1. Introduction
Carbon-derived fuels play a major role in energy conversion applications, despite current efforts to replace them with more sustainable sources. They are used both in the energy and transportation sectors, where pyrolysis or the incomplete combustion of carbon-based fuels occurs regularly, leading to the emission of cancerogenic and mutagenic pollutants. Among these are soot, light hydrocarbons (methane, propane), volatile organic compounds (VOCs), and polyaromatic hydrocarbons (PAH), which are associated with the formation of smog and are harmful to human welfare [
1,
2,
3,
4]. Soot is the amalgamation of numerous substances containing carbon, from amorphous to ordered graphitic structures, polyenes, and graphene. The nanometric size of soot particles allows for their easy emission into the environment, where, as part of a ubiquitous smog, they can penetrate living organisms through the lungs or skin. Long-term exposure to soot particles can lead to many illnesses, including cancero and mutagenic changes or lung and heart problems [
5,
6,
7]. Volatile organic compounds belong to a group of substances with a high vapour pressure at low temperatures. Thus, according to this definition, a wide range of substances belong to this group, ranging from simple alkanes to PAHs, many of which are detrimental for human health and to the environment [
8,
9,
10]. Some of the most harmful non-halogenic VOCs include benzene, benzopyrene, toluene, formaldehyde, propylene, acetone, and styrene [
9,
11,
12,
13]. The materials’ reactivity towards VOCs oxidation can be gauged by testing catalysts in the combustion of light hydrocarbons, as they are thermodynamically more stable and more difficult to activate.
A large source of the aforementioned pollutants constitutes combustion engines, where the complex process of fuel combustion follow several parallel reaction pathways with the formation of many harmful types of pollutants. Among the most promising ways to reduce the emissions of pollutants from combustion engines into the environment is the use of complex catalysts in catalytic converters, which reduce the temperature of soot, light hydrocarbons, VOCs, and PAHs oxidation, allowing for their abatement before their release. Most currently used oxidation catalysts are based on more and more expensive noble metals (Pt, Pd, Rh) deposited onto metal oxide carriers (ceria, alumina) [
9,
14]. The rising costs and decreasing availability of the noble metals present an incentive to search for competitive catalytic solutions. Several classes of materials have been tested as potential replacements, among which are oxides with different structures and compositions, including quite promising perovskites and spinels [
8,
15,
16,
17,
18,
19,
20,
21,
22]. The catalytic activity of these systems can be further modified by adding new noble metals, transition metals, or alkali metals as surface or bulk promoters [
23,
24,
25,
26,
27,
28,
29].
Manganese oxides nanostructured by alkali [
30], such as the layered birnessite (AMn
4O
8), are reported as highly promising candidates for substituting the noble metal-based systems, as they use cheap substrates, are environmentally benign, and are very reactive in oxidation reactions [
12,
13,
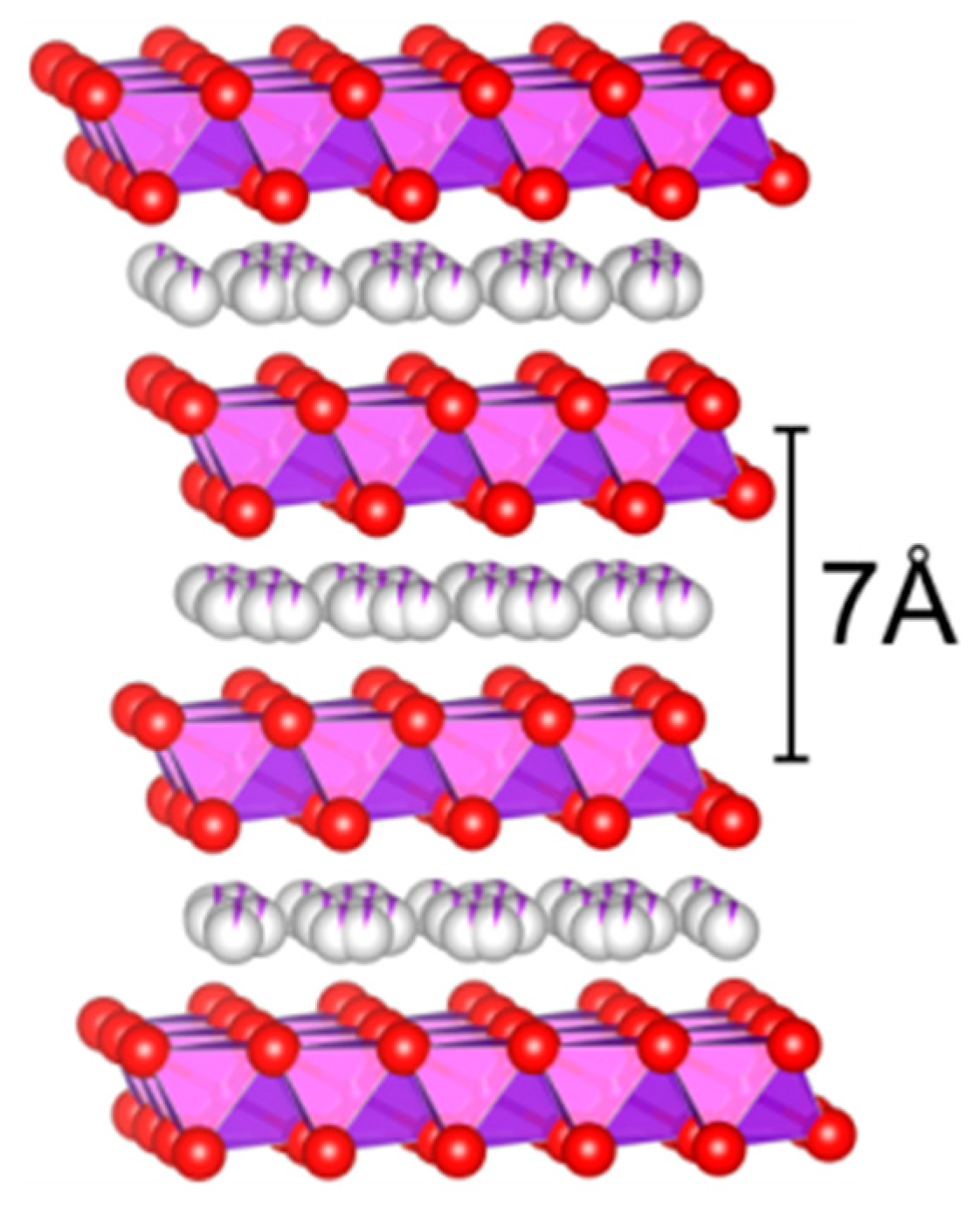
31]. The octahedral-layered birnessite (OL-1) comprises of the edge-sharing MnO
6 octahedral units forming a two-dimensional (2D) layered structure (
Figure 1). The layers are typically ~7 Å apart and host v exchangeable cations (e.g., Na
+, K
+) or molecules in the interlayer space [
32,
33]. Their high reactivity stems from a combination of several factors such as a beneficial porous structure, high oxygen mobility, and good redox properties due to the different oxidation states of lattice manganese ions [
34].
This work aimed to explore the effects of different alkali cations (H+, Li+, Na+, K+, Rb+, or Cs+) located between the MnO6 octahedral layers of birnessite on the catalytic activity of such materials in soot and light hydrocarbon oxidation reactions. It is anticipated that such an investigation will aid in the rational designing of a cheap and efficient noble metal-free catalyst for low-temperature oxidation reactions. The investigated catalysts were synthesized following the same coprecipitation method, introducing the alkali cations to a manganese precursor and then allowing the formation of the layered birnessite structure, thus assuring no contamination with other alkali elements (as occurs when the alkali are introduced via ion exchange). The obtained materials were tested in soot, methane, and propane combustion. Since NO is often present in the exhaust gasses, we further investigated the materials for their ability to convert NO to NO2. The latter gas was evaluated as an effective way to transfer oxygen from the catalyst to soot particles. The effectiveness in NO conversion was sought after, as its presence in the gas feed lowers the temperature of soot combustion, enhancing the loose contact condition’s reactivity by lowering the reaction temperature window towards the tight contact condition.
