
Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer—Current Use and Future Opportunities




Abstract
Simple Summary
Abstract
1. Introduction
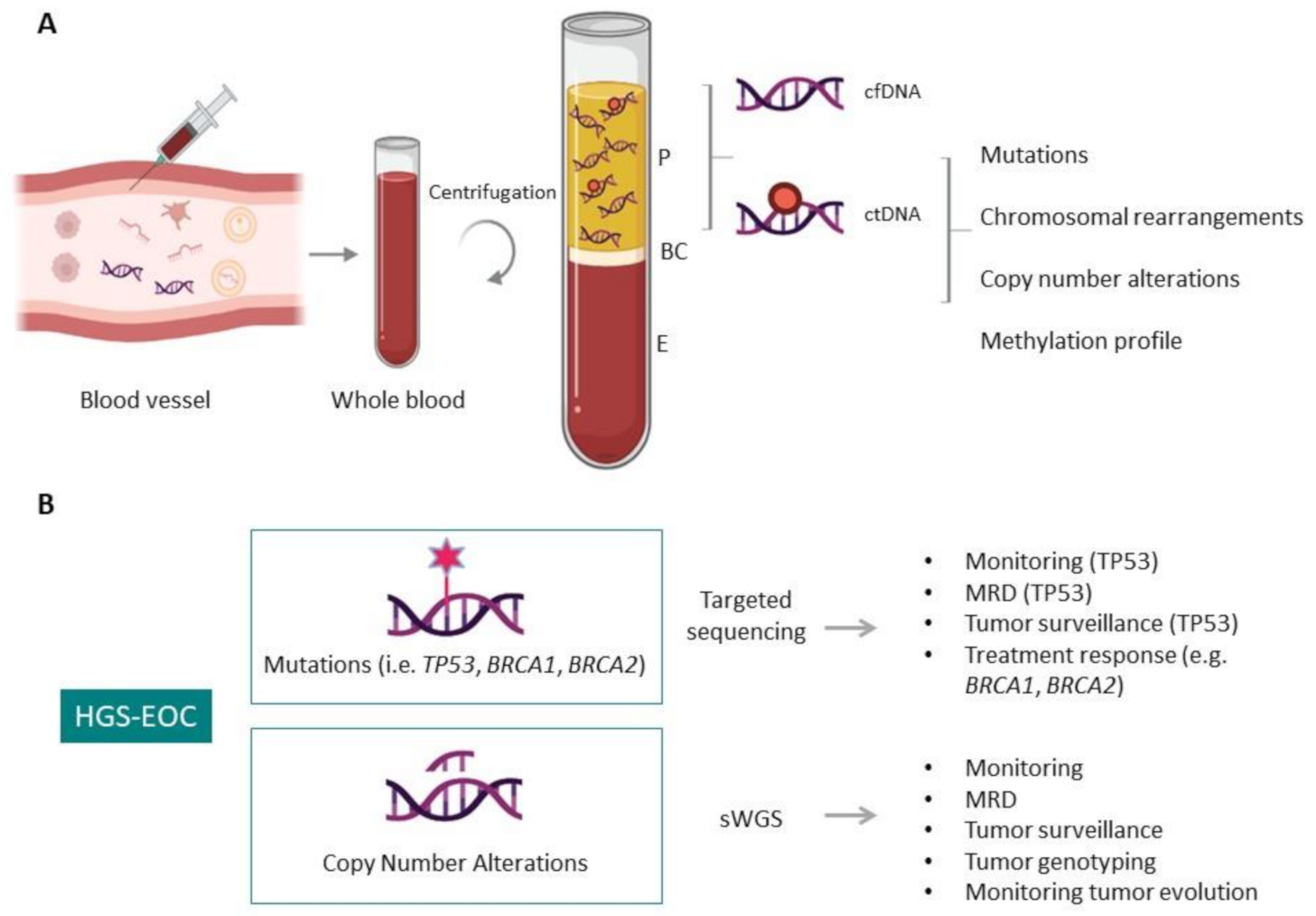
2. Liquid Biopsy: Potentially Useful Marker
3. Liquid Biopsy in Ovarian Cancer
4. CTCs, EVs and cfmiRNAs in HGS-EOC
5. Circulating-Free and Circulating Tumor DNA
6. ctDNA Analysis: The State of the Art for Ovarian Cancer
6.1. Somatic Mutation Detection
6.2. Structural Aberration Detection
7. Conclusion and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Schilsky, R.L. Implementing personalized cancer care. Nat. Rev. Clin. Oncol. 2014, 11, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.; Lin, T.-Y.; Toska, E.; et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Knelson, E.H.; Sequist, L.V. A bright future for KRAS inhibitors. Nat. Rev. Cancer 2020, 1, 25–27. [Google Scholar] [CrossRef]
- Hamid, O.; Cowey, C.L.; Offner, M.; Faries, M.; Carvajal, R.D. Efficacy, safety, and tolerability of approved combination BRAF and MEK inhibitor regimens for BRAF-mutant melanoma. Cancers 2019, 11, 1642. [Google Scholar] [CrossRef]
- Solassol, I.; Pinguet, F.; Quantin, X. FDA- and EMA-approved tyrosine kinase inhibitors in advanced EGFR-mutated non-small cell lung cancer: Safety, tolerability, plasma concentration monitoring, and management. Biomolecules 2019, 9, 668. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Mills, G.B. Overcoming implementation challenges of personalized cancer therapy. Nat. Rev. Clin. Oncol. 2012, 9, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Levine, R.L. Tumor heterogeneity confounds and illuminates: Assessing the implications. Nat. Med. 2014, 20, 342–344. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Beltrame, L.; Di Marino, M.; Fruscio, R.; Calura, E.; Chapman, B.; Clivio, L.; Sina, F.; Mele, C.; Iatropoulos, P.; Grassi, T.; et al. Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: A Retrospective study. Ann. Oncol. 2015, 26, 1363–1371. [Google Scholar] [CrossRef]
- Paracchini, L.; Mannarino, L.; Craparotta, I.; Romualdi, C.; Fruscio, R.; Grassi, T.; Fotia, V.; Caratti, G.; Perego, P.; Calura, E.; et al. Regional and temporal heterogeneity of epithelial ovarian cancer tumor biopsies: Implications for therapeutic strategies. Oncotarget 2016, 5. [Google Scholar] [CrossRef]
- Ballabio, S.; Craparotta, I.; Paracchini, L.; Mannarino, L.; Corso, S.; Pezzotta, M.G.; Vescio, M.; Fruscio, R.; Romualdi, C.; Dainese, E.; et al. Multisite analysis of high-grade serous epithelial ovarian cancers identifies genomic regions of focal and recurrent copy number alteration in 3q26.2 and 8q24.3. Int. J. Cancer 2019, 145, 2670–2681. [Google Scholar] [CrossRef]
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef]
- Duffy, M.J. Tumor markers in clinical practice: A review focusing on common solid cancers. Med. Princ. Pract. 2013, 22, 4–11. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Frenkel, E.P.; Shete, S.; Naftalis, E.Z.; Huth, J.F.; Beitsch, P.D.; Leitch, M.; Hoover, S.; Euhus, D.; et al. Circulating tumor cells in patients with breast cancer dormancy. Clin. Cancer Res. 2004, 10, 8152–8162. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.; Siu, L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef]
