
Structural and Drug Targeting Insights on Mutant p53





Abstract
Simple Summary
Abstract
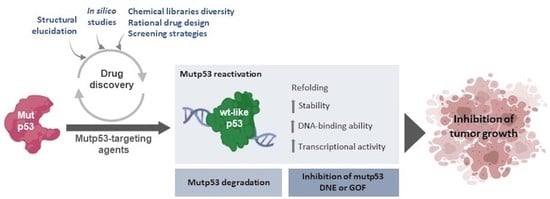
1. Introduction
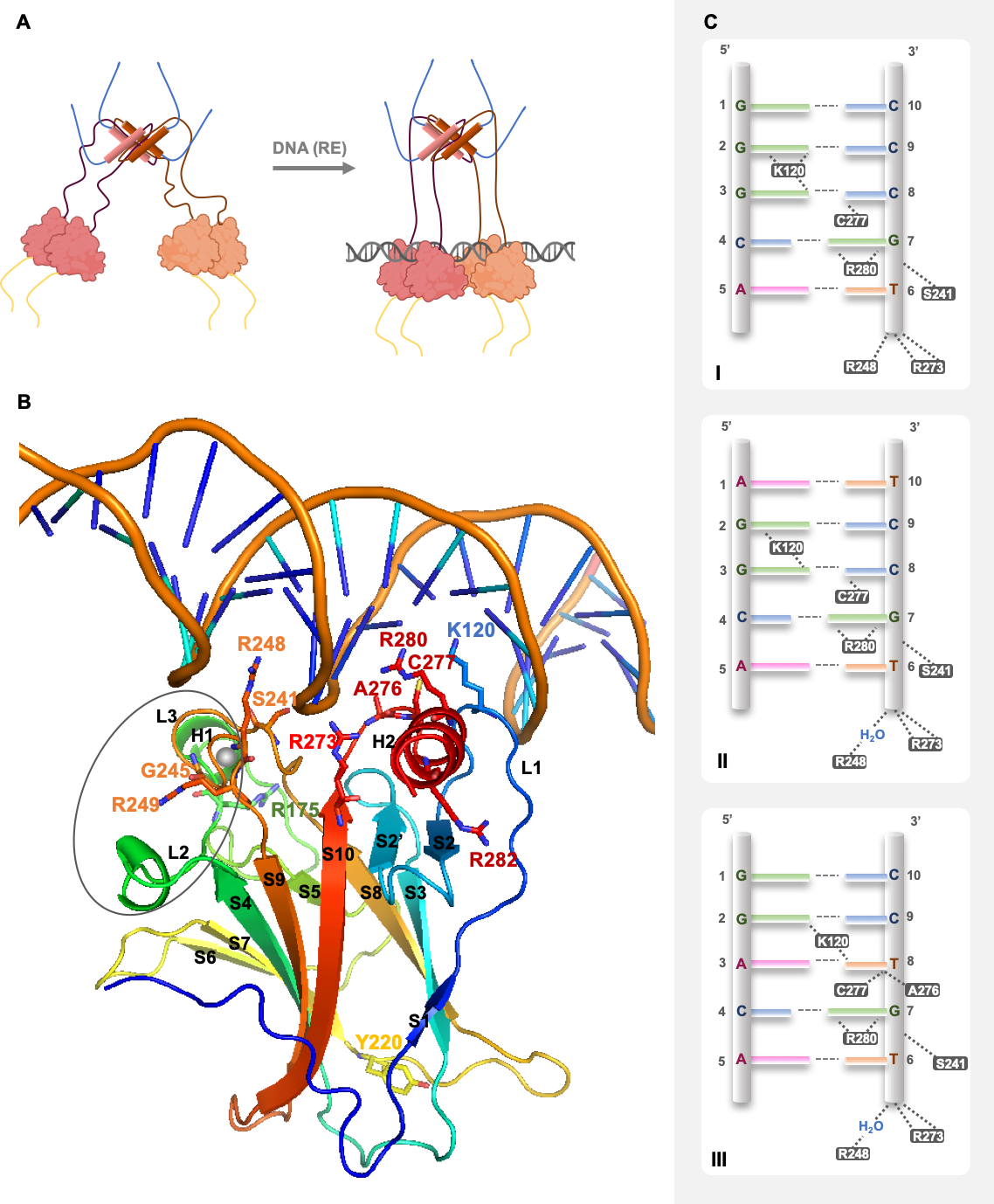
2. p53 Structure and DNA Recognition

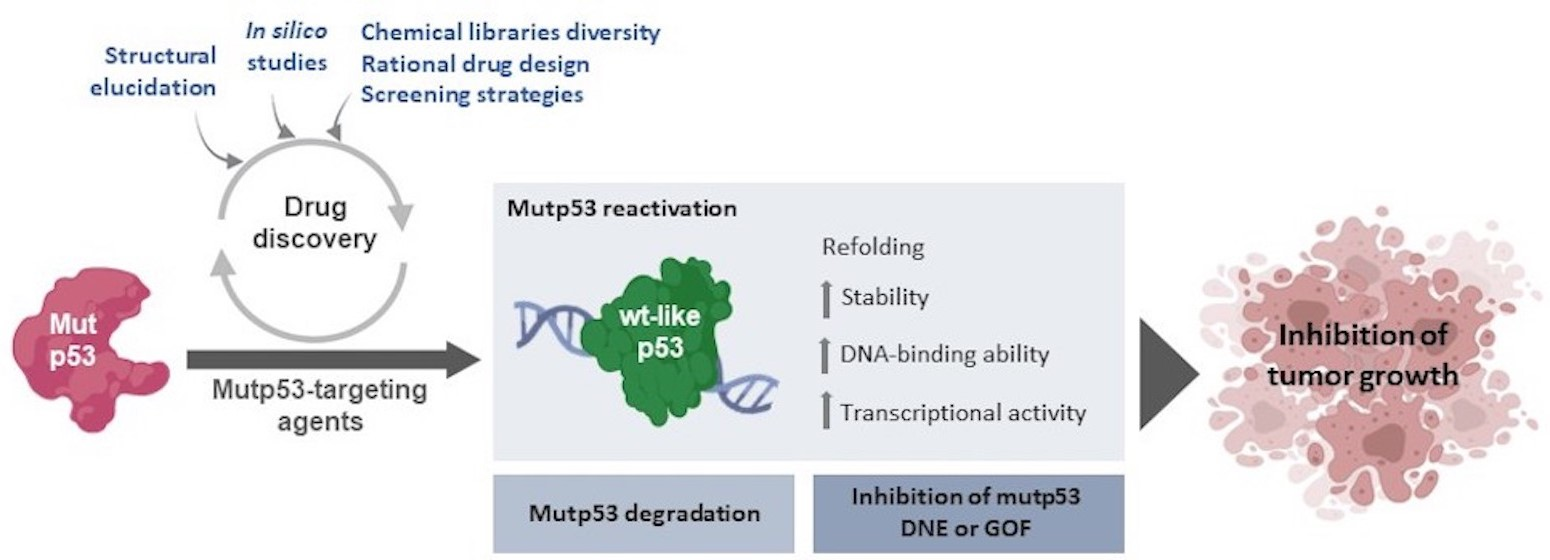
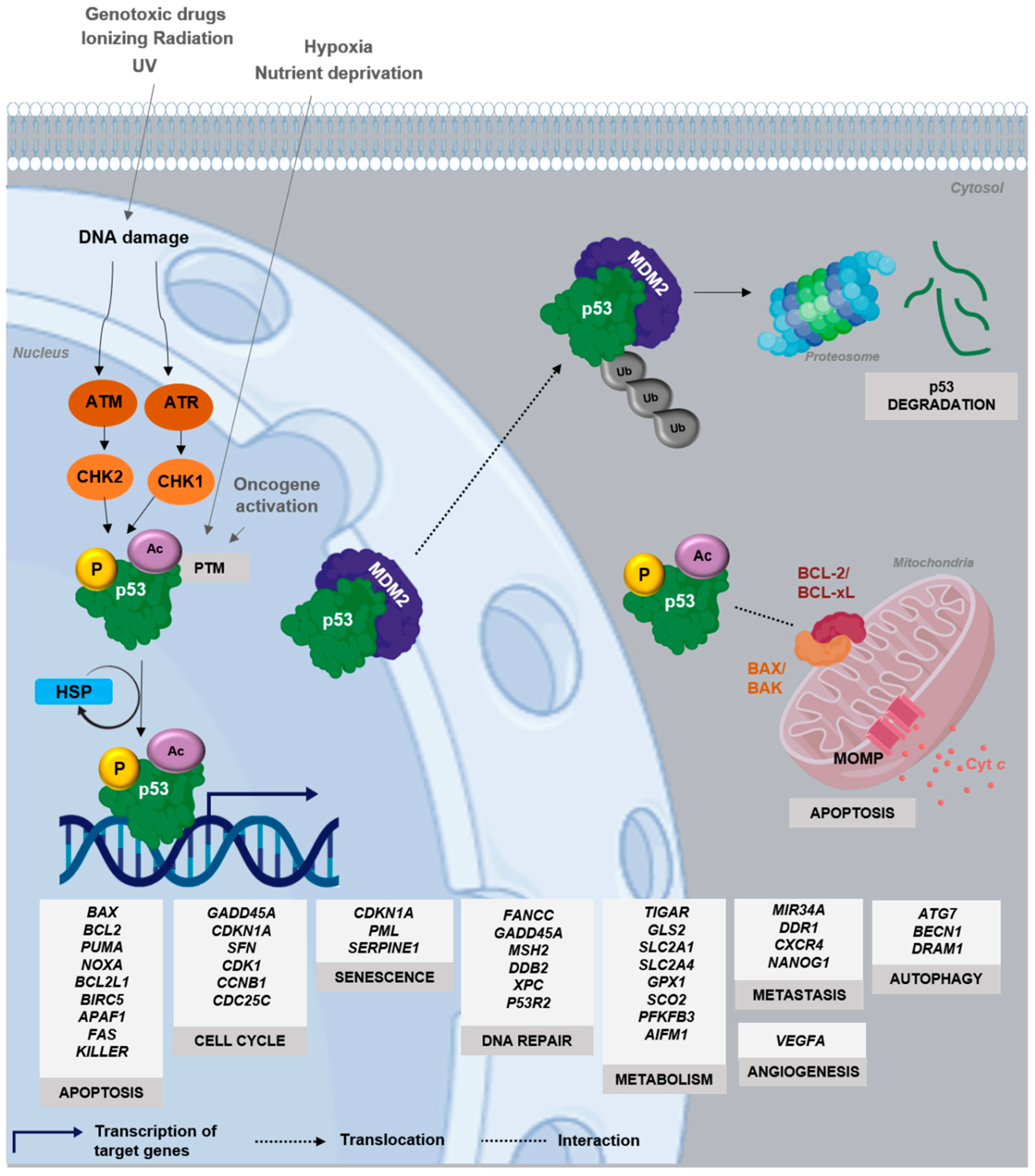
3. Dynamics and Regulation of p53
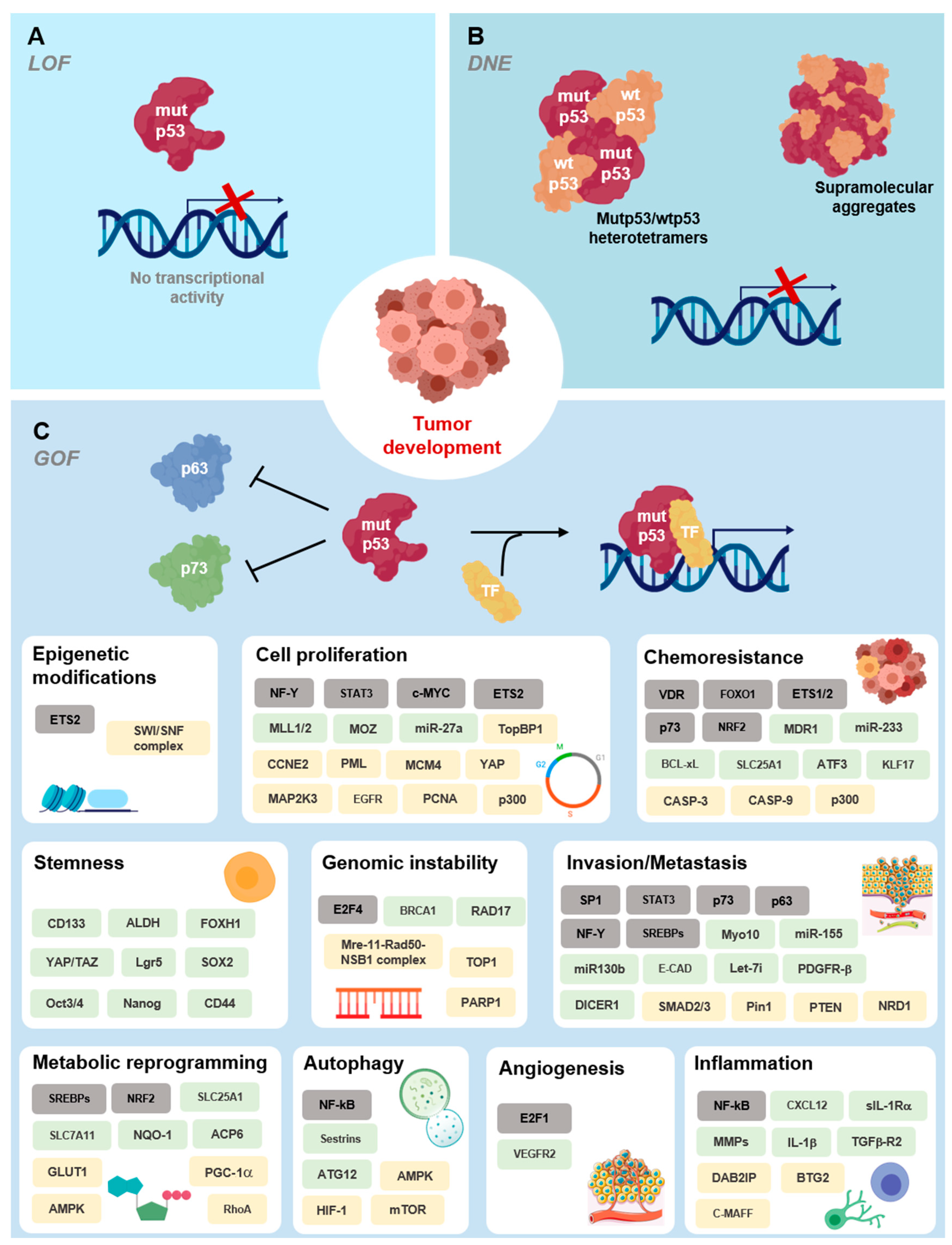
4. Mutant p53 Functions in Tumorigenesis

Deleterious Effects of Mutant p53 on DNA Binding and Protein Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable (DBD) | Amino Acid Location and Biochemical Mutation Type | Mutp53 Structural Modifications | Thermo-Dynamic Stability & | Estimated Folded Protein at 37 °C # | DNA-Binding Affinity at 20 °C # | Structural Elucidation of p53 DBD (PDB ID Code Eample) | Ref. |
|---|---|---|---|---|---|---|---|
| Wtp53 | |||||||
| with DNA | NA | NA | NA | NA | +++ | 1TUP *, 1TSR *, 2AC0, 2ADY, 2AHI, 2ATA | [20,36,42] |
| w/o DNA | NA | NA | + | +++ | NA | 2OCJ, 1UOL * | [20,87] |
| Oncogenic mutp53 | |||||||
| DNA contact region | |||||||
| S241F | L3; uncharged polar to hydrophobic aromatic/small to large residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R248Q | L3; cationic to uncharged polar residue | Alteration of L3 conformation (NMR data) | +/− | +++ | − | ND | [20,76] |
| R248W | L3; cationic to hydrophobic aromatic bulky residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R273C | S10; cationic to uncharged polar/large to small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | ND | ND | ND | 2J20 *, 4IBQ | [24,79] |
| R273H | S10; aliphatic to aromatic/large to bulky-small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | + | +++ | − | 2BIM *, 4IBS, 4IJT | [20,24,48,78] |
| C277F | S10-H2 turn; uncharged polar to hydrophobic aromatic/small to large residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R280K | H2; large to small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | ND | ND | − | 6FF9 | [45,80,81] |
| Structural—DNA region | |||||||
| F134L | S2′; aromatic to aliphatic residue | ND | − | − | − | ND | [20] |
| H168R | L2; aromatic to aliphatic/bulky-small to large residue | Alteration of L2 conformation | ND | ND | ND | 2BIN * | [78] |
| G245S | L3; small to large uncharged polar residue | Small distortion of L3/dimerization interface | +/− | +++ | + | 2J1Y * | [20,48,79] |
| G245D | L3; small to large anionic residue | Small distortion of L3 and LSH (in silico data) | ND | ND | ND | ND | [88] |
| R249S | L3; cationic to uncharged polar/large to small residue | Alteration of L3 conformation affecting R248-mediated DNA anchoring and of dimer interface; increased flexibility of the β-sandwich | +/− | +++ | − | 2BIO *, 3D05, 3DO6, 3DO7 | [20,48,78,82] |
| R282Q | H2; cationic to uncharged polar residue | Flexibility is decreased in L1 and increased in L3 | ND | ND | ND | 2PCX | [89] |
| R282W | H2; aliphatic to aromatic/cationic to hydorphobic bulky residue | Impaired LSH anchoring to β-sandwich, increase of L1 flexibility | − | − | ++ | 2J21 * | [20,48,79] |
| Structural—Zinc region | |||||||
| R175A | L2; cationic to hydrophobic/large to small residue | Interference with zinc-binding (smaller effect than R175H) (theoretical interpretation) | +/− | +++ | + | ND | [20,45] |
| R175H | L2; aliphatic to aromatic/large to bulky-small residue | Alteration of L2 and L3 conformation, loss of zinc-binding (theoretical interpretation) | − | − | − | ND | [20,45,48] |
| M237I | L3; large to small residue/decrease in atom electro-negativity | ND | − | +/− | − | ND | [20] |
| C242S | L3; decrease in atom electro-negativity | Zinc ligand substitution with loss of zinc coordination (theoretical interpretation) | − | +/− | − | ND | [20,45] |
| Structural—-sandwich | |||||||
| V143A | S3; large to small residue | Internal hydrophobic cavity | − | − | + | 2J1W * | [20,48,79] |
| L145Q | S3; hydrophobic to uncharged polar/small to large residue | -sheet and loop-sheet-helix motif destabilization (in silico data) | − | +/− | +/− | ND | [20,90] |
| P151S | S3/S4 turn; hydrophobic to uncharged polar residue | ND | − | − | +/− | ND | [20] |
| V157F | S4; aliphatic to aromatic/small to large-bulky residue | Internal hydrophobic cavity | − | − | ++ | 4KVP | [91] |
| I195T | S5; hydrophobic to uncharged polar/large to small residue | ND | − | − | − | ND | [20] |
| Y220C | S7–S8 turn; large to small residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | − | − | +/− | 2JIX *, 6SHZ * | [20,79,84] |
| Y220H | S7–S8 turn; hydrophobic to cationic residue | Mild alteration of intermolecular interactions on -sandwich surface at S7–S8 turn, no crevice observed | ND | ND | ND | 6SI1 * | [84] |
| Y220N | S7–S8 turn; aromatic to aliphatic/bulky hydrophobic to uncharged polar residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | ND | ND | ND | ND | [84] |
| Y220S | S7–S8 turn; aromatic to aliphatic/bulky hydrophobic to small uncharged polar residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | ND | ND | ND | 6SI2 * | [84] |
| I232T | S8; hydrophobic to uncharged polar/large to small residue | ND | − | +/− | + | ND | [20] |
| I255F | S9; aliphatic to aromatic residue | ND | − | − | +/− | ND | [20] |
