The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas

 , ,
, ,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
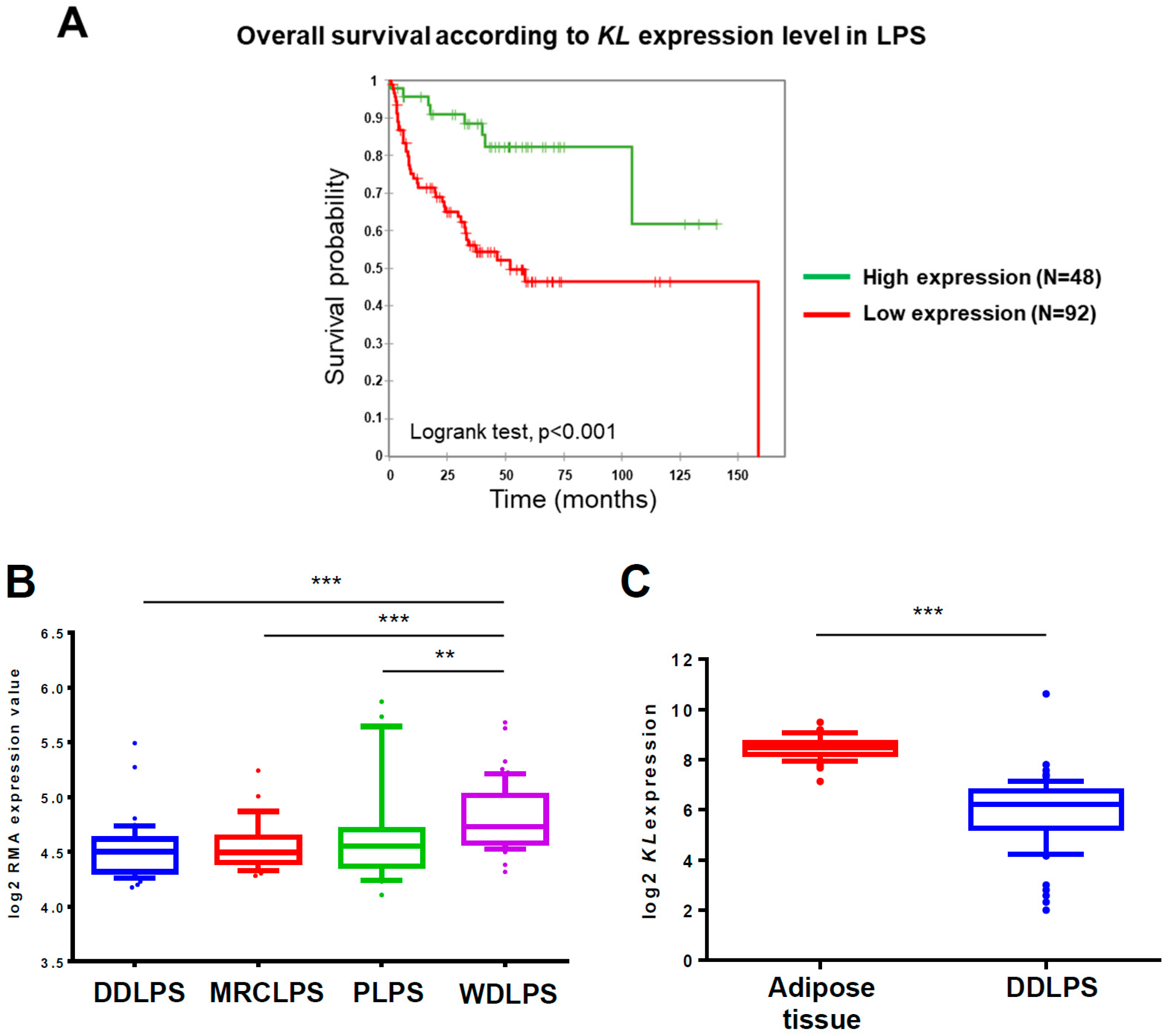
2.1. KL Expression Has a Prognostic Value for Liposarcoma Patients and Is Downregulated in DDLPS Tumors
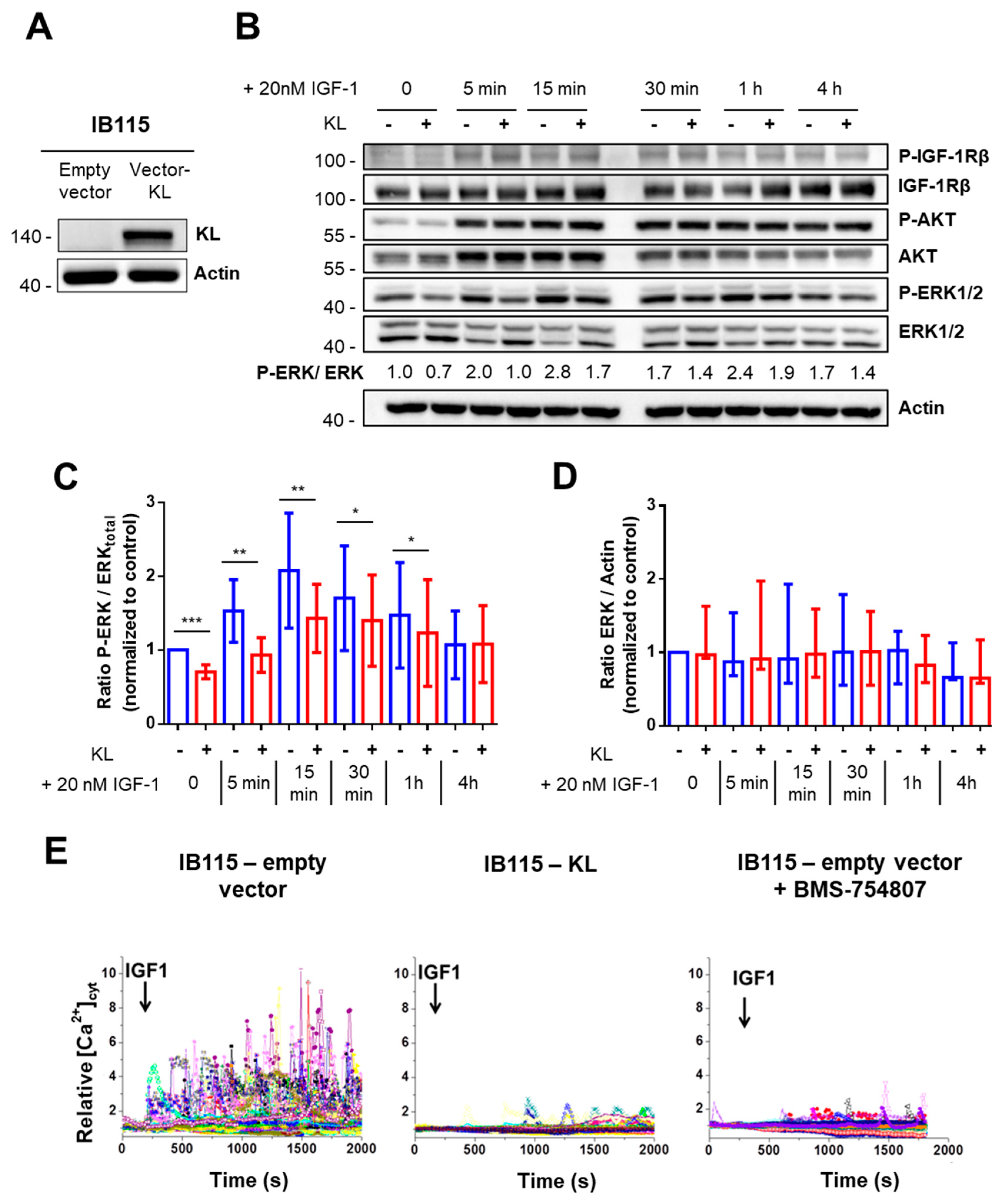
2.2. In the DDLPS Cell Line, Klotho Reduces IGF-1R-Dependent Signaling
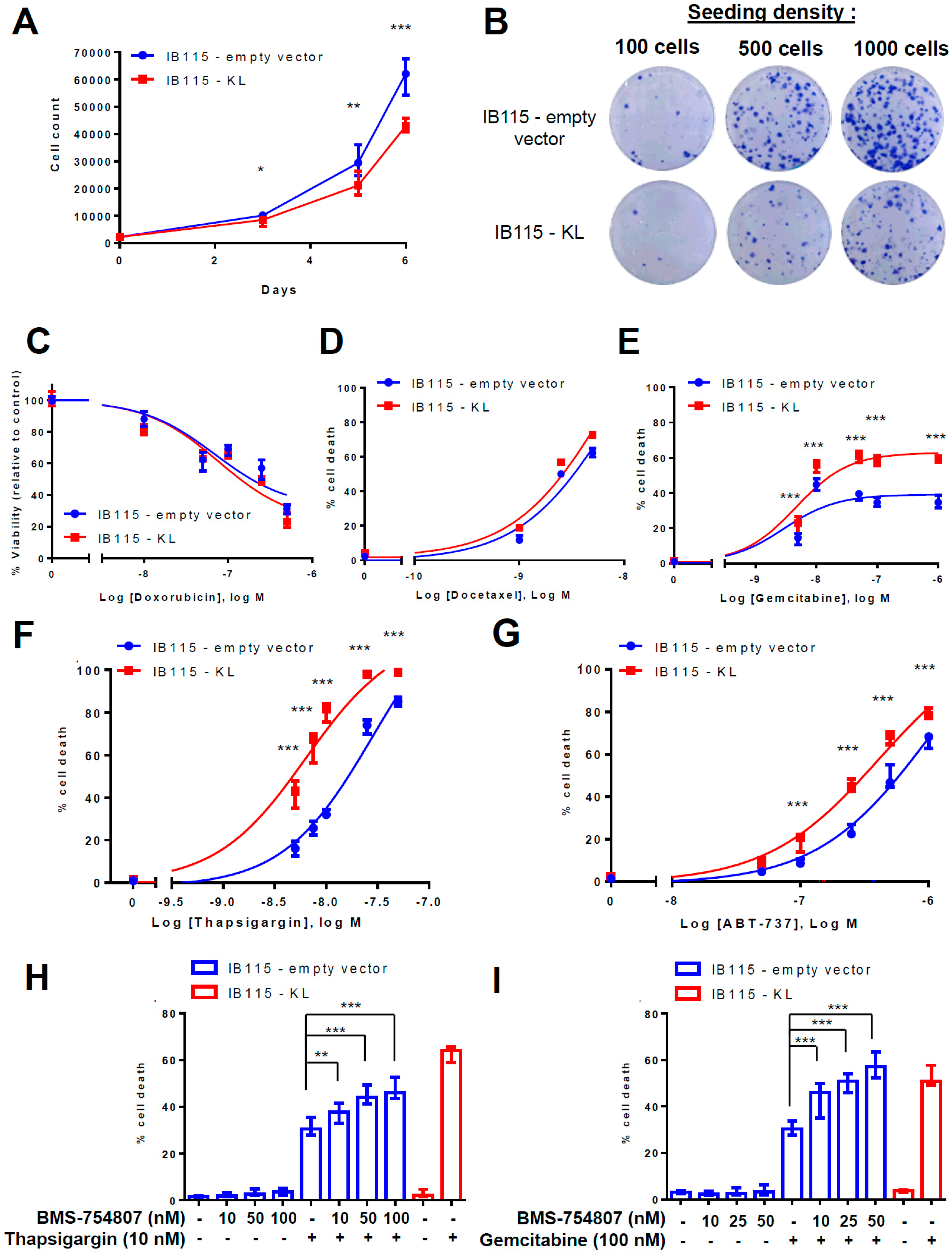
2.3. KL Overexpression in a DDLPS Cell Line Decreases Its Tumorigenic Phenotypes
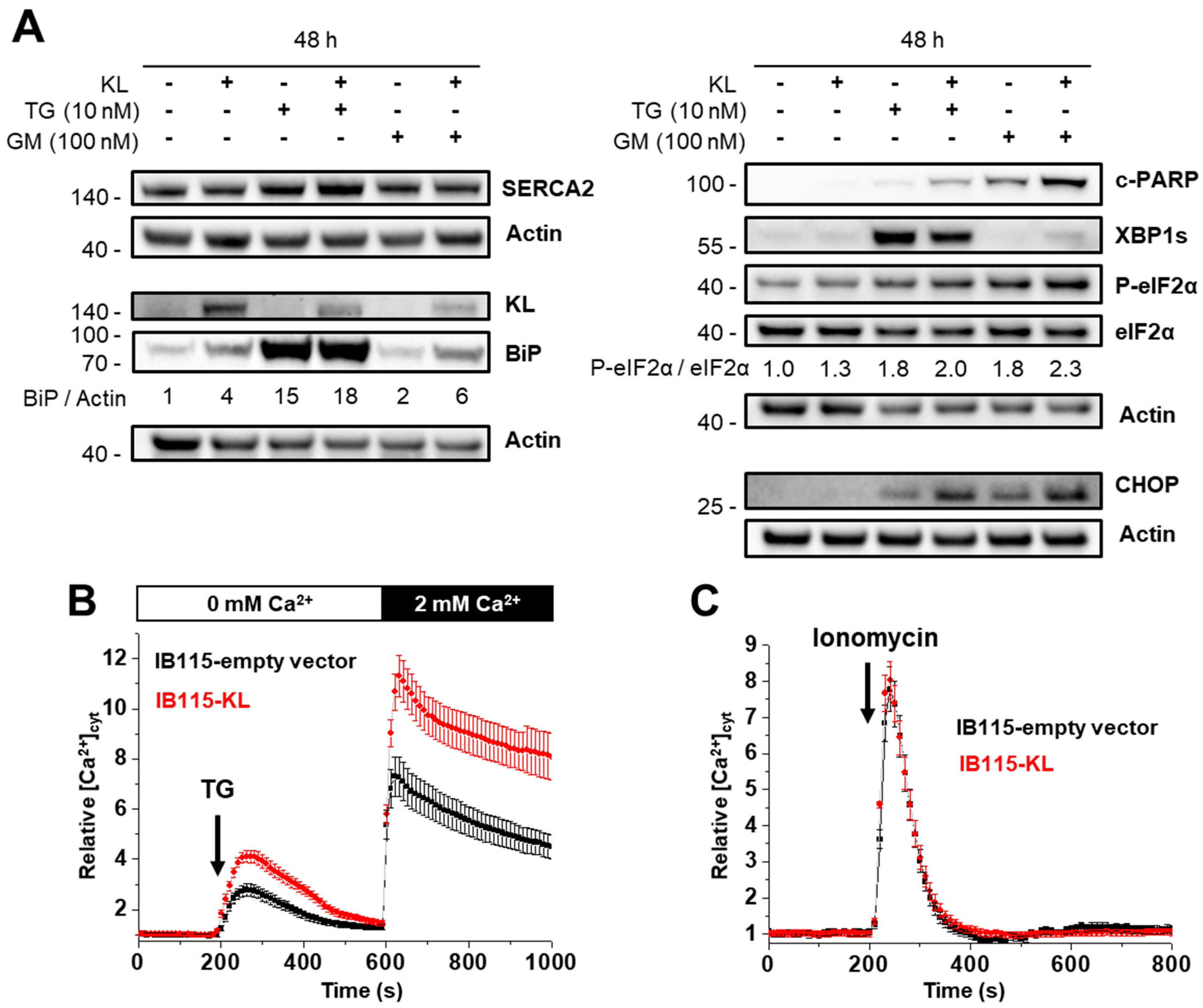
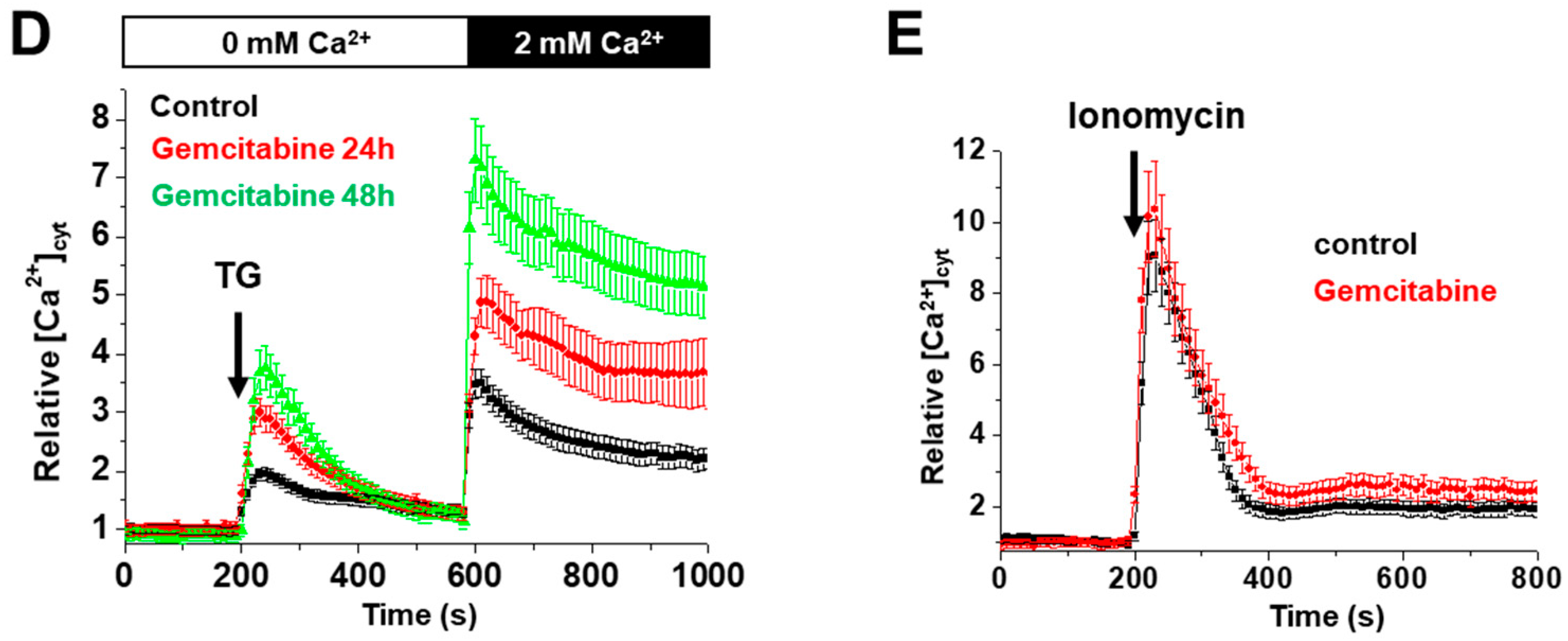
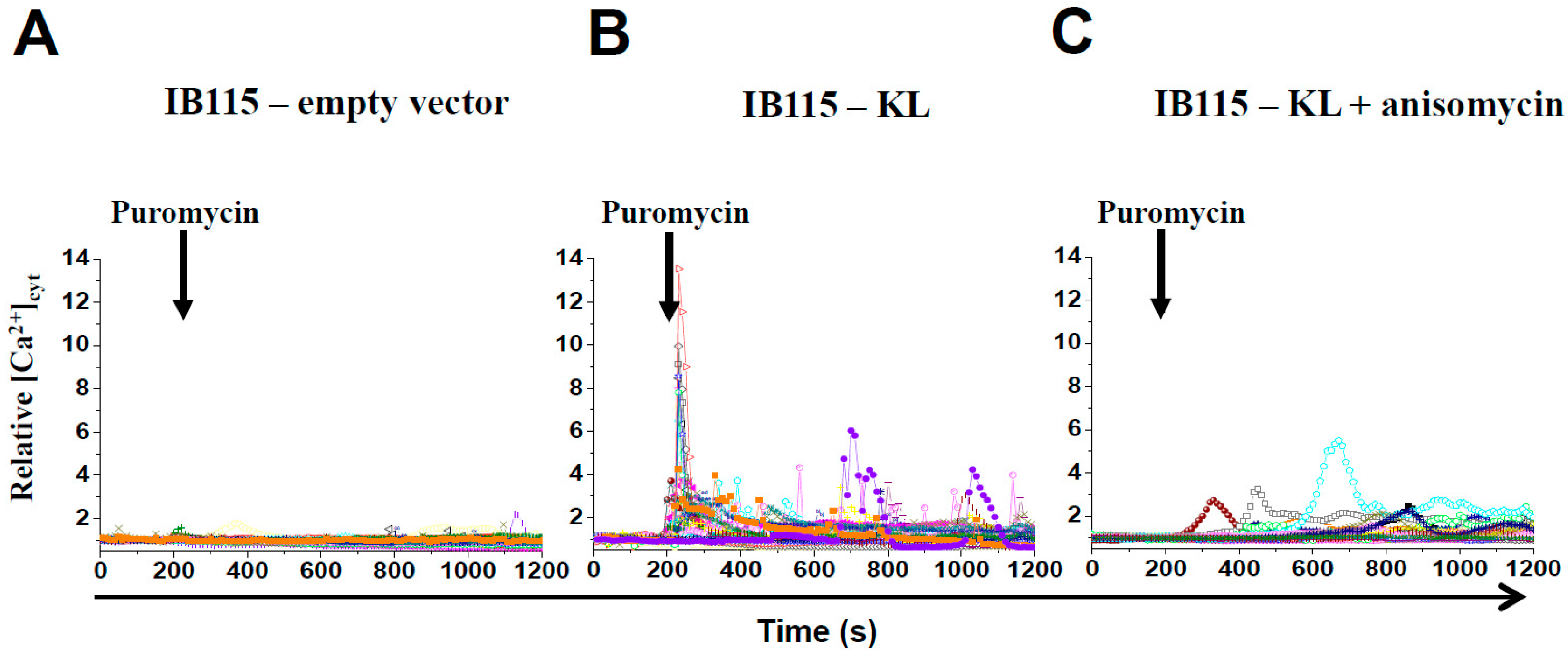
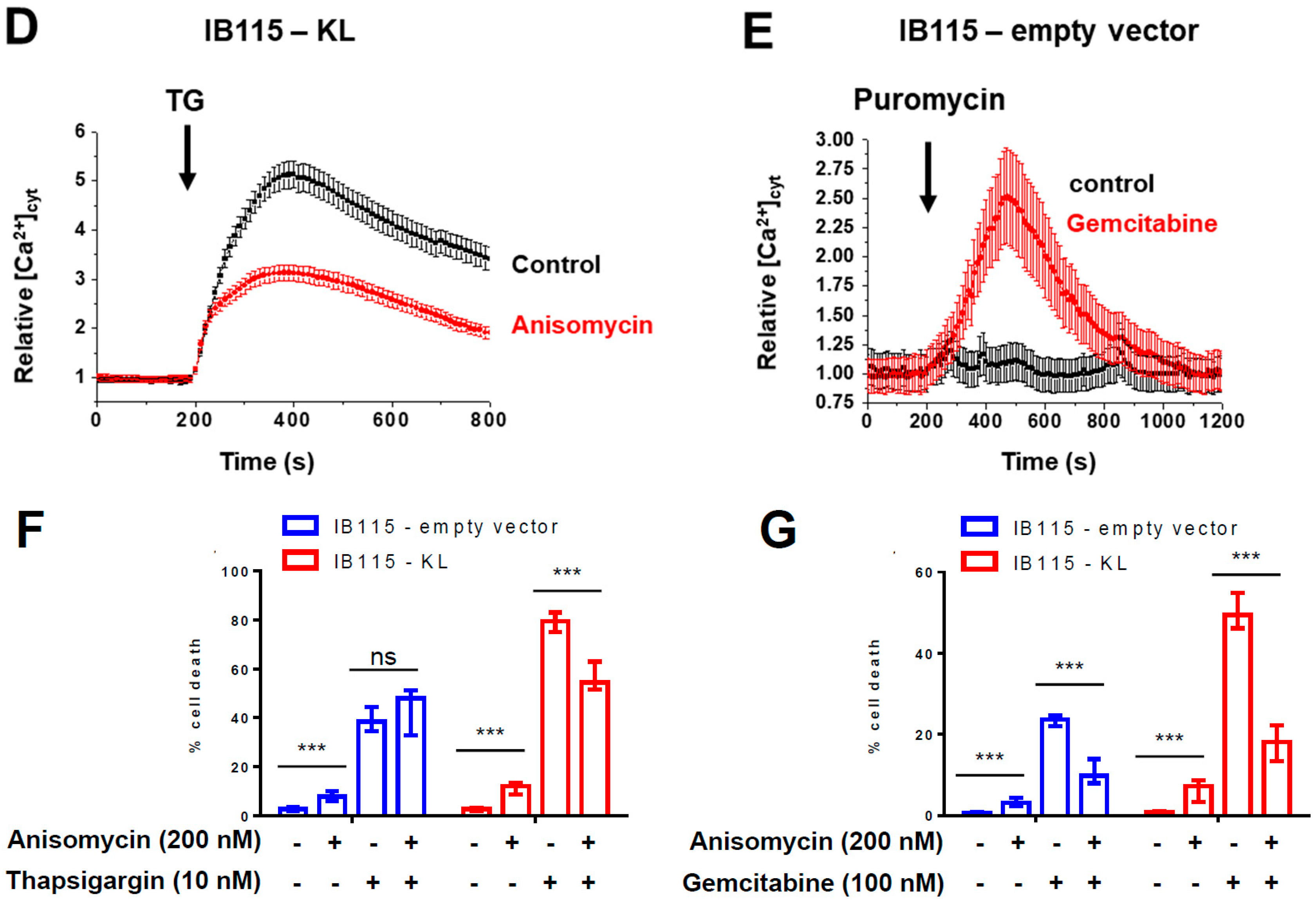
2.4. KL-Overexpression Sensitizes DDLPS to ER Stress by Disrupting Intracellular Calcium Homeostasis
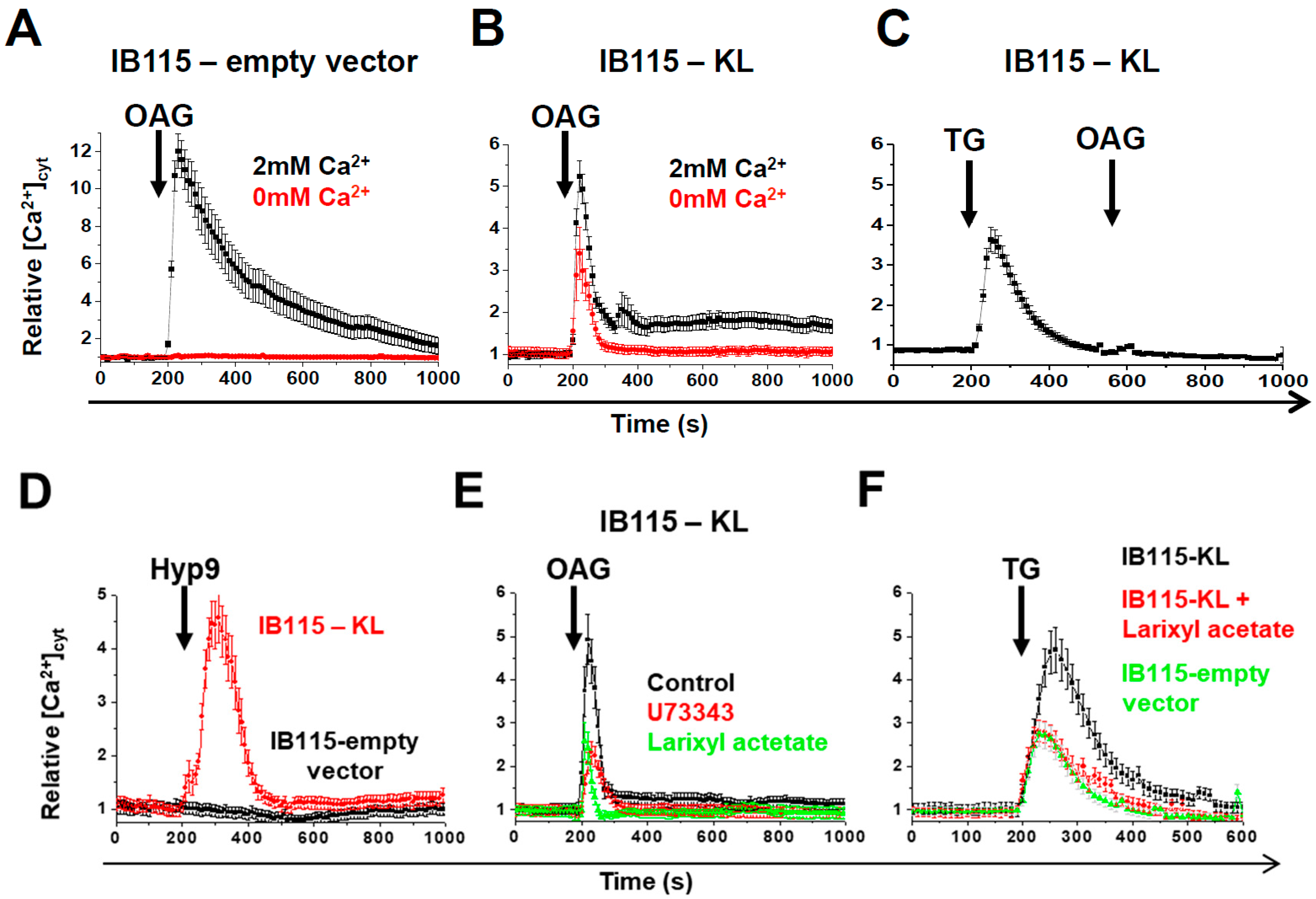
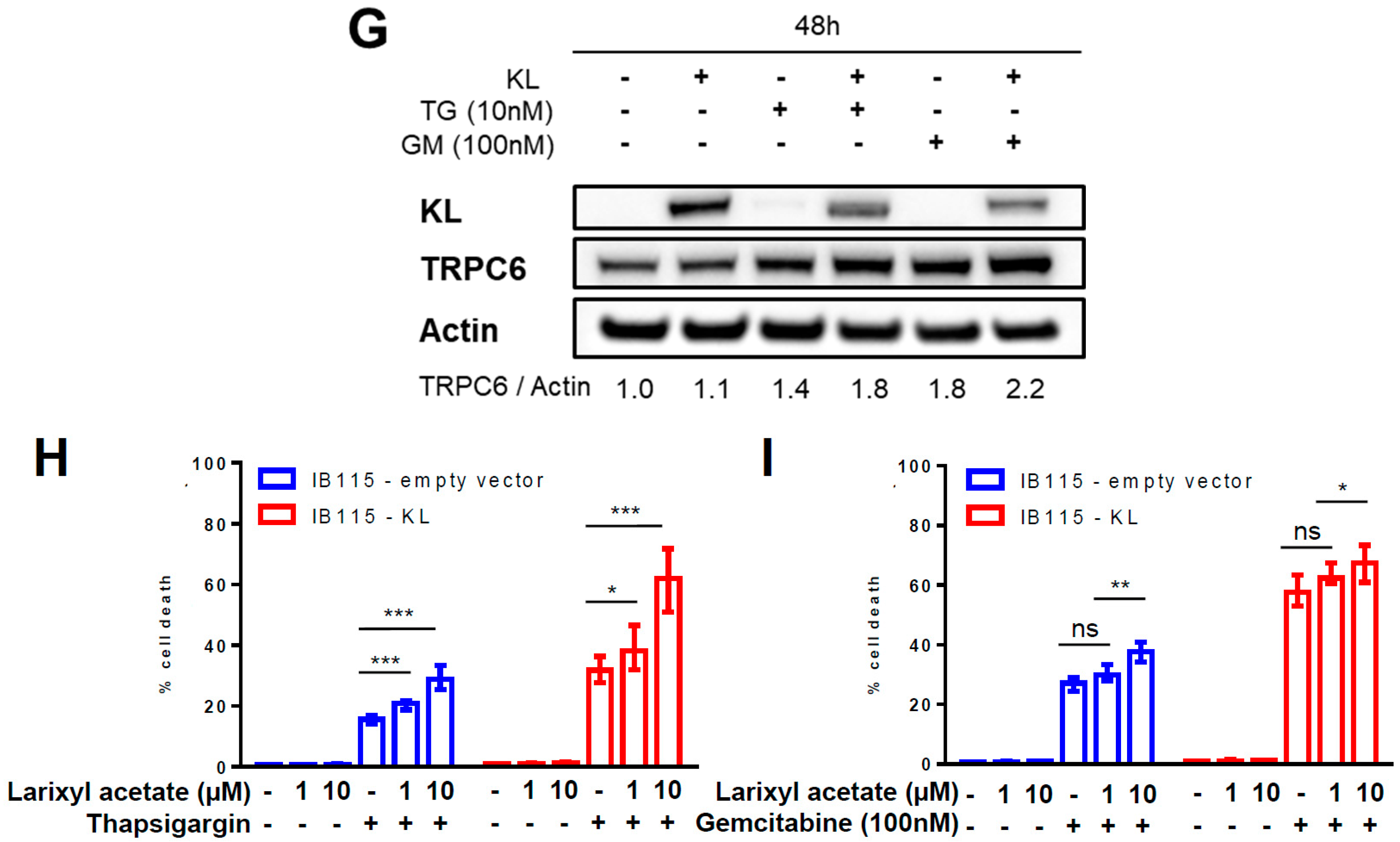
2.5. Intracellular Localization of TRPC6 Contributes to Reticular Ca2+-Leakage in KL-Overexpressing DDLPS Cells
2.6. In DDLPS Cells, Klotho Promotes Reticular Ca2+ Leakage and Apoptosis by Opening the Translocon
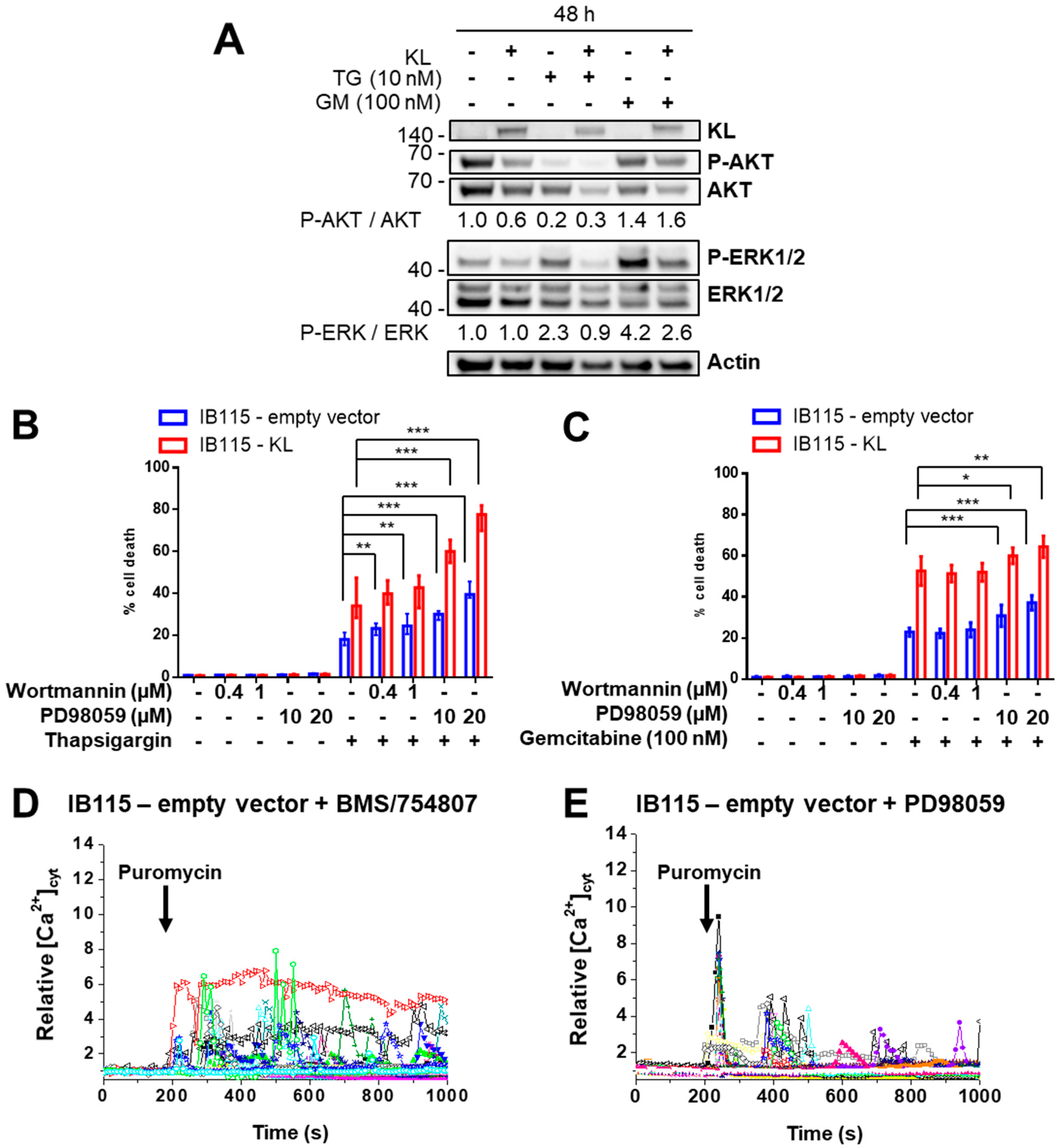
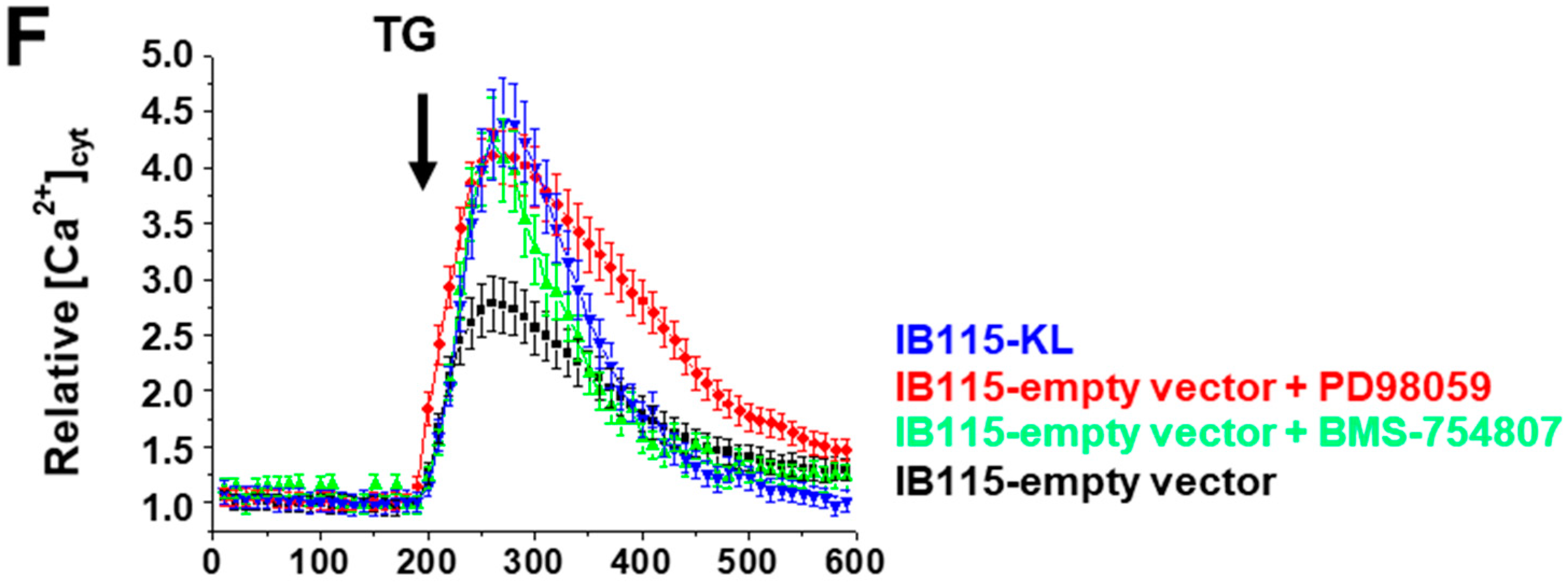
2.7. Klotho Regulates Drug Sensitivity and Reticular Ca2+-Leakage by Inhibiting ERK1/2 Signaling
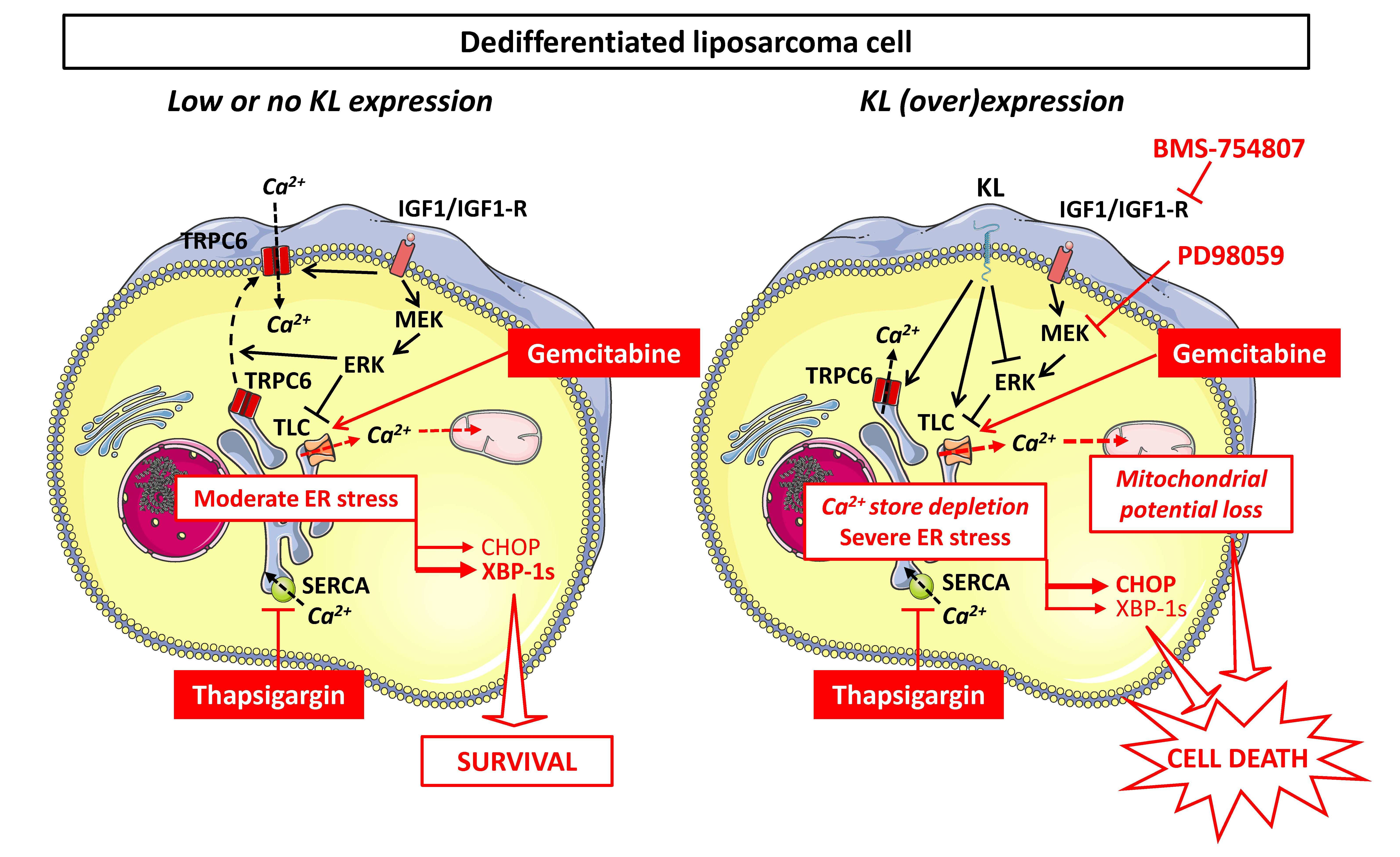
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines
4.3. Western Blot
4.4. Cell Proliferation
4.5. Clonogenic Assay
4.6. MTT Assay
4.7. Mitochondrial Potential Loss Assay (Tetramethylrhodamine, Methyl Ester, TMRM)
4.8. Cytosolic Calcium Imaging
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jo, V.Y.; Fletcher, C.D.M. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology (Phila) 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Abbas Manji, G.; Singer, S.; Koff, A.; Schwartz, G.K. Application of Molecular Biology to Individualize Therapy for Patients with Liposarcoma. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.M.; Kattan, M.W.; Antonescu, C.R.; Brennan, M.F.; Singer, S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann. Surg. 2006, 244, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Henricks, W.H.; Chu, Y.C.; Goldblum, J.R.; Weiss, S.W. Dedifferentiated liposarcoma: A clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am. J. Surg. Pathol. 1997, 21, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Tirumani, S.H.; Tirumani, H.; Jagannathan, J.P.; Shinagare, A.B.; Hornick, J.L.; Ramaiya, N.H.; Wagner, A.J. Metastasis in dedifferentiated liposarcoma: Predictors and outcome in 148 patients. Eur. J. Surg. Oncol. EJSO 2015, 41, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Fisher, C.; Al-Muderis, O.; Judson, I.R. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur. J. Cancer 2005, 41, 2853–2860. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Shen, Z.; Xu, X.; Hu, B.-B.; Meerani, S.; Tang, L.-N.; Zheng, S.-E.; Sun, Y.-J.; Min, D.-L.; Yao, Y. Evaluation of the Expression and Role of IGF Pathway Biomarkers in Human Sarcomas. Int. J. Immunopathol. Pharmacol. 2013, 26, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, B.; Yuan, L.; Ye, Z. Prognostic value of IGF-1R expression in bone and soft tissue sarcomas: A meta-analysis. OncoTargets Ther. 2015, 8, 1949–1955. [Google Scholar]
- Conti, A.; Espina, V.; Chiechi, A.; Magagnoli, G.; Novello, C.; Pazzaglia, L.; Quattrini, I.; Picci, P.; Liotta, L.A.; Benassi, M.S. Mapping protein signal pathway interaction in sarcoma bone metastasis: Linkage between rank, metalloproteinases turnover and growth factor signaling pathways. Clin. Exp. Metastasis 2014, 31, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Morooka, T.; Hoshida, Y.; Zhang, B.; Qiu, Y.; Nakamichi, I.; Hamada, K.; Ueda, T.; Naka, N.; Kudawara, I.; et al. Prognostic Significance of Activated AKT Expression in Soft-Tissue Sarcoma. Clin. Cancer Res. 2006, 12, 3070–3077. [Google Scholar] [CrossRef] [PubMed]
