer was recovered by filtration and purified by repeated dissolution in chloroform and reprecipitation in hexane. This procedure also allowed to separate PCHO from the PCHC due to the different solubility of the two polymers in hexane.
3.6. Typical Procedure for Ring-Opening Copolymerization (ROCOP) between Cyclohexene Oxide and CO2 Promoted by Complex 2 (Table 2, Entry 1)
The following typical procedure is reported as a representative example. Initially, (C6F5)2Zn2(OBn)2 (13.8 mg, 20.3 µmol, 1 equiv.) was weighed and transferred to a Schlenk tube. Toluene-d8 (2 mL) and cyclohexene oxide (1.0 mL, 10 mmol, 500 equiv.) were injected at room temperature, and the resulting mixture was immediately transferred into a 60 mL stainless steel pressure reactor. The pressure of CO2 was set at 2 MPa, the reactor was sealed and transferred to an oil bath equilibrated at the desired temperature. The reaction mixture after 20 h was cooled at room temperature, and 3 mL of chloroform-d was added; a sample of the solution was transferred into an NMR tube and readily analysed by 1H NMR for determining the composition. Poly(cyclohexene carbonate) was recovered from the reaction solution after coagulation in plenty of hexane; the polymer was recovered by filtration and purified by repeated dissolution in chloroform and reprecipitation in hexane.
4. Conclusions
The strong Lewis acid catalyst Zn(C6F5)2(toluene) (1) efficiently catalyses the homopolymerization of CHO, leading to PCHO with a remarkable TOF of 4200 min−1 via AMM. The same catalyst 1 allows the chemoselective ROCOP of CO2 with CHO in the presence of a Bn(OH) initiator, yielding PCHC of low stereoregularity (Pm = 0.60) and with selectivity up to 91%. Investigation of the reaction mechanism prompted us to synthesize the dinuclear zinc complex (C6F5)2Zn2(OBn)2 formed in the reaction of 1 with Bn(OH) and identified as one of the active catalysts formed in this polymerization process; this compound was spectroscopically characterized by 1H, 19F and 13C NMR spectroscopy. The ROCOP of CHO with CO2 catalysed by 2 in solventless conditions produced PCHCs with a polydispersity index corresponding to a single-site catalyst. This study further demonstrates that perfluoroaryl zinc complexes undergo structural rearrangement in the course of solution polymerization processes at high temperatures to produce a complex mixture of catalyst species that deserve careful consideration before a definitive conclusion can be drawn in the assessment of the catalyst structure.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12090970/s1, List of Figures S1–S29.
Author Contributions
Investigation, Y.C.A.S., A.B.; data curation, Y.C.A.S., A.B., C.C., S.D. and A.G.; writing—original draft preparation, Y.C.A.S., A.B. and A.G.; writing—review and editing, Y.C.A.S., A.B., C.C., S.D. and A.G.; funding acquisition, A.B., C.C., S.D. and A.G. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful for the funding from the Ministero dell’Università e della Ricerca MUR (PRIN2017 grant 2017WR2LRS: project title: “Beyond fossil fuels: CO2-only monomers and polymers for a circular economy (CO2-only) ”) and from the Università degli Studi di Salerno (FARB-Buonerba ORSA224812).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The Technological and Economic Prospects for CO2 Utilization and Removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef]