| F270C | S10; hydrophobic aromatic to uncharged polar/large to small residue | Internal hydrophobic cavity (theoretical interpretation) | − | − | +/− | ND | [20] |
| F270L | S10; aromatic to aliphatic/large to small residue | Internal hydrophobic cavity | ND | ND | ND | 2J1Z * | [79] |
5. Targeting Mutant p53
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riley, T.; Sontag, E.; Chen, P.A.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 1–13. [Google Scholar] [CrossRef]
- Chillemi, G.; Kehrloesser, S.; Bernassola, F.; Desideri, A.; Dötsch, V.; Levine, A.J.; Melino, G. Structural Evolution and Dynamics of the p53 Proteins. Cold Spring Harb. Perspect. Med. 2017, 7, a028308. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Chen, Y.; Petersen, R.B.; Zheng, L.; Huang, K. Salvation of the fallen angel: Reactivating mutant p53. Br. J. Pharmacol. 2019, 176, 817–831. [Google Scholar] [CrossRef]
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014, 588, 2628–2638. [Google Scholar] [CrossRef]
- Christophorou, M.A.; Martin-Zanca, D.; Soucek, L.; Lawlor, E.R.; Brown-Swigart, L.; Verschuren, E.; Evan, G.I. Temporal dissection of p53 function In Vitro and In Vivo. Nat. Genet. 2005, 37, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression In Vivo. Nat. Cell Biol. 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The Tumor Suppressor p53: From Structures to Drug Discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Rajagopalan, S.; Settanni, G.; Marsh, R.; Armoogum, D.A.; Nicolaou, N.; Bain, A.J.; Lerner, E.; Haas, E.; Ying, L.; et al. Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2009, 106, 20758–20763. [Google Scholar] [CrossRef]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.V.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef]
- Lubin, D.J.; Butler, J.S.; Loh, S.N. Folding of Tetrameric p53: Oligomerization and Tumorigenic Mutations Induce Misfolding and Loss of Function. J. Mol. Biol. 2010, 395, 705–716. [Google Scholar] [CrossRef]
- Tidow, H.; Veprintsev, D.; Freund, S.M.V.; Fersht, A.R. Effects of Oncogenic Mutations and DNA Response Elements on the Binding of p53 to p53-binding Protein 2 (53BP2). J. Biol. Chem. 2006, 281, 32526–32533. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-H.; Kim, T.J.; Lee, J.H.; Choi, J.-H. Mutant p53 stimulates cell invasion through an interaction with Rad21 in human ovarian cancer cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Rozenberg, H.; Diskin-Posner, Y.; Rohs, R.; Shakked, Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013, 41, 8748–8759. [Google Scholar] [CrossRef]
- Nikolova, P.V.; Henckel, J.; Lane, D.; Fersht, A.R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. USA 1998, 95, 14675–14680. [Google Scholar] [CrossRef] [PubMed]
- Suad, O.; Rozenberg, H.; Brosh, R.; Diskin-Posner, Y.; Kessler, N.; Shimon, L.; Frolow, F.; Liran, A.; Rotter, V.; Shakked, Z. Structural Basis of Restoring Sequence-Specific DNA Binding and Transactivation to Mutant p53 by Suppressor Mutations. J. Mol. Biol. 2009, 385, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Collavin, L.; Lunardi, A.; Del Sal, G. p53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010, 17, 901–911. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, M.R.; Sot, B. The relevance of protein-protein interactions for p53 function: The CPE contribution. Protein Eng. Des. Sel. 2010, 24, 41–51. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Maclaine, N.J.; Hupp, T.R. The regulation of p53 by phosphorylation: A model for how distinct signals integrate into the p53 pathway. Aging 2009, 1, 490–502. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Melero, R.; Rajagopalan, S.; Lázaro, M.; Joerger, A.; Brandt, T.; Veprintsev, D.; Lasso, G.; Gil, D.; Scheres, S.; Carazo, J.M.; et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef]
- Weinberg, L.R.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015, 16, 464. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Funk, W.D.; Pak, D.T.; Karas, R.H.; Wright, W.E.; Shay, J.W. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 1992, 12, 2866–2871. [Google Scholar] [CrossRef]
- Jordan, J.J. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008, 4, e1000104. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.; Pavletich, N. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D.; Noskov, S.Y.; Lim, C. Factors governing loss and rescue of DNA binding upon single and double mutations in the p53 core domain. Nucleic Acids Res. 2002, 30, 1563–1574. [Google Scholar] [CrossRef]
- DeLano, W. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Joerger, A.C.; Fersht, A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.C.; Fitzgerald, M.X.; Marmorstein, R. Structure of the p53 Core Domain Dimer Bound to DNA. J. Biol. Chem. 2006, 281, 20494–20502. [Google Scholar] [CrossRef]
- Kantarci, N.; Doruker, P.; Haliloglu, T. Cooperative Fluctuations Point to the Dimerization Interface of P53 Core Domain. Biophys. J. 2006, 91, 421–432. [Google Scholar] [CrossRef][Green Version]
- Ang, H.C.; Joerger, A.; Mayer, S.; Fersht, A.R. Effects of Common Cancer Mutations on Stability and DNA Binding of Full-length p53 Compared with Isolated Core Domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef]