2. Results and Discussion
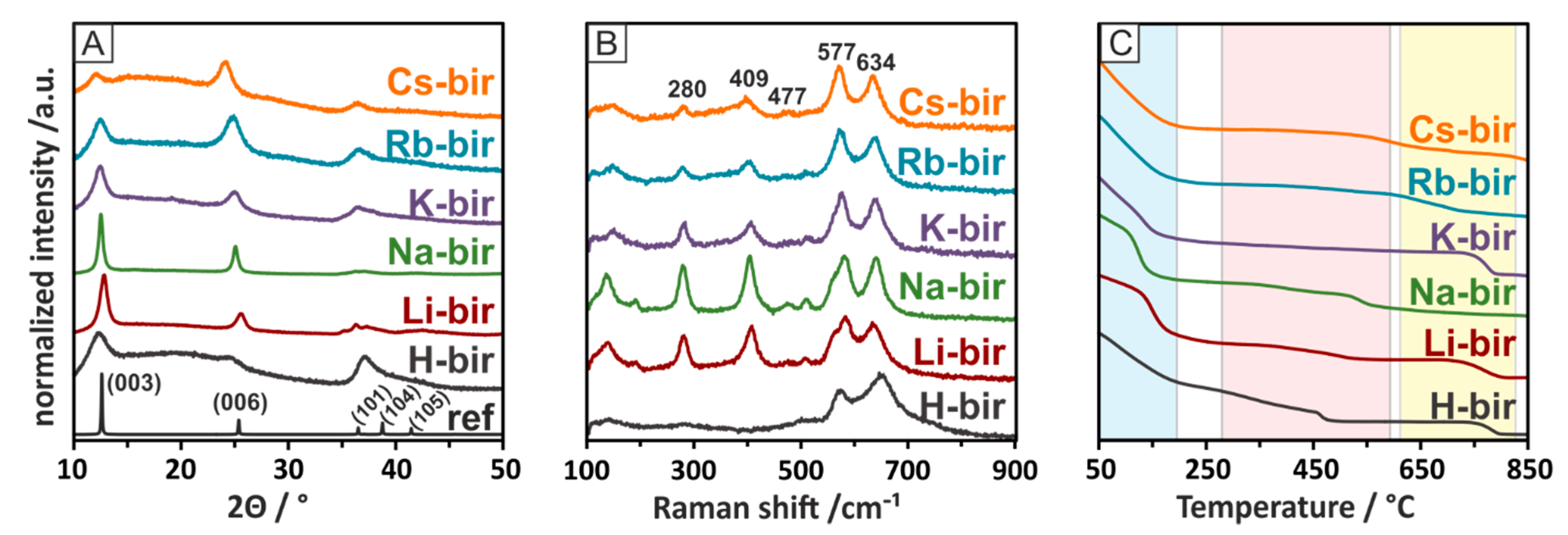
Diffractograms, Raman spectra, and the results of the thermogravimetric analysis of the birnessite series containing different alkali cations are displayed in
Figure 2. The examination of the diffraction patterns reveals the pureness of the prepared materials, as no other reflexes, apart from those assignable to birnessite, are present. A noteworthy difference between the diffractograms is the position of the first reflex at ~12°. The 2
ϴ value of this reflex determines the separation of the layers in the birnessite phase, which is influenced by the introduced alkali cations. The introduction of ions with larger radii increasingly separates the layers. The H-bir sample stands out from this tendency, most likely due to the weak force of attraction the cations may have in pulling the layers together compared to the alkali cations. The position of the reflexes as well as the calculated interlayer distances are summarized in
Table 1. X-ray fluorescence was used to determine the elemental composition of the alkali-modified birnessite catalysts. Due to their light atomic weight, H and Li were undetectable in the current measurements, while the Na content was on the boundary of detection limits. The ratio of metallic components in the birnessite samples was very close to the stoichiometry of the mixed oxide, and the calculated results are collected in
Table 1.
According to the literature, the Raman spectra of birnessite present bands at 280, 409, 477, 506, 577, 634, and 728 cm
−1. An analysis of the recorded Raman spectra revealed similar bands for the investigated samples. The most characteristic bands for birnessite, 634 and 577 cm
−1, are due to the symmetric stretching vibration of MnO
6 groups in the interlayer direction and the Mn−O vibration in the chain framework, respectively. The remaining bands can be assigned to the O-cation vibration [
34,
35,
36]. The Raman spectra of the alkali-modified birnessite are consistent with those found in the literature and display no notable bands assignable to other phases. Of note are the changing intensities of the bands assigned to the cation-oxygen vibrations, most notably those of 280 and 409 cm
−1. These bands vanish entirely for the H-bir as the vibrations of O–H give weak Raman bands [
37].
The XPS analysis was used to determine the surface composition and an average oxidation state of Mn (Mn
x+) in the samples, and the resultant values are collected in
Table 2. The relevant spectra with the fitted bands due to Mn in different oxidation states for the Mn 3s and Mn 2p ranges are collected in
Figures S1 and S2. The respective O 1s ranges and detailed scans of respective alkali bands are collected in
Figures S3 and S4. For the four samples for which the amount of alkali could be quantified, the Mn
x+ is quite well linearly correlated with its amount as determined by the XRF (X-ray fluorescence spectroscopy,
Table 1) but not with the XPS-derived value, as shown in
Figure S5a. A rough correlation of the Mn
x+ with the O/Mn ratio determined from the XPS study can also be observed in
Figure S5b, as could be expected. However, no correlation was found either with the work function or any reactivity descriptor of the methane, propane, or soot catalytic oxidation.
