
Longitudinal Circulating Tumor DNA Analysis in Blood and Saliva for Prediction of Response to Osimertinib and Disease Progression in EGFR-Mutant Lung Adenocarcinoma

 ,
,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Specimen Collection
2.2. Tumor Volumetric Measurements
2.3. ddPCR
2.4. NGS
2.5. EFIRM
2.6. Statistical Considerations
3. Results
3.1. Patients and Clinical Samples
3.2. Volumetric Tumor Measurements
3.3. Baseline Detection of Plasma ctDNA and Association between Baseline Mutant EGFR Plasma ctDNA Levels and Tumor Burden
3.4. Correlations between ddPCR; NGS; eLB, and Calculated Tumor Volume
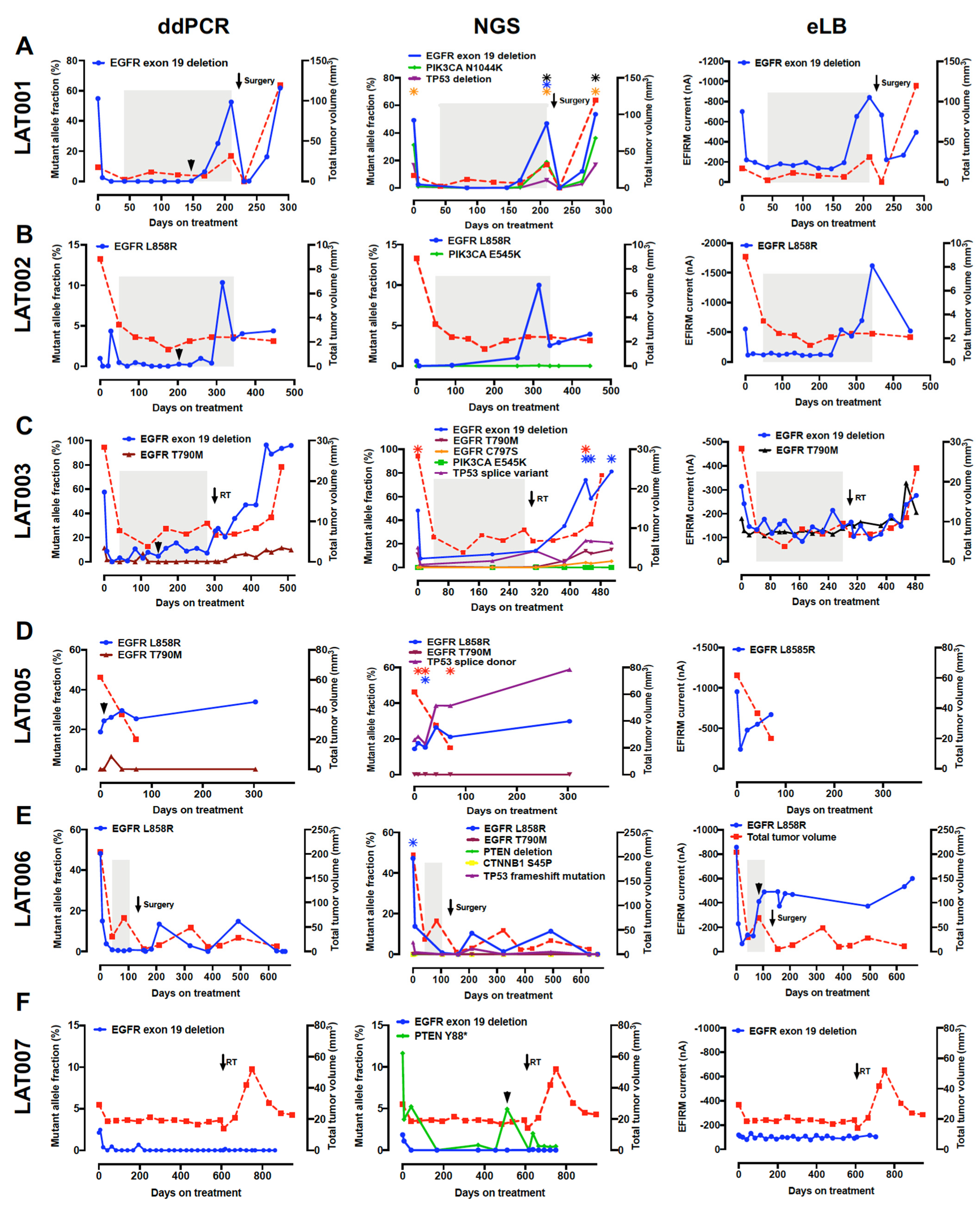
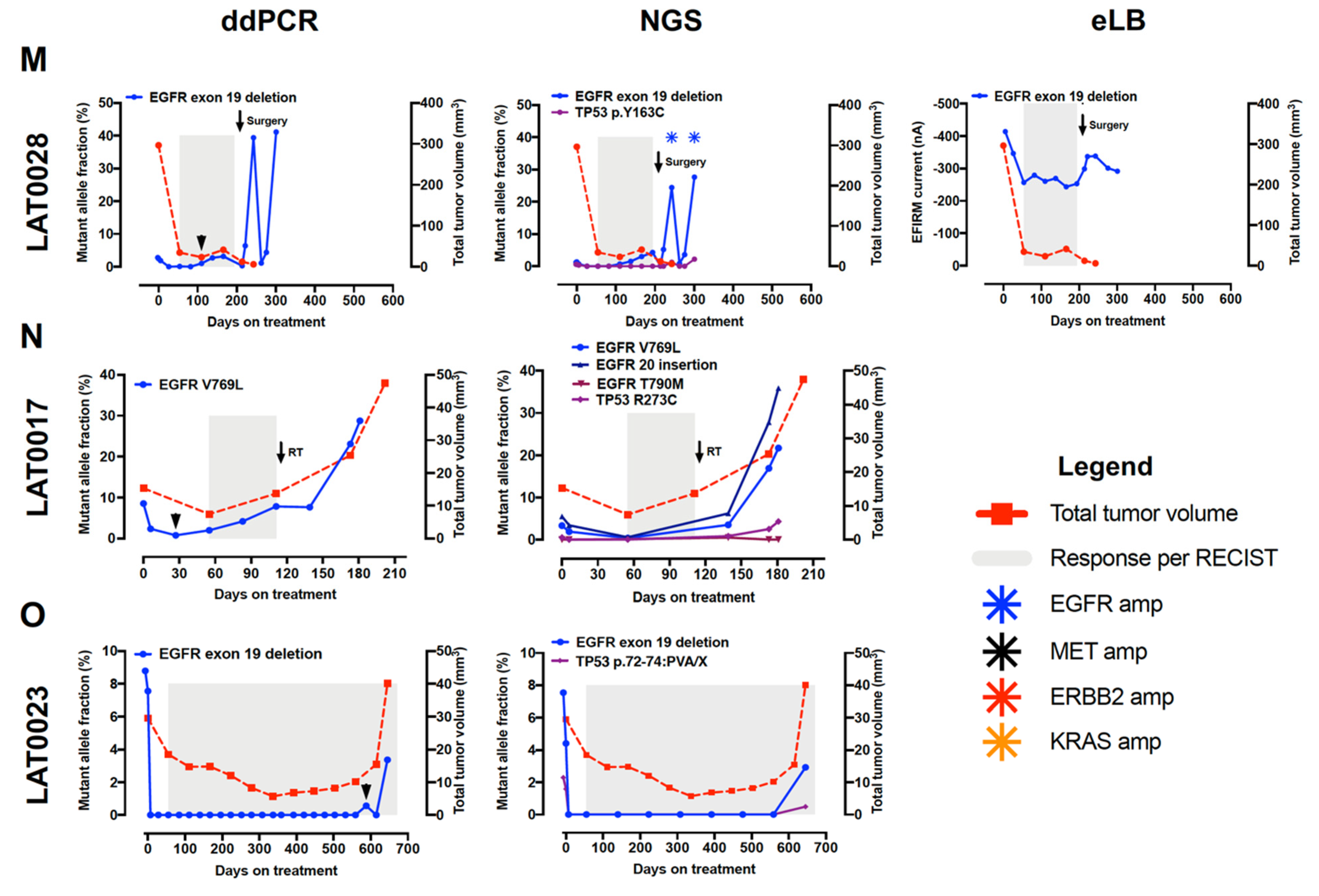
3.5. Dynamic ctDNA Changes Reflect Treatment Response and Emergence of Resistance
3.6. NGS ctDNA Assay (InVisionFirst-Lung) Detects a Broad Array of Genomic Modifications Which May Be Implicated in Osimertinib Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, C.; Giaccone, G. Precision oncology in non-small-cell lung cancer: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 348–349. [Google Scholar] [CrossRef]
- Heerink, W.J.; de Bock, G.H.; de Jonge, G.J.; Groen, H.J.; Vliegenthart, R.; Oudkerk, M. Complication rates of CT-guided transthoracic lung biopsy: Meta-Analysis. Eur. Radiol. 2017, 27, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Roper, N.; Brown, A.L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell. Rep. Med. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer. Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar] [CrossRef]
- Pritchett, M.A.; Camidge, D.R.; Patel, M.; Khatri, J.; Boniol, S.; Friedman, E.K.; Khomani, A.; Dalia, S.; Baker-Neblett, K.; Plagnol, V. Prospective clinical validation of the InVisionFirst-Lung circulating tumor DNA assay for molecular profiling of patients with advanced nonsquamous non–small-cell lung cancer. JCO Precis. Oncol. 2019, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Markovets, A.; Barrett, J.C.; Chmielecki, J.; Goldberg, S.B.; Shepherd, F.A.; Vowler, S.; Oxnard, G.R. Complete clearance of plasma EGFR mutations as a predictor of outcome on osimertinib in the AURA trial. Am. Soc. Clin. Oncol. 2017, 35, 9018. [Google Scholar] [CrossRef]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Janne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Tan, W.; Tang, X.; Ma, J. Variations in EGFR ctDNA Correlates to the Clinical Efficacy of Afatinib in Non Small Cell Lung Cancer with Acquired Resistance. Pathol. Oncol. Res. 2017, 23, 307–315. [Google Scholar] [CrossRef]