- Mader, S.; Pantel, K. Liquid biopsy: Current status and future perspectives. Oncol. Res. Treat. 2017, 40, 404–408. [Google Scholar] [CrossRef]
- Marrugo-Ramírez, J.; Mir, M.; Samitier, J. Blood-based cancer biomarkers in liquid biopsy: A promising non-invasive alternative to tissue biopsy. Int. J. Mol. Sci. 2018, 19, 2877. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Heong, V.; Ye, J.; Lim, D.; Low, J.; Choolani, M.; Scott, C.; Tan, D.S.P.; Huang, R.Y.-J. Decoding transcriptomic intra-tumour heterogeneity to guide personalised medicine in ovarian cancer. J. Pathol. 2019, 247, 305–319. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; DʹAvola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Song, J.-L.; Chen, C.; Yuan, J.-P.; Sun, S.-R. Progress in the clinical detection of heterogeneity in breast cancer. Cancer Med. 2016, 5, 3475–3488. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.-M.; Lheureux, S.; Liu, J.F.; et al. PARP Inhibitors in the management of ovarian cancer: ASCO guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.; George, A.; Banerjee, S. PARP inhibitors in ovarian cancer: An overview of the practice-changing trials. Genes Chromosomes Cancer 2021, 60, 385–397. [Google Scholar] [CrossRef]
- Buamah, P. Benign conditions associated with raised serum CA-125 concentration. J. Surg. Oncol. 2000, 75, 264–265. [Google Scholar] [CrossRef]
- Meyer, T.; Rustin, G.J.S. Role of tumour markers in monitoring epithelial ovarian cancer. Br. J. Cancer 2000, 82, 1535–1538. [Google Scholar] [CrossRef]
- Sölétormos, G.; Duffy, M.J.; Abu Hassan, S.O.; Verheijen, R.H.M.; Tholander, B.; Bast, R.C.; Gaarenstroom, K.N.; Sturgeon, C.M.; Bonfrer, J.M.; Petersen, P.H.; et al. clinical use of cancer biomarkers in epithelial ovarian cancer: Updated guidelines from the european group on tumor markers. Int. J. Gynecol. Cancer 2016, 26, 43–51. [Google Scholar] [CrossRef]
- Testa, A.C.; Di Legge, A.; Bonatti, M.; Manfredi, R.; Scambia, G. Imaging techniques for evaluation of uterine myomas. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 34, 37–53. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; deFazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef]
- Marth, C.; Kisic, J.; Kaern, J.; Tropé, C.; Fodstad, Ø. Circulating tumor cells in the peripheral blood and bone marrow of patients with ovarian carcinoma do not predict prognosis. Cancer 2002, 94, 707–712. [Google Scholar] [CrossRef]
- Judson, P.L.; Geller, M.A.; Bliss, R.L.; Boente, M.P.; Downs, L.S.; Argenta, P.A.; Carson, L.F. Preoperative detection of peripherally circulating cancer cells and its prognostic significance in ovarian cancer. Gynecol. Oncol. 2003, 91, 389–394. [Google Scholar] [CrossRef]
- Aktas, B.; Kasimir-Bauer, S.; Heubner, M.; Kimmig, R.; Wimberger, P. Molecular profiling and prognostic relevance of circulating tumor cells in the blood of ovarian cancer patients at primary diagnosis and after platinum-based chemotherapy. Int. J. Gynecol. Cancer 2011, 21, 822–830. [Google Scholar] [CrossRef]