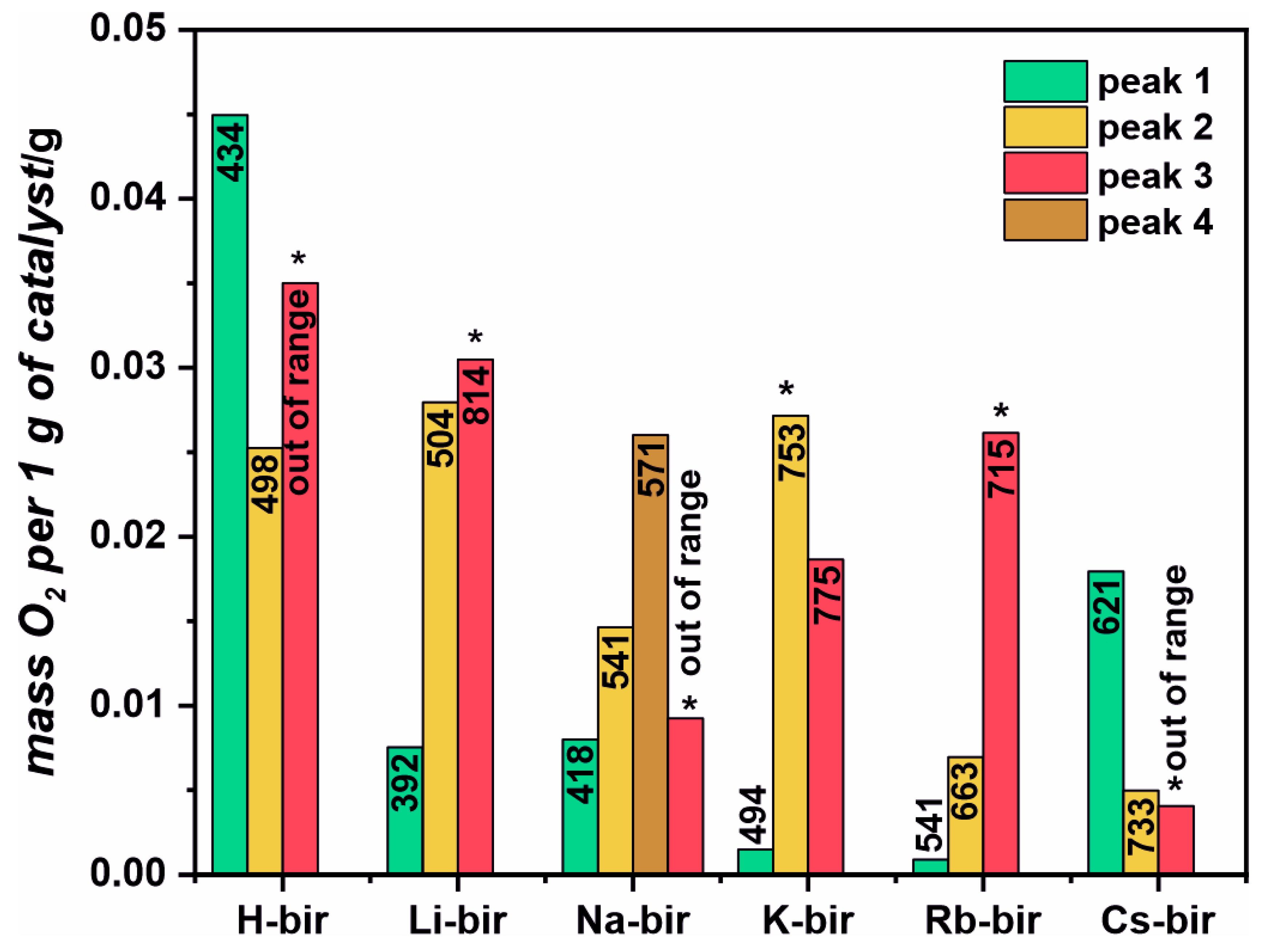
A thermal gravimetric analysis was performed in a He flow from room temperature (RT) to 850 °C. Separately, a similar temperature program was used to follow the evolution of H
2O, CO
2, and O
2 from the catalysts in a flow reactor by using a mass spectrometer as a detector. The combined analysis allowed us to assign the three mass loss zones to the release of particular gas-phase molecules. In the first zone, between room temperature, RT, and ~200 °C, the mass loss is attributed to water desorption from the catalysts. Water is known to be present in the interlayer region of the structure. In the second region, between 300 and 600 °C, the mass loss is attributed to the evolution of oxygen, which, in the case of the H, Li and Na samples, is more profound than in the other samples. Accordingly, in the high-temperature region, from 600 to 800 °C, the K, Rb, and Cs-modified birnessite samples lose more mass due to oxygen evolution than the H, Li, and Na samples do. The total mass loss due to oxygen evolution follows the same sequence as the specific surface area (SSA) of the catalysts, as presented in
Table 1. However, while both the SSA and the amount of released oxygen diminish with the increasing size of the introduced cation, they are not strictly proportional. The SSA ranges from 60 m
2 for H-bir to around 1 m
2 for the Cs-bir, dropping 60 times, while the TGA-derived oxygen-related mass loss only halves. Thus, the amount of oxygen released from various modified birnessite samples is not strictly correlated with the surface area but rather with the chemical composition. The important thing to notice is that after the oxygen release tests, the structure of the material is intact, as evidenced by the Raman spectroscopy and X-ray diffraction. These show that it is mostly labile oxygen, which may partake in the oxidation reaction, that is evaluated. Thus, the total evolved oxygen, as derived from the TGA, is indicative of the redox properties of the birnessite samples. In our samples, the oxygen is released above 300 °C (
Figure S6 and
Figure 3), usually with two peaks related to the subsurface oxygen and one related to the bulk lattice oxygen. The release of the lattice oxygen continues to temperatures higher than the highest temperature attained in the experiments [
38,
39,
40].
Transmission electron microscopy (TEM) images were collected to assess the morphology of the prepared catalysts. The TEM overview images of the bulk alkali-promoted birnessite catalysts are shown in
Figure 4. Depending on the cation presence, different birnessite morphology can be observed varying from the 2D folded birnessite sheets for H-bir and Li-bir, with pronounced elongated and fragmented nanograins for Na-bir and K-bir, following the 3D-compacted denser structures for Rb-bir and Cs-bir comprised of birnessite nanograins. The evolution of the structure detected by TEM is in agreement with the trend of the surface area changes presented in
Table 1.
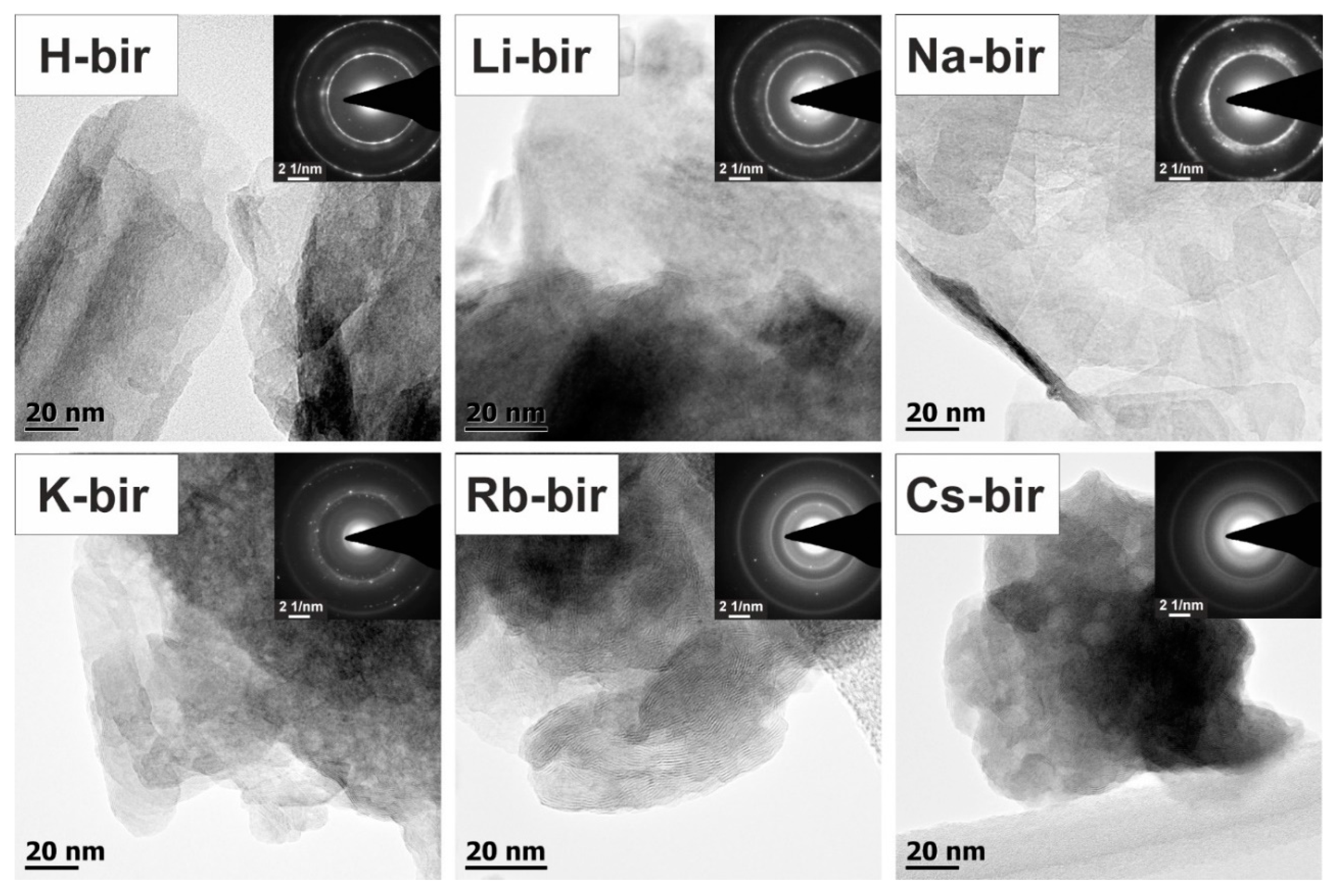
The detail aspect images, which are presented in
Figure 5, show clear birnessite morphology changes depending on the type of bulk alkali cation presence. The revealed sample morphology varied from the 2D folded birnessite sheets for the H-bir and Li-bir loose structures, with pronounced elongated and fragmented nanograins for Na-bir and K-bir, following the 3D compacted and denser structures for Rb-bir and Cs-bir comprised of birnessite nanograins. The separate layers of the MnO
6 octahedra sheets of the birnessite structure can be discerned in all cases. The corresponding selected area electron diffraction (SAED) patterns, shown in the
Figure 5 insets, are consistent with the birnessite crystal structure. The displayed notable blurring of the polycrystalline concentric rings reflects the nanocrystalline birnessite grain size being comparable (~10 nm) within the birnessite catalysts.