- Mok, T.; Wu, Y.L.; Lee, J.S.; Yu, C.J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer. Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riediger, A.L.; Dietz, S.; Schirmer, U.; Meister, M.; Heinzmann-Groth, I.; Schneider, M.; Muley, T.; Thomas, M.; Sultmann, H. Mutation analysis of circulating plasma DNA to determine response to EGFR tyrosine kinase inhibitor therapy of lung adenocarcinoma patients. Sci. Rep. 2016, 6, 33505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Uchida, J.; Kukita, Y.; Kumagai, T.; Nishino, K.; Inoue, T.; Kimura, M.; Oba, S.; Imamura, F. Numerical indices based on circulating tumor DNA for the evaluation of therapeutic response and disease progression in lung cancer patients. Sci. Rep. 2016, 6, 29093. [Google Scholar] [CrossRef] [Green Version]
- Moran, T.; Felip, E.; Bosch-Barrera, J.; de Aguirre, I.; Ramirez, J.L.; Mesia, C.; Carcereny, E.; Roa, D.; Sais, E.; Garcia, Y.; et al. Monitoring EGFR-T790M mutation in serum/plasma for prediction of response to third-generation EGFR inhibitors in patients with lung cancer. Oncotarget 2018, 9, 27074–27086. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Cui, S.; Ding, J.; Sun, Y.; Zhang, L.; Zhao, Y.; Gu, A.; Chu, T.; Wang, H.; Zhong, H.; et al. Dynamics of EGFR mutations in plasma recapitulates the clinical response to EGFR-TKIs in NSCLC patients. Oncotarget 2017, 8, 63846–63856. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Lin, C.C.; Joon, A.; Feng, Z.; Troche, G.; Lira, M.E.; Chia, D.; Mao, M.; Ho, C.L.; Su, W.C.; et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. Am. J. Respir. Crit. Care. Med. 2014, 190, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Strom, C.M.; Cheng, J.; Lin, C.C.; Hsu, C.Y.; Soo Hoo, G.W.; Chia, D.; Kim, Y.; Li, F.; Elashoff, D.; et al. Electric Field-Induced Release and Measurement Liquid Biopsy for Noninvasive Early Lung Cancer Assessment. J. Mol. Diagn. 2018, 20, 738–742. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wei, F.; Huang, W.L.; Lin, C.C.; Li, L.; Shen, M.M.; Yan, Q.; Liao, W.; Chia, D.; Tu, M.; et al. Ultra-Short Circulating Tumor DNA (usctDNA) in Plasma and Saliva of Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2020, 12, 2041. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef]