- Poveda, A.; Kaye, S.B.; McCormack, R.; Wang, S.; Parekh, T.; Ricci, D.; Lebedinsky, C.A.; Tercero, J.C.; Zintl, P.; Monk, B.J. Circulating tumor cells predict progression free survival and overall survival in patients with relapsed/recurrent advanced ovarian cancer. Gynecol. Oncol. 2011, 122, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Obermayr, E.; Castillo-Tong, D.C.; Pils, D.; Speiser, P.; Braicu, I.; Van Gorp, T.; Mahner, S.; Sehouli, J.; Vergote, I.; Zeillinger, R. Molecular characterization of circulating tumor cells in patients with ovarian cancer improves their prognostic significance—A study of the OVCAD consortium. Gynecol. Oncol. 2013, 128, 15–21. [Google Scholar] [CrossRef]
- Chebouti, I.; Kuhlmann, J.D.; Buderath, P.; Weber, S.; Wimberger, P.; Bokeloh, Y.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. ERCC1-expressing circulating tumor cells as a potential diagnostic tool for monitoring response to platinum-based chemotherapy and for predicting post-therapeutic outcome of ovarian cancer. Oncotarget 2016, 8, 24303–24313. [Google Scholar] [CrossRef] [PubMed]
- Obermayr, E.; Bednarz-Knoll, N.; Orsetti, B.; Weier, H.-U.; Lambrechts, S.; Castillo-Tong, D.C.; Reinthaller, A.; Braicu, E.I.; Mahner, S.; Sehouli, J.; et al. Circulating tumor cells: Potential markers of minimal residual disease in ovarian cancer? A study of the OVCAD consortium. Oncotarget 2017, 8, 106415–106428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, H.; Yu, X.; Li, S.; Lei, Z.; Li, C.; Zhang, Q.; Han, Q.; Li, Y.; Zhang, K.; et al. Analysis of circulating tumor cells in ovarian cancer and their clinical value as a biomarker. Cell. Physiol. Biochem. 2018, 48, 1983–1994. [Google Scholar] [CrossRef]
- Kolostova, K.; Matkowski, R.; Jędryka, M.; Soter, K.; Cegan, M.; Pinkas, M.; Jakabova, A.; Pavlasek, J.; Spicka, J.; Bobek, V. The added value of circulating tumor cells examination in ovarian cancer staging. Am. J. Cancer Res. 2015, 5, 3363–3375. [Google Scholar]
- Kolostova, K.; Pinkas, M.; Jakabova, A.; Pospisilova, E.; Svobodova, P.; Spicka, J.; Cegan, M.; Matkowski, R.; Bobek, V. Molecular characterization of circulating tumor cells in ovarian cancer. Am. J. Cancer Res. 2016, 6, 973–980. [Google Scholar]
- Guo, Y.-X.; Neoh, K.H.; Chang, X.-H.; Sun, Y.; Cheng, H.-Y.; Ye, X.; Ma, R.-Q.; Han, R.P.S.; Cui, H. Diagnostic value of HE4+ circulating tumor cells in patients with suspicious ovarian cancer. Oncotarget 2018, 9, 7522–7533. [Google Scholar] [CrossRef]
- Kuhlmann, J.D.; Wimberger, P.; Bankfalvi, A.; Keller, T.; Schöler, S.; Aktas, B.; Buderath, P.; Hauch, S.; Otterbach, F.; Kimmig, R.; et al. ERCC1-positive circulating tumor cells in the blood of ovarian cancer patients as a predictive biomarker for platinum resistance. Clin. Chem. 2014, 60, 1282–1289. [Google Scholar] [CrossRef]
- Pan, C.; Stevic, I.; Müller, V.; Ni, Q.; Oliveira-Ferrer, L.; Pantel, K.; Schwarzenbach, H. Exosomal MicroRNAs as tumor markers in epithelial ovarian cancer. Mol. Oncol. 2018, 12, 1935–1948. [Google Scholar] [CrossRef]