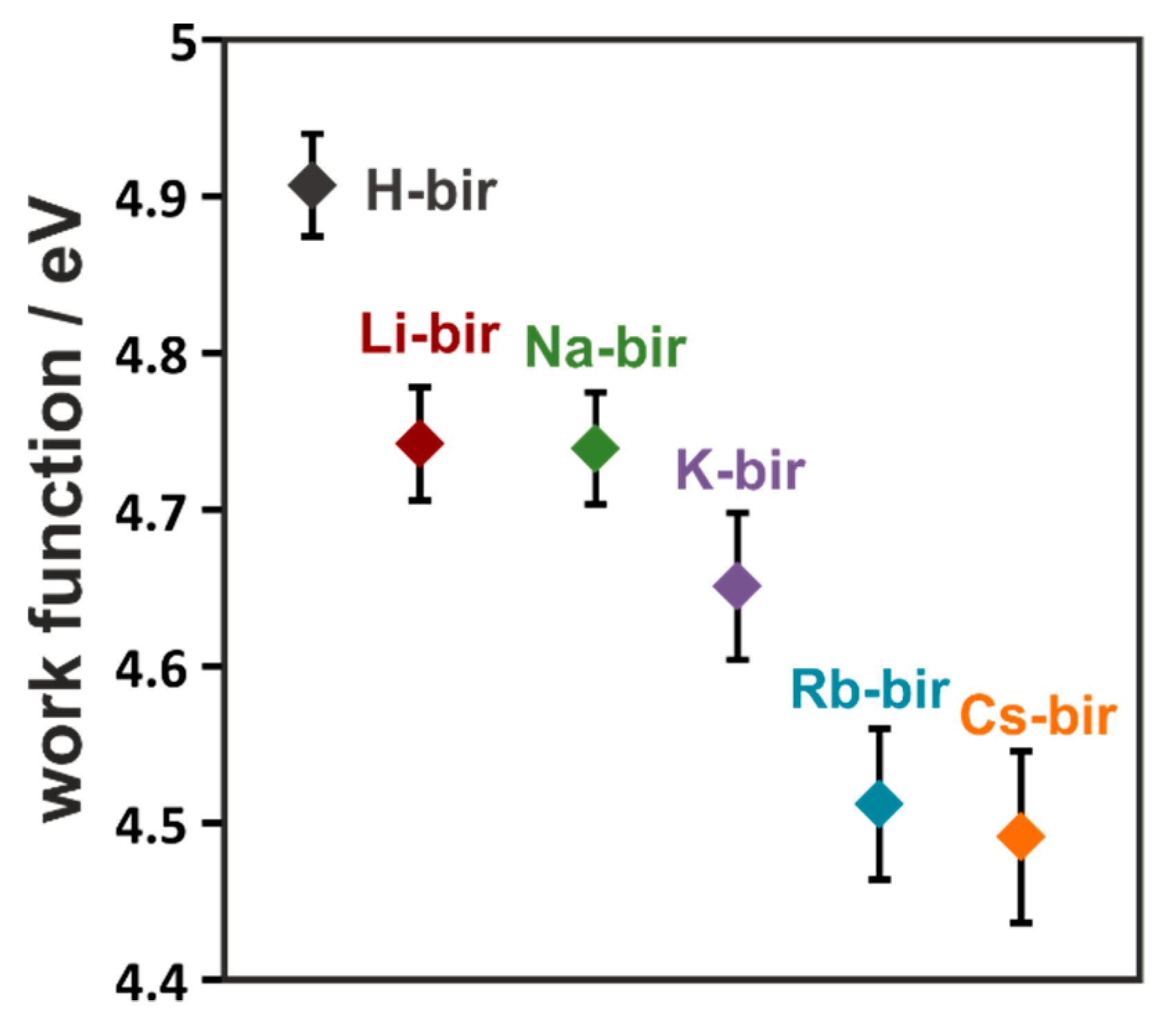
As often found in literature, a correlation between work function and catalytic activity in soot oxidation may be found when electron transfer is the main step of the mechanism of oxidation [
41,
42]. Such is the case when oxygen is activated to reactive forms on the catalyst surface. The typical tendency of the correlation is that of increased activity with lowered work function. To gain insight into the mechanism, the electron-donor properties of the tested catalysts were determined using a Kelvin probe. With the introduction of the alkali cations, the work function of the framework was drastically changed (
Figure 6). The highest work function was found for the H-bir samples (4.91 eV). Following the increasing atomic mass of the cations, the work function systematically decreases, with the lowest value of 4.49 eV measured for Cs-bir. Therefore, the electron-donor properties of these materials depend greatly on the introduced intra-layer cation, as their replacement can modify the electron-donor properties of the manganese dioxide framework by 0.42 eV.