- Friedler, A.; Veprintsev, D.B.; Hansson, L.O.; Fersht, A.R. Kinetic instability of p53 core domain mutants: Implications for rescue by small molecules. J. Biol. Chem. 2003, 278, 24108–24112. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Gutkowska, M.; Klejman, M.P.; Wawrzynow, B.; Tracz-Gaszewska, Z.; Wiech, M.; Zylicz, M.; Zylicz, A. ATP Binding to Hsp90 Is Sufficient for Effective Chaperoning of p53 Protein. J. Biol. Chem. 2010, 285, 32020–32028. [Google Scholar] [CrossRef]
- Walerych, D.; Olszewski, M.B.; Gutkowska, M.; Helwak, A.; Zylicz, M.; Zylicz, A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene 2009, 28, 4284–4294. [Google Scholar] [CrossRef]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1869, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A Master Decider of Cell Fate. Genes 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kohli, S.; Das, S. p53 regulation upon genotoxic stress: Intricacies and complexities. Mol. Cell. Oncol. 2014, 1, e969653. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhu, W.-G. Surf the Post-translational Modification Network of p53 Regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties onTP53mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; White, E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008, 22, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Maurer, U.; Green, D.R.; Schuler, M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 2003, 4, 371–381. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Ano Bom, A.P. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fersht, A.R. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA 2017, 114, E2634–E2643. [Google Scholar] [CrossRef]
- Mantovani, F.; Walerych, D.; Del Sal, G. Targeting mutant p53 in cancer: A long road to precision therapy. FEBS J. 2016, 284, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.; Allen, M.D.; Fersht, A.R. Crystal Structure of a Superstable Mutant of Human p53 Core Domain. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; May, P. Structural aspects of the p53 protein in relation to gene evolution: A second look. J. Mol. Biol. 1996, 260, 623–637. [Google Scholar] [CrossRef]
- Walker, D.R.; Bond, J.P.; Tarone, R.E.; Harris, C.C.; Makalowski, W.; Boguski, M.S.; Greenblatt, M.S. Evolutionary conservation and somatic mutation hotspot maps of p53: Correlation with p53 protein structural and functional features. Oncogene 1999, 18, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-B.; DeDecker, B.S.; Freund, S.M.V.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar] [CrossRef]
- Ng, J.W.K.; Lama, D.; Lukman, S.; Lane, D.; Verma, C.S.; Sim, A.Y.L. R248Q mutation-Beyond p53-DNA binding. Proteins Struct. Funct. Bioinform. 2015, 83, 2240–2250. [Google Scholar] [CrossRef]
- Joerger, A.; Ang, H.C.; Veprintsev, D.; Blair, C.M.; Fersht, A.R. Structures of p53 Cancer Mutants and Mechanism of Rescue by Second-site Suppressor Mutations. J. Biol. Chem. 2005, 280, 16030–16037. [Google Scholar] [CrossRef]
- Joerger, A.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef]
- Malcikova, J.; Tichy, B.; Damborsky, J.; Kabathova, J.; Trbusek, M.; Mayer, J.; Pospisilova, S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. Biol. Chem. 2010, 391, 197–205. [Google Scholar] [CrossRef]
- Gomes, A.S.; Trovão, F.; Pinheiro, B.A.; Freire, F.; Gomes, S.; Oliveira, C.; Domingues, L.; Romão, M.J.; Saraiva, L.; Carvalho, A.L. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. Int. J. Mol. Sci. 2018, 19, 1184. [Google Scholar] [CrossRef] [PubMed]
- Friedler, A.; DeDecker, B.S.; Freund, S.M.; Blair, C.; Rüdiger, S.; Fersht, A.R. Structural Distortion of p53 by the Mutation R249S and its Rescue by a Designed Peptide: Implications for “Mutant Conformation”. J. Mol. Biol. 2004, 336, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Coillie, S.V.; Fang, J.-Y.; Xu, J. Gain of function of mutant p53: R282W on the peak? Oncogenesis 2016, 5, e196. [Google Scholar] [CrossRef]
- Bauer, M.R.; Krämer, A.; Settanni, G.; Jones, R.N.; Ni, X.; Tareque, R.K.; Fersht, A.R.; Spencer, J.; Joerger, A.C. Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: The Y220X Paradigm. ACS Chem. Biol. 2020, 15, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef]
- Ryan, K.M.; Vousden, K.H. Characterization of Structural p53 Mutants Which Show Selective Defects in Apoptosis but Not Cell Cycle Arrest. Mol. Cell. Biol. 1998, 18, 3692–3698. [Google Scholar] [CrossRef]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 276–281. [Google Scholar] [CrossRef]
- Pintus, S.; Ivanisenko, N.; Demenkov, P.; Ramachandran, S.; Kolchanov, N.; Ivanisenko, V. The substitutions G245C and G245D in the Zn2+-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J. Biomol. Struct. Dyn. 2013, 31, 78–86. [Google Scholar] [CrossRef]
- Tu, C.; Tan, Y.-H.; Shaw, G.; Zhou, Z.; Bai, Y.; Luo, R.; Ji, X. Impact of low-frequency hotspot mutation R282Q on the structure of p53 DNA-binding domain as revealed by crystallography at 1.54 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 471–477. [Google Scholar] [CrossRef]