- Zhang, W.; Ou, X.; Wu, X. Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis. Int. J. Oncol. 2019, 54, 1719–1733. [Google Scholar] [CrossRef]
- Schwich, E.; Rebmann, V.; Horn, P.A.; Celik, A.A.; Bade-Döding, C.; Kimmig, R.; Kasimir-Bauer, S.; Buderath, P. Vesicular-bound HLA-G as a predictive marker for disease progression in epithelial ovarian cancer. Cancers 2019, 11, 1106. [Google Scholar] [CrossRef]
- Resnick, K.E.; Alder, H.; Hagan, J.P.; Richardson, D.L.; Croce, C.M.; Cohn, D.E. The detection of differentially expressed micrornas from the serum of ovarian cancer patients using a novel real-time PCR platform. Gynecol. Oncol. 2009, 112, 55–59. [Google Scholar] [CrossRef]
- Todeschini, P.; Salviato, E.; Paracchini, L.; Ferracin, M.; Petrillo, M.; Zanotti, L.; Tognon, G.; Gambino, A.; Calura, E.; Caratti, G.; et al. Circulating MiRNA landscape identifies MiR-1246 as promising diagnostic biomarker in high-grade serous ovarian carcinoma: A validation across two independent cohorts. Cancer Lett. 2017, 388, 320–327. [Google Scholar] [CrossRef]
- Swisher, E.M.; Wollan, M.; Mahtani, S.M.; Willner, J.B.; Garcia, R.; Goff, B.A.; King, M.-C. Tumor-specific P53 sequences in blood and peritoneal fluid of women with epithelial ovarian cancer. Am. J. Obstet. Gynecol. 2005, 193, 662–667. [Google Scholar] [CrossRef]
- Otsuka, J.; Okuda, T.; Sekizawa, A.; Amemiya, S.; Saito, H.; Okai, T.; Kushima, M. Detection of P53 mutations in the plasma dna of patients with ovarian cancer. Int. J. Gynecol. Cancer 2004, 14, 459–464. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory analysis of TP53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high-grade serous ovarian carcinoma: A retrospective study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef]
- Kim, Y.M.; Lee, S.W.; Lee, Y.J.; Lee, H.Y.; Lee, J.E.; Choi, E.K. Prospective study of the efficacy and utility of TP53 mutations in circulating tumor DNA as a non-invasive biomarker of treatment response monitoring in patients with high-grade serous ovarian carcinoma. J. Gynecol. Oncol. 2019, 30, e32. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef]
- Vanderstichele, A.; Busschaert, P.; Smeets, D.; Landolfo, C.; Van Nieuwenhuysen, E.; Leunen, K.; Neven, P.; Amant, F.; Mahner, S.; Braicu, E.I.; et al. Chromosomal instability in cell-free DNA as a highly specific biomarker for detection of ovarian cancer in women with adnexal masses. Clin. Cancer Res. 2017, 23, 2223–2231. [Google Scholar] [CrossRef]
- Paracchini, L.; Beltrame, L.; Grassi, T.; Inglesi, A.; Fruscio, R.; Landoni, F.; Ippolito, D.; Marchette, M.d.; Paderno, M.; Adorni, M.; et al. Genome-wide copy number alterations in circulating tumor dna as a novel biomarker in high grade serous ovarian cancer patients. Clin. Cancer Res. 2020, 27, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Yeung, T.-L.; Leung, C.S.; Yip, K.-P.; Yeung, C.L.A.; Wong, S.T.C.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell. Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Kindelberger, D.; Doyle, C.; Lowe, A.; Barry, W.T.; Matulonis, U.A. Predictive value of circulating tumor cells (CTCs) in newly-diagnosed and recurrent ovarian cancer patients. Gynecol. Oncol. 2013, 131, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Gho, Y.S.; Quesenberry, P. Journal of extracellular vesicles: The seven year itch! J. Extracell. Vesicles 2019, 8, 1654729. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.L.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell. Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Yoshimura, A.; Sawada, K.; Nakamura, K.; Kinose, Y.; Nakatsuka, E.; Kobayashi, M.; Miyamoto, M.; Ishida, K.; Matsumoto, Y.; Kodama, M.; et al. Exosomal MiR-99a-5p Is Elevated in sera of ovarian cancer patients and promotes cancer cell invasion by increasing fibronectin and vitronectin expression in neighboring peritoneal mesothelial cells. BMC Cancer 2018, 18, 1065. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Meng, X.; Müller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and prognostic relevance of circulating exosomal MiR-373, MiR-200a, MiR-200b and MiR-200c in patients with epithelial ovarian cancer. Oncotarget 2016, 7, 16923–16935. [Google Scholar] [CrossRef]
- Zuberi, M.; Mir, R.; Das, J.; Ahmad, I.; Javid, J.; Yadav, P.; Masroor, M.; Ahmad, S.; Ray, P.C.; Saxena, A. Expression of serum MiR-200a, MiR-200b, and MiR-200c as candidate biomarkers in epithelial ovarian cancer and their association with clinicopathological features. Clin. Transl. Oncol. 2015, 17, 779–787. [Google Scholar] [CrossRef]
- Gao, Y.-C.; Wu, J. MicroRNA-200c and MicroRNA-141 as potential diagnostic and prognostic biomarkers for ovarian cancer. Tumor Biol. 2015, 36, 4843–4850. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Kristensen, G.; Embleton, A.; Adusei, C.; Barretina-Ginesta, M.P.; Beale, P.; Helland, Å. Evaluation of prognostic and predictive significance of circulating MicroRNAs in ovarian cancer patients. Dis. Mark. 2017, 2017, 3098542. [Google Scholar] [CrossRef]
- Teeuwssen, M.; Fodde, R. Wnt signaling in ovarian cancer stemness, EMT, and therapy resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef]
- Wang, X.; Kong, D.; Wang, C.; Ding, X.; Zhang, L.; Zhao, M.; Chen, J.; Xu, X.; Hu, X.; Yang, J.; et al. Circulating MicroRNAs as novel potential diagnostic biomarkers for ovarian cancer: A systematic review and updated meta-analysis. J. Ovarian Res. 2019, 12, 24. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Mouliere, F.; Robert, B.; Peyrotte, E.A.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef]
- Breitbach, S.; Sterzing, B.; Magallanes, C.; Tug, S.; Simon, P. Direct measurement of cell-free DNA from serially collected capillary plasma during incremental exercise. J. Appl. Physiol. 2014, 117, 119–130. [Google Scholar] [CrossRef]
- Beiter, T.; Fragasso, A.; Hudemann, J.; Niess, A.M.; Simon, P. Short-term treadmill running as a model for studying cell-free DNA kinetics in vivo. Clin. Chem. 2011, 57, 633–636. [Google Scholar] [CrossRef]