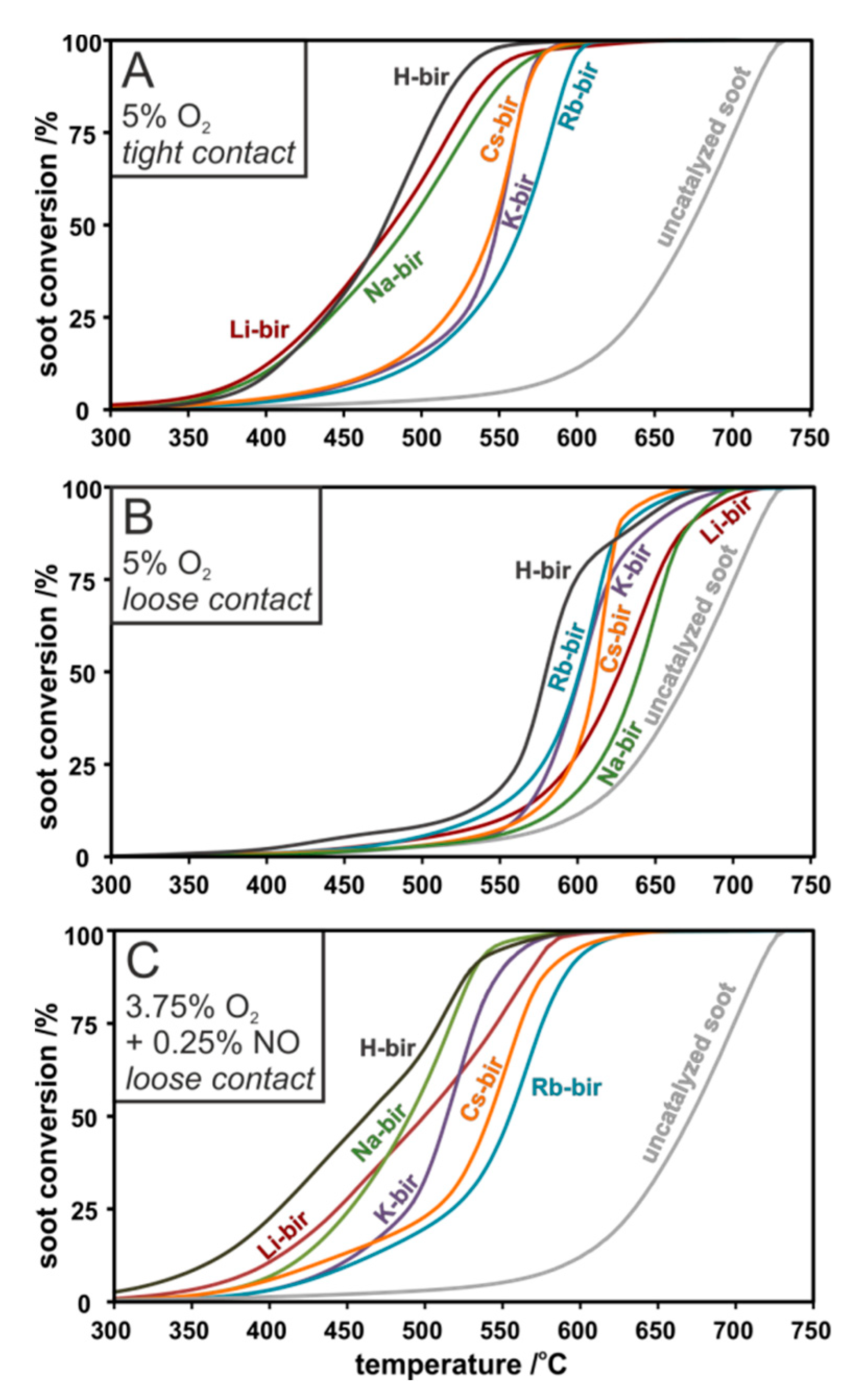
Reactivity curves of the detailed investigations into the catalytic activity in different reaction conditions of the investigated alkali modified series in tight contact, loose contact and loose contact with NO addition are presented in
Figure 7. As a reference, a conversion curve for the non-catalytic combustion of soot is also provided. The progress of soot combustion depends not only on the type of contact between the soot and the catalyst but also on the gas mixture and the type of alkali in the material interlayers. Nevertheless, in all cases, the investigated catalysts lowered the temperature of soot oxidation significantly compared to an uncatalyzed reaction. In tight contact (
Figure 7A), the most active catalysts (H-, Li-, Na-bir) show similar activities, lowering the temperature of 50% soot conversion,
T50%, by 200 °C compared to the uncatalyzed reaction. The remaining K-, Rb-, and Cs-bir, while still active, decrease the temperature by 120 °C. The loose contact measurements revealed a change in the catalytic activity trend. While H-bir remained the most active (Δ
T50% ~ 100 °C), the Li- and Na-bir exhibited a lower activity (Δ
T50% ~ 50 °C), with K, Rb, and Cs in between (
Figure 7B). Upon the NO addition to the gas mixture in loose contact mode, the order was returned to that of tight contact, with the sequence following: highest shift in
T50% = 210 °C for H-bir < Li-bir < Na-bir < K-bir < Cs-bir < Rb-bir to the lowest shift of
T50% = 116 °C (
Figure 7C). In all cases, the H-bir catalyst was found to be the most active material.
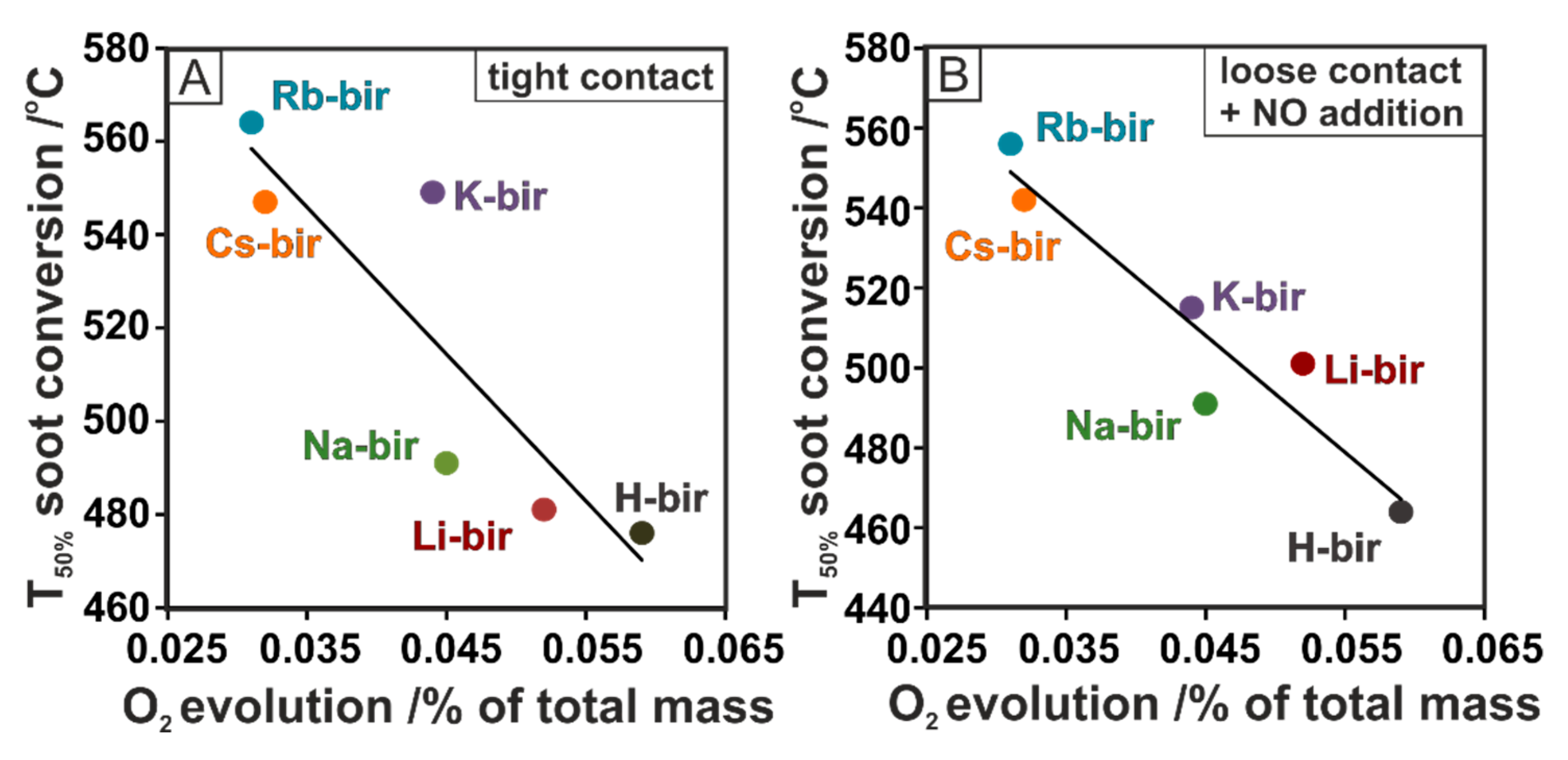
To reveal the origin of the activity of the tested birnessite catalysts, the soot conversion results were correlated with the physicochemical characteristics. The expected correlation between the soot oxidation activity and the electron-donor properties based on our previous works is completely reversed [
42,
43,
44]. The highest activity is observed for the material with the highest work function, showing that the activity is not related to the formation of reactive oxygen species via electron transfer. The alternative mechanism involves bulk oxygen following Mars–van Krevelen reaction scheme. In such a case, the activity should be related to the lattice oxygen mobility. Indeed, as can be seen in
Figure 8A,B, in both tight and loose contact + NO, a strong correlation between the soot conversion and the quantity of evolved oxygen is found. The results show a higher conversion of soot when more oxygen is released by the tested materials. Thus, in both tight and loose contact + NO, the lowest soot conversions are determined for the Cs-bir and Rb-bir samples, from which the smallest amount of oxygen desorbed; H-bir, the most active with the highest conversion, was the material that released the most oxygen. A similar correlation can be discerned for loose contact, as shown in the TG experiment (
Figure 2C). In the high-temperature regime, the most active catalysts (K-, Rb-, Cs-bir) release more oxygen than their counterparts (Li-, Na-bir) in the temperature range of the reaction (550–700 °C). The working hypothesis concerning the high activity of the H-bir material in loose contact can be attributed to a large amount of evolved oxygen, the abundance of which may initiate the combustion process at a lower temperature and thereafter sustain it. This concept is supported by the ability of birnessite to lose bulk oxygen while the vacancies can be filled with oxygen from the gas phase. What is important, the process of releasing and gaining oxygen can occur to a large extent while keeping the structure intact.