- Calhoun, S.; Daggett, V. Structural Effects of the L145Q, V157F, and R282W Cancer-Associated Mutations in the p53 DNA-Binding Core Domain. Biochemistry 2011, 50, 5345–5353. [Google Scholar] [CrossRef]
- Wallentine, B.D.; Wang, Y.; Tretyachenko-Ladokhina, V.; Tan, M.; Senear, D.F.; Luecke, H. Structures of oncogenic, suppressor and rescued p53 core-domain variants: Mechanisms of mutant p53 rescue. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Aramayo, R.; Sherman, M.B.; Brownless, K.; Lurz, R.; Okorokov, A.; Orlova, E.V. Quaternary structure of the specific p53–DNA complex reveals the mechanism of p53 mutant dominance. Nucleic Acids Res. 2011, 39, 8960–8971. [Google Scholar] [CrossRef]
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Nikolova, P.V.; Wong, K.-B.; Dedecker, B.; Henckel, J.; Fersht, A.R. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000, 19, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Baroni, T.E.; Wang, T.; Qian, H.; Dearth, L.R.; Truong, L.N.; Zeng, J.; Denes, A.E.; Chen, S.W.; Brachmann, R.K. A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. USA 2004, 101, 4930–4935. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, R.K.; Yu, K.; Eby, Y.; Pavletich, N.P.; Boeke, J.D. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 1998, 17, 1847–1859. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.-A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- NIH-ClinicalTrials. APR-246 & Azacitidine for the Treatment of TP53 Mutant Myelodysplastic Syndromes (MDS). 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03745716 (accessed on 11 April 2021).
- NIH-ClinicalTrials. Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Malignancies (COTI2-101). 2019. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02433626 (accessed on 11 April 2019).
- Foster, B.A. Pharmacological Rescue of Mutant p53 Conformation and Function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed]
- Madka, V.; Zhang, Y.; Li, Q.; Mohammed, A.; Sindhwani, P.; Lightfoot, S.; Wu, X.-R.; Kopelovich, L.; Rao, C.V. p53-stabilizing Agent CP-31398 Prevents Growth and Invasion of Urothelial Cancer of the Bladder in Transgenic UPII-SV40T Mice. Neoplasia 2013, 15, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Rippin, T.M.; Bykov, V.J.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Selivanova, G.; Wiman, K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: A statistical analysis of information in the National Cancer Institute database. Carcinogenesis 2002, 23, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2009, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Messina, R.L. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef]
- Aryee, D.N.T.; Niedan, S.; Ban, J.; Schwentner, R.; Muehlbacher, K.; Kauer, M.; Kofler, R.; Kovar, H. Variability in functional p53 reactivation by PRIMA-1Met/APR-246 in Ewing sarcoma. Br. J. Cancer 2013, 109, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Zhou, J.; Chan, Z.-L.; Chooi, J.-Y.; Chen, Z.-R.; Chng, W.-J. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 2015, 6, 36689–36699. [Google Scholar] [CrossRef]
- Zhang, W.; Yi, B.; Wang, C.; Chen, D.; Bae, S.; Wei, S.; Guo, R.-J.; Lu, C.; Nguyen, L.; Yang, W.-H.; et al. Silencing of CD24 Enhances the PRIMA-1–Induced Restoration of Mutant p53 in Prostate Cancer Cells. Clin. Cancer Res. 2016, 22, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Joerger, A.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef]
- Madan, E.; Parker, T.M.; Bauer, M.; Dhiman, A.; Pelham, C.J.; Nagane, M.; Kuppusamy, M.L.; Holmes, M.; Holmes, T.R.; Shaik, K.; et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018, 293, 4262–4276. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Ahmed, S.; Dayton, A.; Kuppusamy, M.L.; Tazi, M.; Bratasz, A.; Tong, L.; Rivera, B.K.; Kálai, T.; Hideg, K. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: Differential cytotoxicity in healthy and cancer cells. Free Radic. Biol. Med. 2010, 48, 1228–1235. [Google Scholar] [CrossRef]
- Kaar, J.L.; Basse, N.; Joerger, A.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-Specific p53 Mutant Reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef]
- Silver, N.; Osman, A.; Patel, A.; Tanaka, N.; Tang, L.; Zhou, G.; Myers, J. A Novel Third Generation Thiosemicarbazone, COTI-2, Is Highly Effective in Killing Head and Neck Squamous Cell Carcinomas (HNSCC) Bearing a Variety of TP53 Mutations. Int. J. Radiat. Oncol. 2016, 94, 942. [Google Scholar] [CrossRef]
- Salim, K.; Vareki, S.M.; Danter, W.; Koropatnick, J. COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur. J. Cancer 2016, 69, S19. [Google Scholar] [CrossRef]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines In Vitro and In Vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed]
- Gilleran, J.A.; Yu, X.; Blayney, A.J.; Bencivenga, A.F.; Na, B.; Augeri, D.J.; Blanden, A.R.; Kimball, S.D.; Loh, S.N.; Roberge, J.Y.; et al. Benzothiazolyl and Benzoxazolyl Hydrazones Function as Zinc Metallochaperones to Reactivate Mutant p53. J. Med. Chem. 2021, 64, 2024–2045. [Google Scholar] [CrossRef]
- Boeckler, F.; Joerger, A.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.; Bauer, M.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Roy, D.H.; Joerger, A.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef]