- Vittori, L.N.; Tarozzi, A.; Latessa, P.M. Circulating cell-free DNA in physical activities. Methods Mol. Biol. 2019, 1909, 183–197. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Filho, E.M.R.; Simon, D.; Ikuta, N.; Klovan, C.; Dannebrock, F.A.; de Oliveira, C.O.; Regner, A. Elevated Cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 1639–1646. [Google Scholar] [CrossRef]
- Ohayon, S.; Boyko, M.; Saad, A.; Douvdevani, A.; Gruenbaum, B.F.; Melamed, I.; Shapira, Y.; Teichberg, V.I.; Zlotnik, A. Cell-free DNA as a marker for prediction of brain damage in traumatic brain injury in rats. J. Neurotrauma 2012, 29, 261–267. [Google Scholar] [CrossRef]
- Vajpeyee, A.; Wijatmiko, T.; Vajpeyee, M.; Taywade, O.; Pandey, S.; Chauhan, P.S. Clinical usefulness of cell-free DNA as a prognostic marker in acute ischemic stroke. Neurologist 2020, 25, 11–13. [Google Scholar] [CrossRef]
- Tsai, N.-W.; Lin, T.-K.; Chen, S.-D.; Chang, W.-N.; Wang, H.-C.; Yang, T.-M.; Lin, Y.-J.; Jan, C.-R.; Huang, C.-R.; Liou, C.-W.; et al. The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin. Chim. Acta 2011, 412, 476–479. [Google Scholar] [CrossRef]
- Einbinder, Y.; Shnaider, A.; Ghanayem, K.; Basok, A.; Rogachev, B.; Lior, Y.; Haviv, Y.S.; Cohen-Hagai, K.; Nacasch, N.; Rozenberg, I.; et al. Elevated circulating cell-free DNA in hemodialysis-treated patients is associated with increased mortality. Am. J. Nephrol. 2020, 51, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free dna in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 16, 224ra24. [Google Scholar] [CrossRef]
- Sanchez, C.; Snyder, M.W.; Tanos, R.; Shendure, J.; Thierry, A.R. New Insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. NPJ Genom. Med. 2018, 3, 31. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.; Hjelm, N.M. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef]
- Yu, S.C.Y.; Lee, S.W.Y.; Jiang, P.; Leung, T.Y.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin. Chem. 2013, 59, 1228–1237. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Shao, X.; He, Y.; Ji, M.; Chen, X.; Qi, J.; Shi, W.; Hao, T.; Ju, S. quantitative analysis of cell-free DNA in ovarian cancer. Oncol. Lett. 2015, 10, 3478–3482. [Google Scholar] [CrossRef]
- Kamat, A.A.; Sood, A.K.; Dang, D.; Gershenson, D.M.; Simpson, J.L.; Bischoff, F.Z. Quantification of total plasma cell-free DNA in ovarian cancer using real-time PCR. Ann. N. Y. Acad. Sci. 2006, 1075, 230–234. [Google Scholar] [CrossRef]
- Capizzi, E.; Gabusi, E.; Grigioni, A.D.; De Iaco, P.; Rosati, M.; Zamagni, C.; Fiorentino, M. Quantification of free plasma DNA before and after chemotherapy in patients with advanced epithelial ovarian cancer. Diagn. Mol. Pathol. 2008, 17, 34–38. [Google Scholar] [CrossRef]
- Dann, R.B.; DeLoia, J.A.; Timms, K.M.; Zorn, K.K.; Potter, J.; Flake, D.D.; Lanchbury, J.S.; Krivak, T.C. BRCA1/2 mutations and expression: Response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol. Oncol. 2012, 125, 677–682. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef]