- Tricoli, J.V.; Rall, L.B.; Karakousis, C.P.; Herrera, L.; Petrelli, N.J.; Bell, G.I.; Shows, T.B. Enhanced Levels of Insulin-like Growth Factor Messenger RNA in Human Colon Carcinomas and Liposarcomas. Cancer Res. 1986, 46, 6169–6173. [Google Scholar] [PubMed]
- Peng, T.; Zhang, P.; Liu, J.; Nguyen, T.; Bolshakov, S.; Belousov, R.; Young, E.D.; Wang, X.; Brewer, K.; Terrada, L.L.; et al. An experimental model for the study of well differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab. Investig. 2011, 91, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Softic, S.; Ouaamari, A.E.; Krumpoch, M.T.; Kleinridders, A.; Kulkarni, R.N.; O’Neill, B.T.; Kahn, C.R. Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function. Diabetes 2016, 65, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Molinelli, E.J.; Nair, J.S.; Sheikh, T.; Samy, R.; Jing, X.; He, Q.; Korkut, A.; Crago, A.M.; Singer, S.; et al. Drug synergy screen and network modeling in dedifferentiated liposarcoma identifies CDK4 and IGF1R as synergistic drug targets. Sci. Signal. 2013, 6, ra85. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.A.; Varewijck, A.J. IGF-IR Targeted Therapy: Past, Present and Future. Front. Endocrinol. (Lausanne) 2014, 5, 224. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.W.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance for Research Through Collaboration study. Cancer 2014, 120, 2448–2456. [Google Scholar] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-D.; Tung, T.Y.; Liang, J.; Zeldich, E.; Tucker Zhou, T.B.; Turk, B.E.; Abraham, C.R. Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry (Mosc) 2014, 53, 5579–5587. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Renal and extrarenal actions of Klotho. Semin. Nephrol. 2013, 33, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Sun, Z. The Antiaging Gene Klotho Regulates Proliferation and Differentiation of Adipose-Derived Stem Cells. Stem Cells 2016, 34, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Chen, J.; Liu, B.; Zhan, J. Klotho Acts as a Tumor Suppressor in Cancers. Pathol. Oncol. Res. 2013, 19, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Wolf, I. The Role of Alpha-Klotho as a Universal Tumor Suppressor. Vitam. Horm. 2016, 101, 197–214. [Google Scholar] [PubMed]