- Bauer, M.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Futur. Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wilcken, R.; Joerger, A.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Liu, X.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.; Boeckler, F.M. Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant p53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.; Monteiro, Â.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Gomes, S.; Loureiro, J.; Soares, J.; Barcherini, V.; Monti, P.; Fronza, G.; Oliveira, C.; Domingues, L.; et al. SLMP53-1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129440. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhuang, J.; Zhang, X. 2-[(4-Hydroxybenzyl) Amino] Phenol (HBAP) Restores the Mutated p53 to the Level Similar to That of Wild-Type p53 Protein and Inhibits Breast Cancer Growth in vivo to by Inducing Tumor Cells Apoptosis. Front. Cell Dev. Biol. 2020, 8, 574799. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.; Soares, M.I.; Silva, J.; Raimundo, L.; Calheiros, J.; Gomes, C.; Reis, F.; Monteiro, F.A.; Nunes, C.; Reis, S.; et al. A selective p53 activator and anticancer agent to improve colorectal cancer therapy. Cell Rep. 2021, 35, 108982. [Google Scholar] [CrossRef] [PubMed]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a Small Molecule Activator of Mutant p53, Which Binds p53 DNA Binding Domain (DBD), Restores Growth-suppressive Function to Mutant p53 and Interrupts HDM2-mediated Ubiquitination of Wild Type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Malami, I.; Muhammad, A.; Etti, I.C.; Waziri, P.M.; Alhassan, A.M. An In Silico approach in predicting the possible mechanism involving restoration of wild-type p53 functions by small molecular weight compounds in tumor cells expressing R273H mutant p53. EXCLI J. 2017, 16, 1276–1287. [Google Scholar] [CrossRef]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef]
- Friedler, A.; Hansson, L.O.; Veprintsev, D.; Freund, S.M.V.; Rippin, T.M.; Nikolova, P.V.; Proctor, M.R.; Rüdiger, S.; Fersht, A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 2002, 99, 937–942. [Google Scholar] [CrossRef]
- Issaeva, N.; Friedler, A.; Bozko, P.; Wiman, K.; Fersht, A.R.; Selivanova, G. Rescue of mutants of the tumor suppressor p53 in cancer cells by a designed peptide. Proc. Natl. Acad. Sci. USA 2003, 100, 13303–13307. [Google Scholar] [CrossRef]
- Hiraki, M.; Hwang, S.-Y.; Cao, S.; Ramadhar, T.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef]
- Gomes, S.; Bosco, B.; Loureiro, J.B.; Ramos, H.; Raimundo, L.; Soares, J.; Nazareth, N.; Barcherini, V.; Domingues, L.; Oliveira, C.; et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers 2019, 11, 1151. [Google Scholar] [CrossRef]
- Peng, Y.; Li, C.; Chen, L.; Sebti, S.; Chen, J. Rescue of mutant p53 transcription function by ellipticine. Oncogene 2003, 22, 4478–4487. [Google Scholar] [CrossRef]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; DasMahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef]
- North, S.; Pluquet, O.; Maurici, D.; El-Ghissassi, F.; Hainaut, P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol. Carcinog. 2002, 33, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Prabhu, V.V.; Zhang, S.; Heuvel, A.P.J.V.D.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin Rescues Deficient p53 Signaling and Antitumor Effects via Upregulating p73 and Disrupting Its Interaction with Mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, A.P.A.R.S.V.I.T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.-H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, T.Q.A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nat. Cell Biol. 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Toretsky, J.; Neckers, L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995, 11, 933–939. [Google Scholar]
- Wang, C.; Chen, J. Phosphorylation and hsp90 Binding Mediate Heat Shock Stabilization of p53. J. Biol. Chem. 2003, 278, 2066–2071. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-T.; Chen, Z.-J.; Jiang, G.-M.; Wu, Y.-M.; Liu, T.; Yi, Y.-M.; Zeng, J.; Du, J.; Wang, H.-S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell. Signal. 2016, 28, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, Y.; Zhang, J.; Liu, S.; Cho, S.J.; Chen, X. Mutant p53 Protein Is Targeted by Arsenic for Degradation and Plays a Role in Arsenic-mediated Growth Suppression. J. Biol. Chem. 2011, 286, 17478–17486. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Q.; Qi, Q.; Gu, H.-Y.; Rong, J.-J.; Mu, R.; Zou, M.-J.; Tao, L.; You, Q.-D.; Guo, Q.-L. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J. Cell. Biochem. 2011, 112, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 718–730. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting Mutant p53 by a SIRT1 Activator YK-3-237 Inhibits the Proliferation of Triple-Negative Breast Cancer Cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, L.; Hong, B.; Heuvel, A.P.J.V.D.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.B.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef]