- The Australian Ovarian Cancer Study Group; Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; et al. Whole–Genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Vencken, P.M.L.H.; Kriege, M.; Hoogwerf, D.; Beugelink, S.; van der Burg, M.E.L.; Hooning, M.J.; Berns, E.M.; Jager, A.; Collée, M.; Burger, C.W.; et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann. Oncol. 2011, 22, 1346–1352. [Google Scholar] [CrossRef]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed]
- Ratajska, M.; Koczkowska, M.; Żuk, M.; Gorczyński, A.; Kuźniacka, A.; Stukan, M.; Biernat, W.; Limon, J.; Wasąg, B. Detection of BRCA1/2 mutations in circulating tumor DNA from patients with ovarian cancer. Oncotarget 2017, 8, 101325–101332. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.; Nordentoft, I.; Vang, S.; Birkenkamp-Demtröder, K.; Jensen, J.B.; Agerbæk, M.; Pedersen, J.S.; Dyrskjøt, L. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci. Rep. 2018, 8, 7242. [Google Scholar] [CrossRef] [PubMed]
- Bieg-Bourne, C.C.; Okamura, R.; Kurzrock, R. Concordance between TP53 alterations in blood and tissue: Impact of time interval, biopsy site, cancer type and circulating tumor DNA burden. Mol. Oncol. 2020, 14, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Beck, J.; Braun, D.P.; Bornemann-Kolatzki, K.; Barilla, H.; Cubello, R.; Quan, W.; Sangal, A.; Khemka, V.; Waypa, J.; et al. Tumor cell-free DNA copy number instability predicts therapeutic response to immunotherapy. Clin. Cancer Res. 2017, 23, 5074–5081. [Google Scholar] [CrossRef]
- Van Roy, N.; Van Der Linden, M.; Menten, B.; Dheedene, A.; Vandeputte, C.; Van Dorpe, J.; Laureys, G.; Renard, M.; Sante, T.; Lammens, T.; et al. shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin. Cancer Res. 2017, 23, 6305–6314. [Google Scholar] [CrossRef]
- Davidson, M.; Barber, L.J.; Woolston, A.; Cafferkey, C.; Mansukhani, S.; Griffiths, B.; Moorcraft, S.-Y.; Rana, I.; Begum, R.; Assiotis, I.; et al. Detecting and tracking circulating tumour DNA copy number profiles during first line chemotherapy in oesophagogastric adenocarcinoma. Cancers 2019, 11, 736. [Google Scholar] [CrossRef]
- Stover, D.G.; Parsons, H.A.; Ha, G.; Freeman, S.S.; Barry, W.T.; Guo, H.; Choudhury, A.D.; Gydush, G.; Reed, S.C.; Rhoades, J.; et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J. Clin. Oncol. 2018, 36, 543–553. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome. Med. 2013, 5, 30. [Google Scholar] [CrossRef]
- Chen, X.; Chang, C.-W.; Spoerke, J.M.; Yoh, K.E.; Kapoor, V.; Baudo, C.; Aimi, J.; Yu, M.; Liang-Chu, M.M.Y.; Suttmann, R.; et al. Low-pass whole-genome sequencing of circulating cell-free DNA Demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort. Clin. Cancer Res. 2019, 25, 2254–2263. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| CTCs Studies in Ovarian Cancer | ||||||
|---|---|---|---|---|---|---|
| Author | No. pt | Subtype/Stage | Detection Rate | Prognostic Significance | Year | Ref |
| Marth et al., | 90 | EOC (I-IV) | 12% (BS) | NS | 2002 | [38] |
| Judson et al., | 53 | EOC (I-IV) | 19% (BS) | NS | 2003 | [39] |
| Aktas et al., | 122 | EOC (I-IV) | 19% (BS), 27% (AC) | OS (p = 0.005 BS and p = 0.004 AC). PFS, NS | 2011 | [40] |
| Poveda et al., | 216 | EOC (I-IV) | CTCs ≥ 2 (12%) CTCs < 2 (88%) (BS) | OS (p = 0.0017) PFS (p = 0.00024) | 2011 | [41] |