- Pescarmona, P.P. Cyclic Carbonates Synthesised from CO2: Applications, Challenges and Recent Research Trends. Curr. Opin. Green Sustain. Chem. 2021, 29, 100457. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed Coupling of Carbon Dioxide with Epoxides for the Synthesis of Cyclic Carbonates: Catalyst Design and Mechanistic Studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Grignard, B.; Gennen, S.; Jérôme, C.; Kleij, A.W.; Detrembleur, C. Advances in the Use of CO2 as a Renewable Feedstock for the Synthesis of Polymers. Chem. Soc. Rev. 2019, 48, 4466–4514. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S. Rise of the Zinc Age in Homogeneous Catalysis? ACS Catal. 2013, 3, 150–158. [Google Scholar] [CrossRef]
- Huang, J.; Worch, J.C.; Dove, A.P.; Coulembier, O. Update and Challenges in Carbon Dioxide-Based Polycarbonate Synthesis. ChemSusChem 2020, 13, 469–487. [Google Scholar] [CrossRef]
- Cheng, M.; Moore, D.R.; Reczek, J.J.; Chamberlain, B.M.; Lobkovsky, E.B.; Coates, G.W. Single-Site β-Diiminate Zinc Catalysts for the Alternating Copolymerization of CO2 and Epoxides: Catalyst Synthesis and Unprecedented Polymerization Activity. J. Am. Chem. Soc. 2001, 123, 8738–8749. [Google Scholar] [CrossRef]
- Ellis, W.C.; Jung, Y.; Mulzer, M.; Di Girolamo, R.; Lobkovsky, E.B.; Coates, G.W. Copolymerization of CO2 and Meso Epoxides Using Enantioselective β-Diiminate Catalysts: A Route to Highly Isotactic Polycarbonates. Chem. Sci. 2014, 5, 4004–4011. [Google Scholar] [CrossRef]
- Kissling, S.; Lehenmeier, M.W.; Altenbuchner, P.T.; Kronast, A.; Reiter, M.; Deglmann, P.; Seemann, U.B.; Rieger, B. Dinuclear Zinc Catalysts with Unprecedented Activities for the Copolymerization of Cyclohexene Oxide and CO2. Chem. Commun. 2015, 51, 4579–4582. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.K.; Romain, C.; Williams, C.K. Dinuclear Metal Catalysts: Improved Performance of Heterodinuclear Mixed Catalysts for CO2—Epoxide Copolymerization. Chem. Commun. 2014, 50, 4164–4167. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Knight, P.D.; Reung, P.T.R.; Williams, C.K. Highly Active Dizinc Catalyst for the Copolymerization of Carbon Dioxide and Cyclohexene Oxide at One Atmosphere Pressure. Angew. Chem. 2009, 121, 949–951. [Google Scholar] [CrossRef]
- Martínez, J.; Castro-Osma, J.A.; Lara-Sánchez, A.; Otero, A.; Fernández-Baeza, J.; Tejeda, J.; Sánchez-Barba, L.F.; Rodríguez-Diéguez, A. Ring-Opening Copolymerisation of Cyclohexene Oxide and Carbon Dioxide Catalysed by Scorpionate Zinc Complexes. Polym. Chem. 2016, 7, 6475–6484. [Google Scholar] [CrossRef]
- Shao, H.; Reddi, Y.; Cramer, C.J. Modeling the Mechanism of CO2/Cyclohexene Oxide Copolymerization Catalyzed by Chiral Zinc β-Diiminates: Factors Affecting Reactivity and Isotacticity. ACS Catal. 2020, 10, 8870–8879. [Google Scholar] [CrossRef]
- Deacy, A.C.; Durr, C.B.; Williams, C.K. Heterodinuclear Complexes Featuring Zn(Ii) and M = Al(Iii), Ga(Iii) or In(Iii) for Cyclohexene Oxide and CO2 Copolymerisation. Dalton Trans. 2020, 49, 223–231. [Google Scholar] [CrossRef]
- Anderson, T.S.; Kozak, C.M. Ring-Opening Polymerization of Epoxides and Ring-Opening Copolymerization of CO2 with Epoxides by a Zinc Amino-Bis(Phenolate) Catalyst. Eur. Polym. J. 2019, 120, 109237. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, L.; He, C.-T.; Qin, J.; Li, Z.; Wang, S.; Xiao, M.; Meng, Y. Kinetic and Mechanistic Investigation for the Copolymerization of CO2 and Cyclohexene Oxide Catalyzed by Trizinc Complexes. Polym. Chem. 2017, 8, 3632–3640. [Google Scholar] [CrossRef]