- Plagnol, V.; Woodhouse, S.; Howarth, K.; Lensing, S.; Smith, M.; Epstein, M.; Madi, M.; Smalley, S.; Leroy, C.; Hinton, J.; et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE 2018, 13, e0193802. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Papadimitrakopoulou, V.; Mok, T.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Sebastian, M.; Theelen, W.; et al. Early clearance of plasma EGFR mutations as a predictor of response to osimertinib in the AURA3 trial. J. Clin. Oncol. 2018, 36, 9027. [Google Scholar] [CrossRef]
- Zhou, C.; Imamura, F.; Cheng, Y.; Okamoto, I.; Cho, B.C.; Lin, M.C.; Majem, M.; Gautschi, O.; Gray, J.E.; Boyer, M.J.; et al. Early clearance of plasma EGFR mutations as a predictor of response to osimertinib and comparator EGFR-TKIs in the FLAURA trial. J. Clin. Oncol. 2019, 37, 9020. [Google Scholar] [CrossRef]
- Hartmaier, R.; Han, J.-Y.; Ahn, M.-J.; Cho, B.C.; Cantarini, M.; Frewer, P.; Frigault, M.M.; Oxnard, G. Abstract CT303: The Effect of Savolitinib Plus Osimertinib on ctDNA Clearance in Patients with EGFR Mutation Positive (EGFRm) MET-Amplified NSCLC in the TATTON Study; AACR: Philadelphia, PA, USA, 2020. [Google Scholar]
- Gray, J.E.; Okamoto, I.; Sriuranpong, V.; Vansteenkiste, J.; Imamura, F.; Lee, J.S.; Pang, Y.K.; Cobo, M.; Kasahara, K.; Cheng, Y.; et al. Tissue and Plasma EGFR Mutation Analysis in the FLAURA Trial: Osimertinib versus Comparator EGFR Tyrosine Kinase Inhibitor as First-Line Treatment in Patients with EGFR-Mutated Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6644–6652. [Google Scholar] [CrossRef] [Green Version]
- Nishino, M.; Dahlberg, S.E.; Cardarella, S.; Jackman, D.M.; Rabin, M.S.; Hatabu, H.; Janne, P.A.; Johnson, B.E. Tumor volume decrease at 8 weeks is associated with longer survival in EGFR-mutant advanced non-small-cell lung cancer patients treated with EGFR TKI. J. Thorac. Oncol. 2013, 8, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, M.; Dahlberg, S.E.; Fulton, L.E.; Digumarthy, S.R.; Hatabu, H.; Johnson, B.E.; Sequist, L.V. Volumetric Tumor Response and Progression in EGFR-mutant NSCLC Patients Treated with Erlotinib or Gefitinib. Acad. Radiol. 2016, 23, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Pantel, K. Liquid biopsies, what we do not know (yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert. Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Yang, S.; Lim, S.; Heo, S.; Daniel, S.; Markovets, A.; Rafati, M.; Park, C.; Yun, J.; Pyo, K. P76. 18 Tissue-and Plasma-Based Landscape of Resistance to Osimertinib. J. Thorac. Oncol. 2021, 16, S593–S594. [Google Scholar] [CrossRef]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-dependent beta-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, G.; Zhuo, M.; Fan, Z.; Miao, L.; Chen, L.; Zeng, A.; Yin, R.; Ou, Y.; Shi, Z.; et al. Next-generation sequencing based mutation profiling reveals heterogeneity of clinical response and resistance to osimertinib. Lung Cancer 2020, 141, 114–118. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, C.; Xi, L.; Cultraro, C.M.; Wei, F.; Jones, G.; Cheng, J.; Shafiei, A.; Pham, T.H.-T.; Roper, N.; Akoth, E.; et al. Longitudinal Circulating Tumor DNA Analysis in Blood and Saliva for Prediction of Response to Osimertinib and Disease Progression in EGFR-Mutant Lung Adenocarcinoma. Cancers 2021, 13, 3342. https://doi.org/10.3390/cancers13133342
Kim C, Xi L, Cultraro CM, Wei F, Jones G, Cheng J, Shafiei A, Pham TH-T, Roper N, Akoth E, et al. Longitudinal Circulating Tumor DNA Analysis in Blood and Saliva for Prediction of Response to Osimertinib and Disease Progression in EGFR-Mutant Lung Adenocarcinoma. Cancers. 2021; 13(13):3342. https://doi.org/10.3390/cancers13133342
Chicago/Turabian StyleKim, Chul, Liqiang Xi, Constance M. Cultraro, Fang Wei, Gregory Jones, Jordan Cheng, Ahmad Shafiei, Trinh Hoc-Tran Pham, Nitin Roper, Elizabeth Akoth, and et al. 2021. "Longitudinal Circulating Tumor DNA Analysis in Blood and Saliva for Prediction of Response to Osimertinib and Disease Progression in EGFR-Mutant Lung Adenocarcinoma" Cancers 13, no. 13: 3342. https://doi.org/10.3390/cancers13133342
APA StyleKim, C., Xi, L., Cultraro, C. M., Wei, F., Jones, G., Cheng, J., Shafiei, A., Pham, T. H.-T., Roper, N., Akoth, E., Ghafoor, A., Misra, V., Monkash, N., Strom, C., Tu, M., Liao, W., Chia, D., Morris, C., Steinberg, S. M., ... Guha, U. (2021). Longitudinal Circulating Tumor DNA Analysis in Blood and Saliva for Prediction of Response to Osimertinib and Disease Progression in EGFR-Mutant Lung Adenocarcinoma. Cancers, 13(13), 3342. https://doi.org/10.3390/cancers13133342