- Demir, Ö.; Barros, E.P.; Offutt, T.L.; Rosenfeld, M.; Amaro, R.E. An integrated view of p53 dynamics, function, and reactivation. Curr. Opin. Struct. Biol. 2021, 67, 187–194. [Google Scholar] [CrossRef]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.; De Oliveira, G.A.P. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2017, 51, 181–190. [Google Scholar] [CrossRef]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef] [PubMed]


| Chemical Name (Class) | Discovery Strategy | Targeted * Mutp53 | Mechanism of Action | Observations | Ref. |
|---|---|---|---|---|---|
| Reactivators—Cysteine-targeting alkylation | |||||
| CP-31398 (Styrylquinazoline)  | Chemical library; protein-based screening assay | V173A; S241F; R249S; R273H | Michael addition # Binding: ND | Transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [104,105,106,107] |
| MIRA-1 (Maleimide)  | Chemical library; cellular screening assay | R175H; P176Y/R248W; R248Q; R248W; R273H; R273H/P309S; R280K; R282W | Michael addition # Binding: ND | Rescue of wt conformation to mutp53 R248W, R175H; restoration of DNA binding to mutp53 R175H, R248Q, P176Y/R248W, R280K, R282W; restoration of transcriptional activity to mutp53 R175H, R273H, R273H/P309S; p53-dependent and -independent in vitro and in vivo antitumor activity | [108] |
| STIMA-1 (Styrylquinazoline)  | Chemical library; cellular screening assay | R175H; R273H | Michael addition # Binding: ND | Restoration of DNA binding to mutp53 R175H; restoration of transcriptional activity to mutp53 R175H, R273H; p53-dependent and -independent in vitro antitumor activity | [107] |
| KSS-9 (Piperlongumine derivative)  | Rational design | R175H | Michael addition # Binding: ND | Rescue of wt conformation; restoration of DNA binding to mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [109] |
| PRIMA-1 PRIMA-1MET (Quinuclidinone)  | Chemical library; cellular screening assay | R175H; R273H; D259Y/K286E; K286E; S241F; R273C; P223L/V274F | Metabolized to methylene quinuclidinone (active metabolite), Michael addition Binding: mutp53 R175H, R273H | Enhanced thermal stability of wtp53, mutp53 R175H, R273H; rescue of wt conformation to mutp53 R175H; restoration of transcriptional activity to mutp53 R175H, R273H, D259Y/K286E, K286E, S241F, R273C, P223L/V274F; p53-dependent and -independent in vitro and in vivo antitumor activity | [110,111,112,113,114,115,116,117,118,119] |
| PK11007 (Sulfonylpyrimidine)  | Chemical library; protein thermal stability-based screening assay | Y220C; V143A | Binds p53 by nucleophilic aromatic substitution Binding: mutp53 Y220C | Enhanced thermal stability of mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [120] |
| HO-3867 (Diarylidenyl piperidone curcumin analogue)  | Chemical library; cellular screening assay | K132Q; R156P; Y163H; R175H; H193R; L194F; Y205F; P223L/V274F; C238Y; N239D; S241F; G245S; G245V; M246I; R248Q; R248W; R249S; R273H; C277F; R280K; E285K | Binds p53 by Michael addition Binding: mutp53 Y220C | Rescue of wt conformation to mutp53 P223L/V274F, R273H, R280K; restoration of transcriptional activity to mutp53 K132Q, R156P, Y163H, R175H, H193R, L194F, Y205F, C238Y, N239D, S241F, G245S, G245V, M246I, R248W, R248Q, R249S, R273H, C277F, R280K, E285K; p53-dependent in vitro and in vivo antitumor activity | [121,122] |
| Thermal stabilizers | |||||
| 3-Benzoylacrylic acid (Benzoylacrylate)  | Chemical library; protein thermal stability-based screening assay | Y220C; R175H; G245D; R249S; R282W | Michael addition Binding: mutp53 Y220C | Enhanced thermal stability of mutp53 Y220C, R175H, G245D, R249S, R282W; absence of in vitro antitumor activity evaluation | [123] |
| Reactivators—Zinc chelators | |||||
| ZMC1/NSC319726 (Thiosemicarbazone)  | Database analysis; cellular screening assay | R175H; C176F; C238S; C242S; C242F; G245S | Zn2+ chelator Binding: mutp53 R175H | Increased cellular zinc concentration; rescue of wt conformation to mutp53 R175H, C176F, C238S, C242S; restoration of transcriptional activity to mutp53 R175H, C176F, C238S, C242S, G245S; p53-dependent and -independent in vitro and in vivo antitumor activity | [124,125] |
| COTI-2 (Thiosemicarbazone)  | Rational drug design; virtual drug screening | R175H | Zn2+ chelator Binding: ND | p53-dependent and -independent in vitro and in vivo antitumor activity | [126,127,128] |
| Benzothiazolyl, Benzoxazolyl Hydrazones (C85)  | Chemical library; biophysical and cellular screening assay | R175H | Zn2+ chelator Binding: ND | Increased cellular zinc concentration; rescue of wt conformation to mutp53 R175H, zinc-deficient p53-dependent in vitro and in vivo antitumor activity | [129] |
| Reactivators—Non-covalent binding | |||||
| PK083 and analogs (Carbazole)  | Rational drug design; virtual drug screening | Y220C, Y220N, Y220S | Binds to a cleft in C-terminal of mutp53 Y220C, Y220N, and Y220S DBD | Enhanced thermal stability (Y220C, Y220N, Y200S); rescue of wt conformation to mutp53 (PK083-Y220C); inhibition of aggregation (PK9318 analog-Y220C); transcription of p53 target genes (PK9318 analog-Y220C); p53-dependent in vitro antitumor activity | [84,130,131,132] |
| PK7088 (Pyrazole)  | Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; rescue of wt conformation to mutp53; transcription of p53 target genes; weak in vitro antitumor activity | [131,133] |