- Nagae, H.; Aoki, R.; Akutagawa, S.; Kleemann, J.; Tagawa, R.; Schindler, T.; Choi, G.; Spaniol, T.P.; Tsurugi, H.; Okuda, J.; et al. Lanthanide Complexes Supported by a Trizinc Crown Ether as Catalysts for Alternating Copolymerization of Epoxide and CO2: Telomerization Controlled by Carboxylate Anions. Angew. Chem. Int. Ed. 2018, 57, 2492–2496. [Google Scholar] [CrossRef]
- Reiter, M.; Vagin, S.; Kronast, A.; Jandl, C.; Rieger, B. A Lewis Acid β-Diiminato-Zinc-Complex as All-Rounder for Co- and Terpolymerisation of Various Epoxides with Carbon Dioxide. Chem. Sci. 2017, 8, 1876–1882. [Google Scholar] [CrossRef]
- Romain, C.; Garden, J.A.; Trott, G.; Buchard, A.; White, A.J.P.; Williams, C.K. Di-Zinc–Aryl Complexes: CO2 Insertions and Applications in Polymerisation Catalysis. Chem. Eur. J. 2017, 23, 7367–7376. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, E.J. Organometallic Catalysts for Polymerizing Monosubstituted Epoxides. J. Polym. Sci. 1960, 47, 486–489. [Google Scholar] [CrossRef]
- Childers, M.I.; Longo, J.M.; Van Zee, N.J.; LaPointe, A.M.; Coates, G.W. Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev. 2014, 114, 8129–8152. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.R.; Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Electronic and Steric Effects on Catalysts for CO2/Epoxide Polymerization: Subtle Modifications Resulting in Superior Activities. Angew. Chem. Int. Ed. 2002, 41, 2599–2602. [Google Scholar] [CrossRef]
- Pankhurst, J.R.; Paul, S.; Zhu, Y.; Williams, C.K.; Love, J.B. Polynuclear Alkoxy–Zinc Complexes of Bowl-Shaped Macrocycles and Their Use in the Copolymerisation of Cyclohexene Oxide and CO2. Dalton Trans. 2019, 48, 4887–4893. [Google Scholar] [CrossRef]
- Sobrino, S.; Navarro, M.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Lara-Sánchez, A.; Garcés, A.; Castro-Osma, J.A.; Rodríguez, A.M. Efficient Production of Poly(Cyclohexene Carbonate) via ROCOP of Cyclohexene Oxide and CO2 Mediated by NNO-Scorpionate Zinc Complexes. Polymers 2020, 12, 2148. [Google Scholar] [CrossRef]
- Walker, D.A.; Woodman, T.J.; Hughes, D.L.; Bochmann, M. Reactions of Zinc Dialkyls with (Perfluorophenyl)Boron Compounds: Alkylzinc Cation Formation vs C6F5 Transfer. Organometallics 2001, 20, 3772–3776. [Google Scholar] [CrossRef]
- Spassky, N.; Dumas, P.; Sepulchre, M.; Sigwalt, P. Properties and Methods of Synthesis of Several Optically Active Polyoxiranes and Polythiiranes. J. Polym. Sci. Polym. Symp. 1975, 52, 327–349. [Google Scholar] [CrossRef]
- Cheng, M.; Darling, N.A.; Lobkovsky, E.B.; Coates, G.W. Enantiomerically-Enriched Organic Reagents Polymer Synthesis: Enantioselective Copolymerization of Cycloalkene Oxides and CO Using Homogeneous, Zinc-Based Catalysts. Chem. Commun. 2000, 20, 2007–2008. [Google Scholar] [CrossRef]
- Yahiaoui, A.; Belbachir, M.; Soutif, J.C.; Fontaine, L. Synthesis and Structural Analyses of Poly(1,2-Cyclohexene Oxide) over Solid Acid Catalyst. Mater. Lett. 2005, 59, 759–767. [Google Scholar] [CrossRef]
- Allen, G.; Booth, C.; Hurst, S.J. The Preparation and Characterization of Poly(t-Butyl Ethylene Oxide) and Poly(Styrene Oxide). Polymer 1967, 8, 385–391. [Google Scholar] [CrossRef]