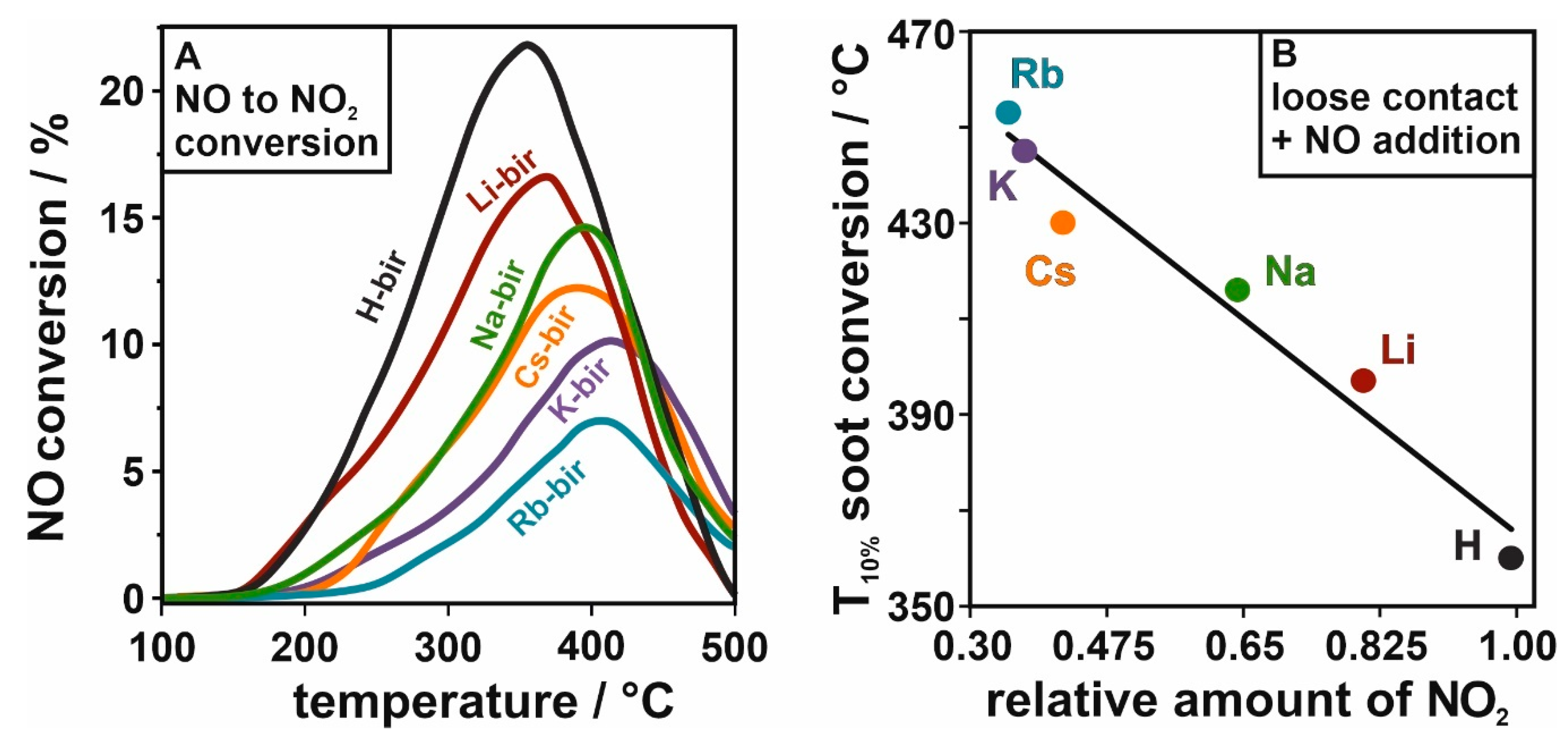
Furthermore, the mobile oxygen in the birnessite structure is of great importance for the activity upon NO addition, which can be attributed to the material’s ability to convert NO to NO
2. The latter can act as an oxygen carrier from the catalyst surface to the soot particles and facilitate combustion in loose contact conditions. This is due to the better oxidative properties of NO
2 compared to O
2 molecules. Therefore, experiments concerning the conversion of NO to NO
2 were carried out using the mass spectrometer as a detector, with the results presented in
Figure 9A. The results show the lowest concentrations of NO
2 were obtained by the birnessite with heavier intra-layer cations (Rb
+ < K
+ < Cs
+), while, with the lighter elements, the NO
2 is produced at lower temperatures and in greater amounts (Na
+ < Li
+ < H
+). The formation of highly oxidative NO
2 can be successfully correlated with soot conversion in loose contact + NO (
Figure 9B). In
Figure 9B, to keep the comparison of the soot oxidation characteristic parameter in the same temperature window as the NO conversion, we decided to use the
T10% instead of
T50%. The results show the important transformation of NO to NO
2 for the efficient soot oxidation reaction, as the NO addition allows the catalysts to reach similar conversions in the temperature window characteristic for tight contact conditions.