| Obermayr et al., | 216 | EOC (I-IV) | 25% (BS) | OS (p = 0.001) PFS (p = 0.001) (AC) | 2013 | [42] |
| Chebouti et al., | 65 | EOC (I-IV) | 17% (BS) | OS (p = 0.0008) PFS (p = 0.0293) (AC) | 2017 | [43] |
| Obermayr et al., | 266 | EOC (I-IV) | 27% | OS (p = 0.007) PFS (p = 0.008) (AC) | 2017 | [44] |
| Zhang et al., | 109 | EOC (I-IV) | 90% | OS (p = 0.041) PFS, NS | 2018 | [45] |
| Kolostova et al., | 118 | EOC (I-IV) | 65% | NS | 2015 | [46] |
| Kolostova et al., | 56 | EOC | 58% | NS | 2016 | [47] |
| Guo et al., | 30 | EOC (I-IV) | 73% | NS | 2018 | [48] |
| Kuhlamann et al., | 143 | EOC (I-IV) | 14% | OS (p = 0.026) PFS (p = 0.009) (BS) | 2014 | [49] |
| EVs and cfmiRNAs Studies in Ovarian Cancer | ||||||
| Author | No. pt | Subtype/Stage | Biomarker | Prognostic Significance | Year | Ref |
| Pan et al., | 106 | EOC (I-IV) | miRNAs: miR-21, miR-100, miR-200b, miR-320, | NA | 2018 | [50] |
| miR-16, miR-93, miR-126, miR-223 | ||||||
| Zhang et al., | 40 | EOC (I-IV) | proteins: LBP, FGG, FGA, GSN | FGG: (OS p = 0.0012) (PFS p = 0.00038) | 2019 | [51] |
| LBP: (OS p = 0.0029) (PFS p = 0.00023) | ||||||
| Schwich et al., | 78 | EOC (I-IV) | protein: HLA-G | PFS 3-years (p = 0.029) PFS 10-years (p = 0.006). OS, NS | 2019 | [52] |
| Resnick et al., | 28 | EOC (I-IV) | miRNAs: miR-21, miR-92, miR-93, miR-126, | NA | 2008 | [53] |
| miR-29a, miR-155, miR-127, miR-99b | ||||||
| Todeschini et al., | 168 | HGS-EOC | miRNA: miR-1246 | NA | 2017 | [54] |
| ctDNA Studies in Ovarian Cancer | ||||||
| Author | No. pt | Subtype/Stage | Biomarker | Prognostic Significance | Year | Ref |
| Swisher et al., | 137 | EOC (I-IV) | TP53 | OS (p = 0.02) PFS, NS | 2005 | [55] |
| Otsuka et al., | 27 | EOC (I-IV) | TP53 | NA | 2004 | [56] |
| Parkinson et al., | 40 | HGS-EOC | TP53 | PFS (p = 0.008) | 2016 | [57] |
| Kim et al., | 61 | HGS-EOC | TP53 | PFS (p = 0.008) | 2019 | [58] |
| Lin et al., | 112 | HGS-EOC | BRCA1/BRCA2 | Rucaparib PFS (p < 0.0001) | 2019 | [59] |
| Vanderstichele et al., | 68 | Adnexal masses | CNA profiling | NA | 2017 | [60] |
| Paracchini et al., | 46 | HGS-EOC | CNA profiling | PFS (p = 0.011) | 2020 | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paracchini, L.; D’Incalci, M.; Marchini, S. Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer—Current Use and Future Opportunities. Cancers 2021, 13, 2386. https://doi.org/10.3390/cancers13102386
Paracchini L, D’Incalci M, Marchini S. Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer—Current Use and Future Opportunities. Cancers. 2021; 13(10):2386. https://doi.org/10.3390/cancers13102386
Chicago/Turabian StyleParacchini, Lara, Maurizio D’Incalci, and Sergio Marchini. 2021. "Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer—Current Use and Future Opportunities" Cancers 13, no. 10: 2386. https://doi.org/10.3390/cancers13102386
APA StyleParacchini, L., D’Incalci, M., & Marchini, S. (2021). Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer—Current Use and Future Opportunities. Cancers, 13(10), 2386. https://doi.org/10.3390/cancers13102386