- Rabagliati, F.M.; López, F. Epoxide Polymerization, 6. Structure of Poly(Propylene Oxide) Prepared with Diphenylzinc/Water as Initiating System. Makromol. Chem. Rapid Commun. 1985, 6, 141–144. [Google Scholar] [CrossRef]
- Rabagliati, F.M.; Lopez-Carrasquero, F. Epoxide Polymerization—VII. Propylene Oxide Polymerization Using the Diphenylzinc-Water System in Benzene at 60°. Eur. Polym. J. 1985, 21, 1061–1065. [Google Scholar] [CrossRef]
- Thiam, M.; Spassky, N. Polymerization of Cyclohexene Oxide Using Yttrium Isopropoxide and a Bimetallic Yttrium-Aluminium Isopropoxide as Initiators. Macromol. Chem. Phys. 1999, 200, 2107–2110. [Google Scholar] [CrossRef]
- Plommer, H.; Reim, I.; Kerton, F.M. Ring-Opening Polymerization of Cyclohexene Oxide Using Aluminum Amine–Phenolate Complexes. Dalton Trans. 2015, 44, 12098–12102. [Google Scholar] [CrossRef] [PubMed]
- Della Monica, F.; Maity, B.; Pehl, T.; Buonerba, A.; De Nisi, A.; Monari, M.; Grassi, A.; Rieger, B.; Cavallo, L.; Capacchione, C. [OSSO]-Type Iron(III) Complexes for the Low-Pressure Reaction of Carbon Dioxide with Epoxides: Catalytic Activity, Reaction Kinetics, and Computational Study. ACS Catal. 2018, 8, 6882–6893. [Google Scholar] [CrossRef]
- Della Monica, F.; Buonerba, A.; Capacchione, C. Homogeneous Iron Catalysts in the Reaction of Epoxides with Carbon Dioxide. Adv. Synth. Catal. 2019, 361, 265–282. [Google Scholar] [CrossRef]
- Buonerba, A.; De Nisi, A.; Grassi, A.; Milione, S.; Capacchione, C.; Vagin, S.; Rieger, B. Novel Iron(Iii) Catalyst for the Efficient and Selective Coupling of Carbon Dioxide and Epoxides to Form Cyclic Carbonates. Catal. Sci. Technol. 2015, 5, 118–123. [Google Scholar] [CrossRef]
- Buonerba, A.; Della Monica, F.; De Nisi, A.; Luciano, E.; Milione, S.; Grassi, A.; Capacchione, C.; Rieger, B. Thioether-Triphenolate Bimetallic Iron(III) Complexes as Robust and Highly Efficient Catalysts for Cycloaddition of Carbon Dioxide to Epoxides. Faraday Discuss. 2015, 183, 83–95. [Google Scholar] [CrossRef]
- Della Monica, F.; Vummaleti, S.V.C.C.; Buonerba, A.; Nisi, A.D.; Monari, M.; Milione, S.; Grassi, A.; Cavallo, L.; Capacchione, C. Coupling of Carbon Dioxide with Epoxides Efficiently Catalyzed by Thioether-Triphenolate Bimetallic Iron(III) Complexes: Catalyst Structure–Reactivity Relationship and Mechanistic DFT Study. Adv. Synth. Catal. 2016, 358, 3231–3243. [Google Scholar] [CrossRef]
- Della Monica, F.; Leone, M.; Buonerba, A.; Grassi, A.; Milione, S.; Capacchione, C. CO2 Cycloaddition to Epoxides Promoted by Bis-Thioether-Phenolate Fe(II) and Fe(III) Complexes. Mol. Catal. 2018, 460, 46–52. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Gallucci, J.C.; Yin, H.; Zhen, H. Arylzinc Alkoxides: [ArZnOCHP]2 and Ar2Zn3(OCHP)4 When Ar = C6H5, p-CF3C6H4, 2,4,6-Me3C6H2, and C6F5. Inorg. Chem. 2005, 44, 4777–4785. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Hiyama, T.; Nozaki, K. Asymmetric Amplification in Asymmetric Alternating Copolymerization of Cyclohexene Oxide and Carbon Dioxide. Chem. Commun. 2005, 14, 1871–1873. [Google Scholar] [CrossRef] [PubMed]
- Mąkolski, Ł.; Szejko, V.; Zelga, K.; Tulewicz, A.; Bernatowicz, P.; Justyniak, I.; Lewiński, J. Unravelling Structural Mysteries of Simple Organozinc Alkoxides. Chem. Eur. J. 2021, 27, 5666–5674. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).