- Olauson, H.; Mencke, R.; Hillebrands, J.-L.; Larsson, T.E. Tissue expression and source of circulating αKlotho. Bone 2017, 100, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef] [PubMed]
- Takasu, N.; Takasu, M.; Komiya, I.; Nagasawa, Y.; Asawa, T.; Shimizu, Y.; Yamada, T. Insulin-like growth factor I stimulates inositol phosphate accumulation, a rise in cytoplasmic free calcium, and proliferation in cultured porcine thyroid cells. J. Biol. Chem. 1989, 264, 18485–18488. [Google Scholar] [PubMed]
- Valdés, J.A.; Flores, S.; Fuentes, E.N.; Osorio-Fuentealba, C.; Jaimovich, E.; Molina, A. IGF-1 induces IP3 -dependent calcium signal involved in the regulation of myostatin gene expression mediated by NFAT during myoblast differentiation. J. Cell. Physiol. 2013, 228, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Dalton, G.; An, S.-W.; Al-Juboori, S.I.; Nischan, N.; Yoon, J.; Dobrinskikh, E.; Hilgemann, D.W.; Xie, J.; Luby-Phelps, K.; Kohler, J.J.; et al. Soluble klotho binds monosialoganglioside to regulate membrane microdomains and growth factor signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Xie, J.; Hwang, K.-H.; Wu, Y.-L.; Oliver, N.; Eom, M.; Park, K.-S.; Barrezueta, N.; Kong, I.-D.; Fracasso, R.P.; et al. Klotho May Ameliorate Proteinuria by Targeting TRPC6 Channels in Podocytes. J. Am. Soc. Nephrol. JASN 2017, 28, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cha, S.-K.; An, S.-W.; Kuro-o, M.; Birnbaumer, L.; Huang, C.-L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [PubMed]
- Cappella, P.; Tomasoni, D.; Faretta, M.; Lupi, M.; Montalenti, F.; Viale, F.; Banzato, F.; D’Incalci, M.; Ubezio, P. Cell cycle effects of gemcitabine. Int. J. Cancer 2001, 93, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [PubMed]