| PK5196 (Halogen-phenol derivative)  | Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; in vitro antitumor activity | [131,134] |
| MB725 (Aminobenzothiazole)  | Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; transcription of p53 target genes; p53-dependent in vitro antitumor activity | [131] |
| SLMP53-1 (Tryptophan-derived isoindolinone)  | Chemical library; yeast-targeted screening assay | R175H, G245D, R248Q, R248W, R273H, R280K, R282W | Binds to mutp53 R280K DBD; in silico proposes that SLMP53-1 bridges DNA-binding surface of mutp53 R280K to DNA minor groove | Enhanced thermal stability of wt- and mutp53 R280K; restoration of DNA binding to mutp53 R280K; restoration of transcriptional activity to mutp53 R280K; p53-dependent in vitro and in vivo antitumor activity | [135,136] |
| HBAP (2-[(4-hydroxybenzyl) amino]phenol)  | Chemical library; cellular screening assay | R280K; R273H | Binds to mutp53 R280K and R273H DBD | Transcription of p53 target genes; in vitro and in vivo antitumor activity | [137] |
| MANIO (Thiazole derivative)  | Chemical library; cellular screening assay | Y126C; R175H; G245D; G245S; R248Q; R248W; R280K; R282W; R273C; R273H | Binds to wt- and mutp53 R248W DBD; in silico proposes that MANIO fits between the DNA molecule and the protein pocket at the dimer interface | Enhanced thermal stability of wt- and mutp53 R248W, Y126C, and R273H; increase of protein DNA-binding ability; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [138] |
| SCH529074 (Piperazinylquinazoline)  | Chemical library; DNA-binding assay | R175H; S241F; R248W; R249S; R273H | Binds to wtp53 nearby DNA binding surface | Protected wtp53 conformation from thermal denaturation; rescue of wt conformation to mutp53 S241F, R248W, R273H; restoration of DNA binding to mutp53 R175H, R249S, R273H; transcription of p53 target genes by mutp53s R175H, S241F R248W, R249S, R273H; blocked MDM2-mediated ubiquitination of p53; p53-dependent in vitro and in vivo antitumor activity | [139] |
Stictic acid | Chemical library; virtual screening | R175H; G245S | In silico binding to wtp53 and mutp53 R175H, R273H, G245S to a transiently open pocket (L1/S3) | Enhanced thermal stability of mutp53 R175H, G245S; transcription of p53 target genes by mutp53 R175H, G245S; p53-dependent in vitro antitumor activity | [140] |
Curcumin, Flavokawain B, Alpinetin Curcumin  Flavokawain B  Alpinetin | Crude extracts; cellular screening assay | R273H | In silico binding to mutp53 R273H bridging DNA-binding surface to DNA sequence | p53-dependent and -independent in vitro antitumor activity | [141] |
| pCAPs (Peptides) pCAP221 sequence: RRKHNKHRPEPDSDER pCAP242 sequence: RRLIVRILKLPNPPER pCAP250 sequence: RRHSTPHPD | Phage peptide display-protein screening assay | V135A; S241F; R249S; R280K | Binding to unknown local of mutp53 R175H and R249S | Rescue of wt conformation to mutp53 R175H, R249S; restoration of DNA binding to mutp53 R175H, R249S; transcription of p53 target genes by mutp53 V135A, S241F, R280K; p53-dependent in vitro and in vivo antitumor activity | [142] |
| CDB3 (Peptide) Sequence: REDEDEIEW | Rational drug design; NMR protein-based screening assay | R175H; I195T; R249S; R273H | Binding to wt and mutp53 R249S, R273H, nearby DNA binding surface and G245S, R175H unknown local | Enhanced thermal stability of wt and mutp53s R175H, R249S, R273H; rescue of wt conformation to mutp53 R175H, R249S, R273H; enhanced DNA binding to mutp53 I195T; transcription of p53 target genes by mutp53 R175H, R273H; p53-dependent in vitro antitumor activity | [82,143,144] |
| Reactivators—Chaperone-mediated effect | |||||
| Chetomin (Epidithiodioxopiperazine)  | Natural products database; cellular luciferase reporter screening assay | R175H | Binds to HSP40 | Rescue of wt conformation to mutp53; transcription of p53 target genes; MDM2 negative regulation; p53-dependent and -independent in vitro and in vivo antitumor activity | [145] |
| SLMP53-2 (Tryptophanol-derived oxazoloisoindolinone)  | Chemical library; cellular screening assay | Y220C | Enhances the mutp53 Y220C interaction with HSP70 | Rescue of wt conformation to mutp53; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [146] |
| Reactivators—Unknown binding | |||||
| Ellipticine (Alkaloid)  | Chemical library; cellular screening assay | R175H; L194F; S241F; R249S; R273C; R273H | ND | Restoration of DNA binding to mutp53 R175H, S241F; rescue of wt conformation to mutp53 S241F; restoration of transcriptional activity to mup53 R175H; L194F, S241F, R249S, R273C, R273H; p53-dependent in vitro antitumor activity | [147] |
| P53R3 (Piperazinylquinazoline)  | Chemical library; DNA-binding assay | M237I; R175H; R273H R248W | ND | Restoration of DNA binding to mutp53 R175H, M237I, R273H; restoration of transcriptional activity to mup53 M237I; p53-dependent in vitro antitumor activity | [148] |
| PEITC (Phenethyl isothiocyanate)  | Cellular screening assay | R175H | ND | Rescue of wtp53 conformation to mutp53; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [149] |
| WR-1065 (Aminothiol)  | Active metabolite of amifostine; cellular screening assay | V272M | ND | Restoration of DNA binding; rescue of wt conformation to mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [150] |