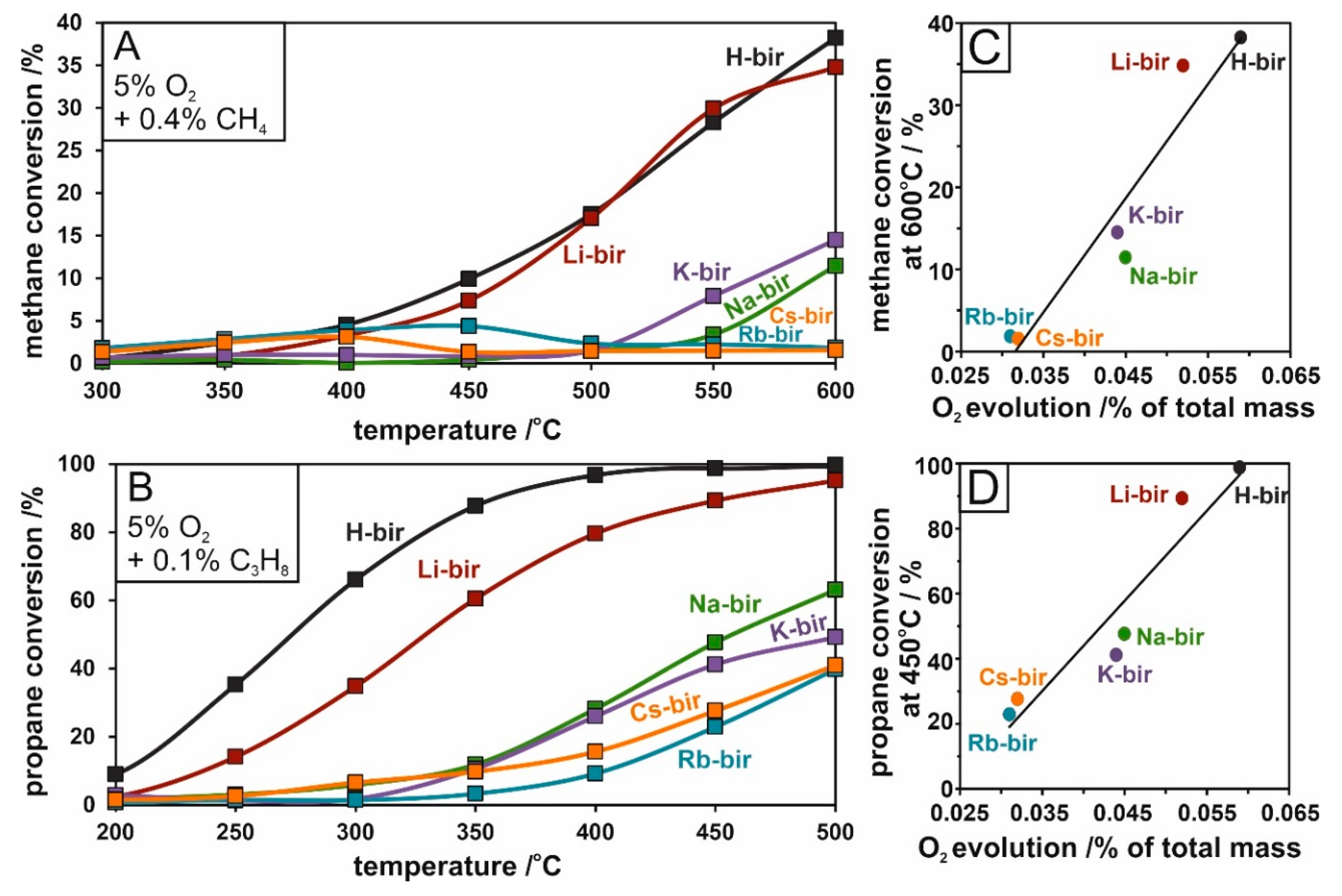
The catalytic activity of the alkali-modified birnessite in light hydrocarbon combustion was ascertained by propane and methane reaction tests. The differences between catalysts can be easily distinguished. In both methane and propane oxidation, the H- and Li- intercalated materials exhibited much higher activities than their counterparts (Na-, K-, Rb-, Cs-bir). In propane oxidation, H-bir achieved a 98% conversion at 400 °C compared to 80% for Li-bir and ~20% for the remaining materials (
Figure 10A). In methane oxidation, the combustion process is shifted to higher temperatures and at lower conversions. At 600 °C, both H and Li-bir converted ~40% of methane, ~10% Na- and K-bir, while Rb and Cs-bir converted negligible amounts (
Figure 10B). Similarly to soot combustion, the activity of the tested alkali-modified catalysts in light hydrocarbon oxidation can be correlated with the amount of evolved oxygen, which is presented in
Figure 10C,D for methane and propane oxidation respectively. The plots follow a similar sequence and slope, revealing an increase in light hydrocarbon conversion following larger amounts of evolved oxygen from the catalyst. It can be thus concluded that the methane and propane oxidation, like soot combustion, make use of oxygen mobility in the birnessite matrix following the Mars–van Krevelen mechanism.
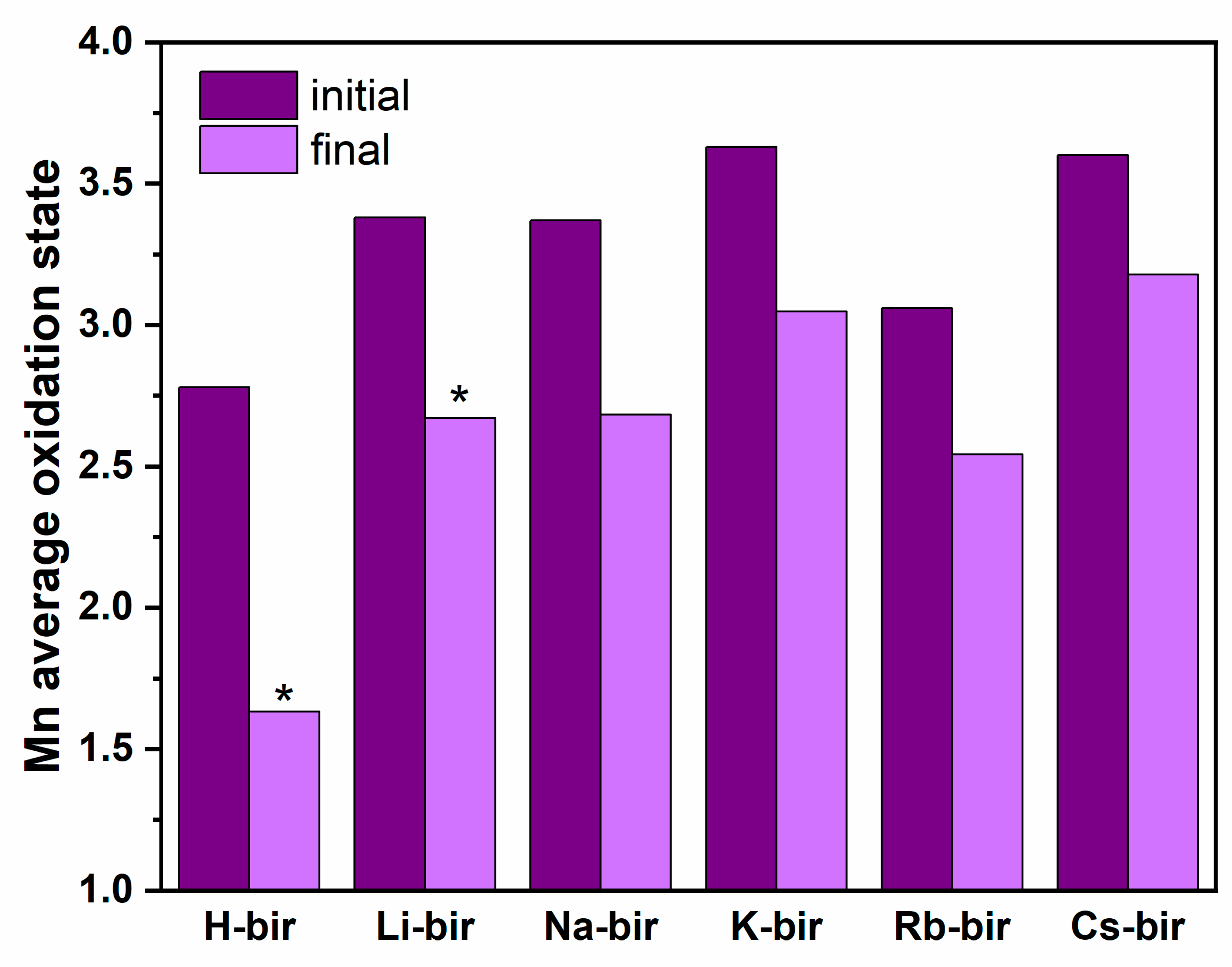
The obtained results revealed a strong effect of alkali cations on the birnessite catalytic activity in oxidation reactions both for soot and light hydrocarbons. Contrary to the previously observed correlations of activity with work function, the results revealed that the electron transfer mechanism and formation of reactive oxygen species is not efficient for the birnessite catalysts. Instead, the functional correlation between the activity and oxygen release was established based on the catalytic tests and by monitoring the oxygen evolution. The highest observed reactivity for H-bir among the studied samples was correlated with the easy oxygen removal from this sample, both in terms of low desorption temperature and the highest quantity of released O
2. Following this trend, the least active samples with K, Rb, and Cs in the structure do not release oxygen below 400 °C. As a result, the Mars–van Krevelen mechanism is proposed as the main reaction pathway. The highest relative amount of the oxygen which may be thermally removed from the H-bir sample results in the greatest change of the average Mn oxidation state at the end of this process. For all other samples, the changes are much less pronounced (
Figure 11). Since the obtained materials exhibit activity comparable to platinum group metal-based catalysts, this study provides a suitable background for the development of noble-metal free active phases for the catalytic elimination of carbonaceous pollutants [
45,
46].