- Novosyadlyy, R.; Kurshan, N.; Lann, D.; Vijayakumar, A.; Yakar, S.; LeRoith, D. Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 2008, 15, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Ding, L.-W.; Sun, Q.-Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Wang, W.; Zhu, Y.; Chen, Y.; Tian, B. Gemcitabine treatment induces endoplasmic reticular (ER) stress and subsequently upregulates urokinase plasminogen activator (uPA) to block mitochondrial-dependent apoptosis in Panc-1 cancer stem-like cells (CSCs). PLoS ONE. 2017, 12, e0184110. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.T.; Poole, A.W. Bcl-xL-inhibitory BH3 mimetic ABT-737 depletes platelet calcium stores. Blood 2012, 119, 4337–4338. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Song, R.; Shen, Y.; Sun, Y.; Gu, Y.; Shu, Y.; Xu, Q. Targeting Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase 2 by Curcumin Induces ER Stress-Associated Apoptosis for Treating Human Liposarcoma. Mol. Cancer Ther. 2011, 10, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, W.; Yang, Y.; Hu, Y.; Gu, Y.; Shu, Y.; Sun, Y.; Wu, X.; Shen, Y.; Xu, Q. High expression of sarcoplasmic/endoplasmic reticulum Ca(2+)-ATPase 2b blocks cell differentiation in human liposarcoma cells. Life Sci. 2014, 99, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Sadighi Akha, A.A.; Harper, J.M.; Salmon, A.B.; Schroeder, B.A.; Tyra, H.M.; Rutkowski, D.T.; Miller, R.A. Heightened Induction of Proapoptotic Signals in Response to Endoplasmic Reticulum Stress in Primary Fibroblasts from a Mouse Model of Longevity. J. Biol. Chem. 2011, 286, 30344–30351. [Google Scholar] [CrossRef] [PubMed]