| Disruptors of protein-protein interaction | |||||
| RETRA (Thiazolthiophenyl ethanone)  | Chemical library; cellular screening assay | R273H | Disrupts mutp53-TAp73 complexes | Increased TAp73 expression; transcription of p53-shared target genes; TAp73-dependent in vitro and in vivo antitumor activity | [151] |
| Prodigiosin (Pyrrolyl pyrromethane)  | Chemical library; cellular screening assay | R273H; S241F; R248Q | Disrupts mutp53-p73 complexes | Transcription of p53-shared target genes; p73-dependent in vitro antitumor activity | [152] |
| LEM2 (Xanthone)  | Chemical library; yeast-targeted screening assay | R273H | Disrupts mutp53-TAp73 and MDM2-TAp73 complexes | Enhanced thermal stability of TAp73; transcription of TAp73- and p53-shared target genes; TAp73-dependent in vitro antitumor activity | [153] |
| Statins (Lovastatin)  | Cellular screening assay | R156P; V157F; R175H; Y220C; R248W | Inhibition of the mevalonate pathway, with CHIP-mediated mutp53 degradation | In vitro and in vivo suppression of mutp53-expressing cancer cell growth | [154] |
| ATRA (Retinoic acid; tretinoin)  | Chemical library; mechanism-based screening assay (protein active site) | R273H; R280K | Disrupts mutp53-Pin1 interaction (via Pin1 inhibition and degradation) | In vitro and in vivo antitumor activity | [155] |
| ReACp53 (Peptide) Sequence: RRRRRRRRRRPILTRITLE | Structure-based rational design; cellular screening assay | R175H; R248Q | Binds to mutp53 aggregation prone region (S9) | Inhibits mutp53 aggregates; shifts the folding equilibrium toward the wt conformation; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [156] |
| Inducers of mutp53 degradation | |||||
| 17-AAG; 17-DMAG (Demethoxygeldanamycin derivatives)  17-AAG  17-DMAG | HSP90 inhibitor; cellular evaluation | L194F; R273H; R273H/P309S; R280K | Inhibits HSP90 with increase of MDM2 and CHIP function | In vivo antitumor activity in synergism with SAHA | [157,158] |
| Geldanamycin (Benzoquinone ansanamycin)  | HSP90 inhibitor; cellular evaluation | R175H; L194F; R248Q; R273H; R280K; R172H (mouse) | Inhibits HSP90 with increase of MDM2 and CHIP function | In vitro antitumor activity | [159,160] |
| Ganetespib (Phenylindolyl triazolone)  | HSP90 inhibitor; cellular evaluation | C124R; R172H; L194F; S241F; R248Q; R273H; C275F | Inhibits HSP90 with mutp53 degradation | In vivo and in vitro antitumor activity | [158] |
| SAHA (Suberoyl-anilide hydroxamic acid)  | HDAC inhibitor; cellular evaluation | L194F; P223L/V274F; R249S; R273H; R273H/P309S; R280K | Inhibits HDAC6/8 (HSP90 machinery) with mutp53 CHIP-ubiquitin/proteasome-mediated degradation; decreases association with YY-1 transcription factor halting GOF | In vivo and in vitro antitumor activity | [158,161,162,163] |
Sodium butyrate | HDAC inhibitor; cellular evaluation | R249S; R280K | Inhibits HDAC8 (HSP90 machinery) with decreased association with YY-1 transcription factor halting GOF | In vitro antitumor activity | [163] |
Arsenic trioxide | Cellular evaluation | R175H; H179Y/R282W; R248W; R270H; R273H; R273H/P309S | Induces mutp53 nuclear proteasome-mediated degradation | Besides inducing mutp53 degradation, arsenic compounds stabilized wtp53 levels in cancer cells; in vitro antitumor activity | [164] |
| Gambogic acid (Xanthone)  | Cellular evaluation | R175H; G266E; R273H; R280K | Depletes mutp53 via HSP90-CHIP ubiquitin/proteasome-mediated degradation | In vitro antitumor activity | [165] |
| Spautin-1 (Fluorobenzylquinazolin amine)  | USP inhibitor; cellular evaluation | P98S; P151H; S158inF; A161T; R175C/D/H; L194F; S227K/R; S241F; G245C; R248L/Q/W; E258K; G266E; R273H/L; R280K; R282W | Inhibits deubiquitinating enzymes leading to mutp53 lysosome-mediated degradation | In vitro antitumor activity | [166] |
| YK-3-237 (Chalcone)  | Chemical library; cellular evaluation | V157F; M237I; R249S; R273H; R280K | Activates deacetylase SIRT1 reducing p53 levels | Transcription of p53 target genes; p53-dependent in vitro antitumor activity | [167] |
| NSC59984 (Methylpiperazinylnitrofuranyl propenone)  | Chemical library; p53-reporter gene cellular screen | R175H/L; S241F; R273H/P309F | Induces MDM2-ubiquitin-proteasome-mediated degradation | Transcription of p53 target genes; p73-dependent in vitro and in vivo antitumor activity | [168] |
| Disulfiram (Tetraethylthiuram disulfide)  | Cellular evaluation | R273H | Thiol-conjugation with mutp53 proteasome-mediated degradation | In vivo and in vitro antitumor activity | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. https://doi.org/10.3390/cancers13133344
Gomes AS, Ramos H, Inga A, Sousa E, Saraiva L. Structural and Drug Targeting Insights on Mutant p53. Cancers. 2021; 13(13):3344. https://doi.org/10.3390/cancers13133344
Chicago/Turabian StyleGomes, Ana Sara, Helena Ramos, Alberto Inga, Emília Sousa, and Lucília Saraiva. 2021. "Structural and Drug Targeting Insights on Mutant p53" Cancers 13, no. 13: 3344. https://doi.org/10.3390/cancers13133344
APA StyleGomes, A. S., Ramos, H., Inga, A., Sousa, E., & Saraiva, L. (2021). Structural and Drug Targeting Insights on Mutant p53. Cancers, 13(13), 3344. https://doi.org/10.3390/cancers13133344