3. Materials and Methods
3.1. Material Synthesis
The birnessite-type catalysts (AMn4O8) with alkali-metal cations intercalated into the interlayer space were prepared by the oxidation of Mn2+ in an alkaline solution [
32]. In the standard procedure of synthesis, a mixture of 3% H2O2 and 0.6 M AOH (where A is Li, Na, K, Rb, or Cs) was poured into a 0.3 M Mn(NO3)2 solution and left under stirring for 10 min. The obtained dark precipitate was kept in the mother liquid in mild room conditions for 24 h. Afterwards, the precipitate was separated from the reaction solution by filtration, washed generously with deionized water, and subsequently air-dried at 80 °C. Thereafter, the obtained products were heated at 4 °C/min and calcined in 450 °C for 2 h. The material reported as H-birnessite was obtained by ion exchange. In this case, 3 g of synthetic Na-birnessite was stirred in an acidic solution (pH = 1) for 21 h [
31]. The product was then filtered, dried, and annealed as above. Reagents: H
2O
2 30%; Mn(NO
3)
2∙4H
2O, pure p.a.; KOH ≥ 85%, pellets; NaOH, microgranular, ≥98.8%; HCl 35–38%, pure p.a. (POCH S.A., Gliwice, Poland), LiOH∙H
2O, ≥99.995%; RbOH∙H
2O, ≥85%; CsOH∙H
2O, ≥90%(Merck KGaA, Darmstadt, Germany).
3.2. Characterization Techniques
The actual bulk content of metallic elements was examined using an Energy-Dispersive X-ray fluorescence spectrometer (XRF, ARL QUANT’X, Thermo Scientific, Waltham, MA, USA), with a Rhode-anode as a source of X-rays and a detecting system of Si(Li) crystal with Peltier cooling. In a typical measurement, 0.085 g of the studied sample was pelletized under 6 MPa for 1 min. Oxygen was not analysed with the used spectrometer. In the calculated formulas, the oxygen content was arbitrarily set to 8.
The XRD patterns were collected using a Rigaku MiniFlex powder diffractometer (Tokyo, Japan) with a Cu K
α radiation source (λ
CuKα = 0.15406 nm) at voltage-current settings: 40 kV, 15 mA. The step scan of 0.02° progressing every 0.8 s for the range of 10–50 degrees was established. The birnessite phase was identified based on ICSD-152290 [
47].
The Raman spectra were registered in standard conditions using a Renishaw InVia spectrometer equipped with a Leica DMLM confocal microscope and a CCD detector. The laser (λ = 785 nm) power at the sample surface was 3 mW (1% of available power) with a magnification of 20×. To provide a relatively high signal to noise ratio, at least 9 accumulations of scattered light in a spectral range of 100 to 900 cm−1 were collected.
An X-ray photoelectron spectroscopic (XPS, Prevac Poland) analysis was performed using an R3000 analyser and a MX-650 Al Kα source with a SAX 100 monochromator by Scienta (Uppsala, Sweden). The analyser was used with a 100 eV pass energy and 0.15 eV sampling. The approximate lamp output reaching the sample was ~220–230 W. High-resolution scans were taken for C 1s, O 1s, Mn 3s, Mn 2p, and relevant alkali bands: Li 1s—which overlaps with the Mn 3p bands and hinders the Li quantitative analysis—and Na 1s, K 2p, Rb 3d, and Cs 3d and 4d, where the latter overlaps with the Mn 3s bands, rendering it impossible to use this range for Mn average oxidation state determination. To determine the average oxidation state of Mn and Mn
x+ in the samples, a procedure described by Ilton et al. was used. [
48] Briefly, relevant sets of bands originating from Mn
2+, Mn
3+, and Mn
4+ were fitted with CasaXPS processing software (Version 2.3.18PR1.0, Casa Software LTd., 1999–2017) to the Mn 3s and Mn 2p detailed scans, with constraints set to their relative positions, intensities, and full widths at half maximum (FWHM). After an initial fit, the positions of the main bands were allowed to relax to account for the shift in the energy scale due to the semiconductive properties of the samples, even though the main C 1s peak was arbitrarily set at 285 eV. The cumulative area of the set of peaks from one of the oxidation states determined its contribution to the average Mn oxidation state. Preferably, the Mn 3s range was used to determine Mn
x+ as the 3s electrons, having a higher kinetic energy than 2p, represent more bulk-like values.
The crystallinity and crystal appearance of the synthesized materials were evaluated by Transmission Electron Microscopy (TEM, Thermo Scientific, Waltham, MA, USA) measurements using an FEI Tecnai Osiris microscope equipped with an X-FEG Schottky high brightness electron source (accelerating voltage = 200 kV). The SAED (Selected Area Electron Diffraction) pattern method was used for performing the TEM structural analysis. In a standard measurement procedure, highly dispersed powder samples were deposited on a lacey carbon film supported on a Cu TEM grid.
The thermal stability of the catalysts was determined by thermogravimetric measurements using a TGA/DSC1 (Mettler Toledo, Greifensee, Switzerland). In a typical measurement, approximately 5 mg of the sample was placed in a 150 µL alumina crucible and heated in Ar flow (60 mL min−1) in the temperature range of 30–850 °C (10 °C min−1 heating rate). The work function of the catalysts was calculated in the basis of contact potential difference (CPD) measurements. The evaluation of the electron-donor properties was conducted using a Kelvin probe (KP6500, McAllister Technical Services, Coeur d’Alene, ID, USA), equipped with a stainless-steel reference electrode: d = 3mm, Φref. = 4.32 eV). In the standard measurements, 0.1 g of a pelletized sample (6 MPa, 1 min, 10 mm in diameter) was placed in the chamber. The pre-treatment in case of surface standardization included heating at 400 °C for 15 min in a vacuum (10−6 Pa), which was followed by cooling to 150 °C and the measurement of the actual CPD of the sample.
The catalytic activity of the synthesized catalysts was determined by performing soot, methane, and propane oxidation tests. The catalytic activity in soot oxidation was determined due to temperature-programmed oxidation (TPO) using model soot (Printex 80, Degussa GmbH, München, Germany). Measurements were conducted in different modes (loose or tight contact) and with different compositions of the flowing gas (5% O2 in He, 3.75% O2 + 0.25% NO in He). The catalyst–soot mixture (mass ratio 8:1), was placed in a quartz fixed-bed reactor and was heated from room temperature to 700 °C at 10 °C min−1, with the flow rate of the proper gas mixture set at 60 mL min−1. The activity of the materials in the NO conversion to NO2 was tested by depositing the same amount of catalyst as in the soot combustion tests and washing it with an O2-NO mixture following the aforementioned heating program and flow rate. The progress of the reaction was tracked by a quadrupole mass spectrometer (RGA200, SRS, Sunnyvale, CA, USA) following the lines for m/z = 18 (H2O), 28 (CO/N2), 30 (NO), 32 (O2), 44 (CO2), and 46 (NO2).
The catalytic oxidation of methane and propane was carried out at normal pressure in a fixed-bed stainless steel tubular flow reactor. These molecules were chosen because of their simplicity and high resistance to oxidation. An amount of 50 mg of the catalyst was placed between quartz wool packs in a steel rod. The proper gas mixture, including 5% O2 + 0.4% CH4 or 5% O2 + 0.1% C3H8, was passed through the reactor. The reaction temperature was increased by incremental steps from RT to 500 °C or 600 °C for propane and methane, respectively, allowing for the temperature to stabilize every 50 °C to measure the catalytic activity at a steady state. The reactions were tracked by a FT-IR analyser (Gasmet, Vantaa, Finland ) following the products (H2O, CO, CO2), possible by-products (C3H6), and VOC gases (CH4, C3H8). The major product of the examined short-chain hydrocarbons oxidation reactions was CO2, with an estimated selectivity of over 99%.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}