- Albarrán, L.; Dionisio, N.; Lopez, E.; Salido, G.M.; Redondo, P.C.; Rosado, J.A. STIM1 regulates TRPC6 heteromultimerization and subcellular location. Biochem. J. 2014, 463, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Bréchard, S.; Melchior, C.; Plançon, S.; Schenten, V.; Tschirhart, E.J. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium 2008, 44, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Jardín, I.; Redondo, P.C.; Salido, G.M.; Rosado, J.A. Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim. Biophys. Acta 2008, 1783, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Salido, G.M.; Jardín, I.; Rosado, J.A. The TRPC ion channels: Association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 2011, 704, 413–433. [Google Scholar] [PubMed]
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiDTM lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar] [PubMed]
- Yang, X.L.; Lin, F.J.; Guo, Y.J.; Shao, Z.M.; Ou, Z.L. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. OncoTargets Ther. 2014, 7, 1033–1042. [Google Scholar]
- Zhang, L.J.; Chen, S.; Wu, P.; Hu, C.S.; Thorne, R.F.; Luo, C.M.; Hersey, P.; Zhang, X.D. Inhibition of MEK blocks GRP78 up-regulation and enhances apoptosis induced by ER stress in gastric cancer cells. Cancer Lett. 2009, 274, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Ohsugi, M.; Liu, Z.; Fatrai, S.; Bernal-Mizrachi, E.; Permutt, M.A. Endoplasmic Reticulum Stress–Induced Apoptosis Is Partly Mediated by Reduced Insulin Signaling Through Phosphatidylinositol 3-Kinase/Akt and Increased Glycogen Synthase Kinase-3β in Mouse Insulinoma Cells. Diabetes 2005, 54, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Chen, R.; Li, H. Cross-talk between PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum stress-induced cell cycle progression and cell death in human hepatocellular carcinoma cells. Int. J. Oncol. 2009, 34, 1749–1757. [Google Scholar] [PubMed]
- Lee, J.; Jeong, D.-J.; Kim, J.; Lee, S.; Park, J.-H.; Chang, B.; Jung, S.-I.; Yi, L.; Han, Y.; Yang, Y.; et al. The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol. Cancer 2010, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Shulman, M.; Israeli, S.; Bose, S.; Avraham, A.; Zundelevich, A.; Evron, E.; Gal-Yam, E.N.; Kaufman, B.; Wolf, I. Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res. Treat. 2012, 133, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Prevarskaya, N. Targeting apoptosis by the remodelling of calcium-transporting proteins in cancerogenesis. FEBS J. 2013, 280, 5500–5510. [Google Scholar] [CrossRef] [PubMed]
- Schäuble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, Ö.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Zhao, Y.; Sarkar, P.S.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho ameliorates chemically induced endoplasmic reticulum (ER) stress signaling. Cell. Physiol. Biochem. 2013, 31, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Stress resistance in long-lived mouse models. Exp. Gerontol. 2006, 41, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Pong, M.; Morgan, T.E.; Wang, H.; Ott, K.; Zhou, B.; Longo, V.D.; Lee, A.S. GRP78/BiP is a novel downstream target of IGF-1 receptor mediated signaling. J. Cell. Physiol. 2012, 227, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, L.; Lub, S.; Fostier, K.; Maes, K.; Van Valckenborgh, E.; Menu, E.; Johnsen, H.E.; Overgaard, M.T.; Larsson, O.; Axelson, M.; et al. The IGF-1 receptor inhibitor picropodophyllin potentiates the anti-myeloma activity of a BH3-mimetic. Oncotarget 2014, 5, 11193–11208. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Guisado, E.; Casas-Rua, V.; Tomas-Martin, P.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Martin-Romero, F.J. Phosphorylation of STIM1 at ERK1/2 target sites regulates interaction with the microtubule plus-end binding protein EB1. J. Cell Sci. 2013, 126, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, M.; Matsuyama, A.; Nakamoto, M. Aberrant calreticulin expression is involved in the dedifferentiation of dedifferentiated liposarcoma. Am. J. Pathol. 2012, 180, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, P.; Brulard, C.; Pérot, G.; Mauduit, O.; Delespaul, L.; Neuville, A.; Stoeckle, E.; Le Guellec, S.; Rochaix, P.; Coindre, J.M.; et al. Stable Instability of Sarcoma Cell Lines Genome Despite Intra-Tumoral Heterogeneity: A Genomic and Transcriptomic Study of Sarcoma Cell Lines. Austin J. Genet. Genomic Res. 2015, 2, 1014. [Google Scholar]
- Hammadi, M.; Delcroix, V.; Vacher, A.-M.; Ducret, T.; Vacher, P. CD95-Mediated Calcium Signaling. Methods Mol. Biol. 2017, 1557, 79–93. [Google Scholar] [PubMed]
- Gobble, R.M.; Qin, L.-X.; Brill, E.R.; Angeles, C.V.; Ugras, S.; O’Connor, R.B.; Moraco, N.H.; Decarolis, P.L.; Antonescu, C.; Singer, S. Expression profiling of liposarcoma yields a multigene predictor of patient outcome and identifies genes that contribute to liposarcomagenesis. Cancer Res. 2011, 71, 2697–2705. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delcroix, V.; Mauduit, O.; Tessier, N.; Montillaud, A.; Lesluyes, T.; Ducret, T.; Chibon, F.; Van Coppenolle, F.; Ducreux, S.; Vacher, P. The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas. Cancers 2018, 10, 439. https://doi.org/10.3390/cancers10110439
Delcroix V, Mauduit O, Tessier N, Montillaud A, Lesluyes T, Ducret T, Chibon F, Van Coppenolle F, Ducreux S, Vacher P. The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas. Cancers. 2018; 10(11):439. https://doi.org/10.3390/cancers10110439
Chicago/Turabian StyleDelcroix, Vanessa, Olivier Mauduit, Nolwenn Tessier, Anaïs Montillaud, Tom Lesluyes, Thomas Ducret, Frédéric Chibon, Fabien Van Coppenolle, Sylvie Ducreux, and Pierre Vacher. 2018. "The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas" Cancers 10, no. 11: 439. https://doi.org/10.3390/cancers10110439
APA StyleDelcroix, V., Mauduit, O., Tessier, N., Montillaud, A., Lesluyes, T., Ducret, T., Chibon, F., Van Coppenolle, F., Ducreux, S., & Vacher, P. (2018). The Role of the Anti-Aging Protein Klotho in IGF-1 Signaling and Reticular Calcium Leak: Impact on the Chemosensitivity of Dedifferentiated Liposarcomas. Cancers, 10(11), 439. https://doi.org/10.3390/cancers10110439