
Untargeted Metabolome Analysis Reveals Reductions in Maternal Hepatic Glucose and Amino Acid Content That Correlate with Fetal Organ Weights in a Mouse Model of Fetal Alcohol Spectrum Disorders

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Data Analysis and Statistical Analysis
3. Results
3.1. Litter Characteristics
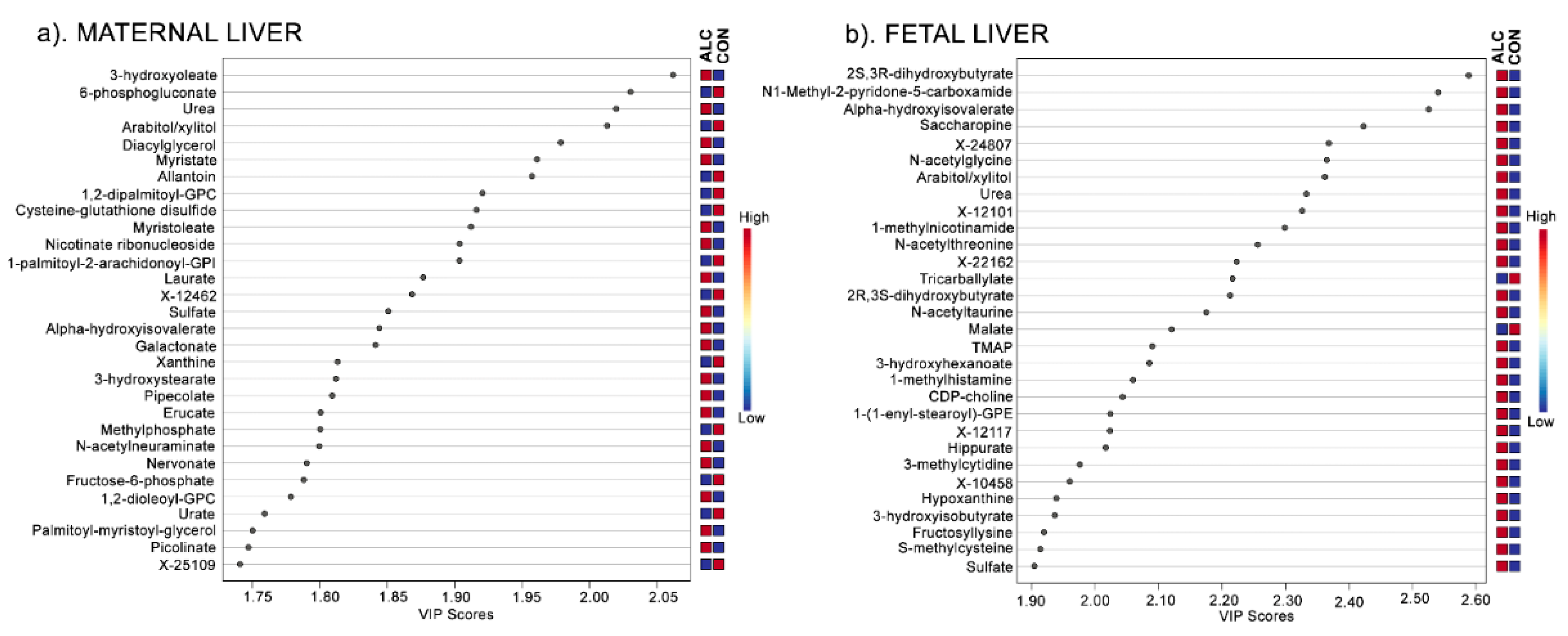
3.2. Metabolite Profiles Distinctly Separate ALC and CON Dams but Not Their Fetuses
3.3. Amino Acid Catabolites Are Enriched in Alcohol-Exposed Maternal and Fetal Liver
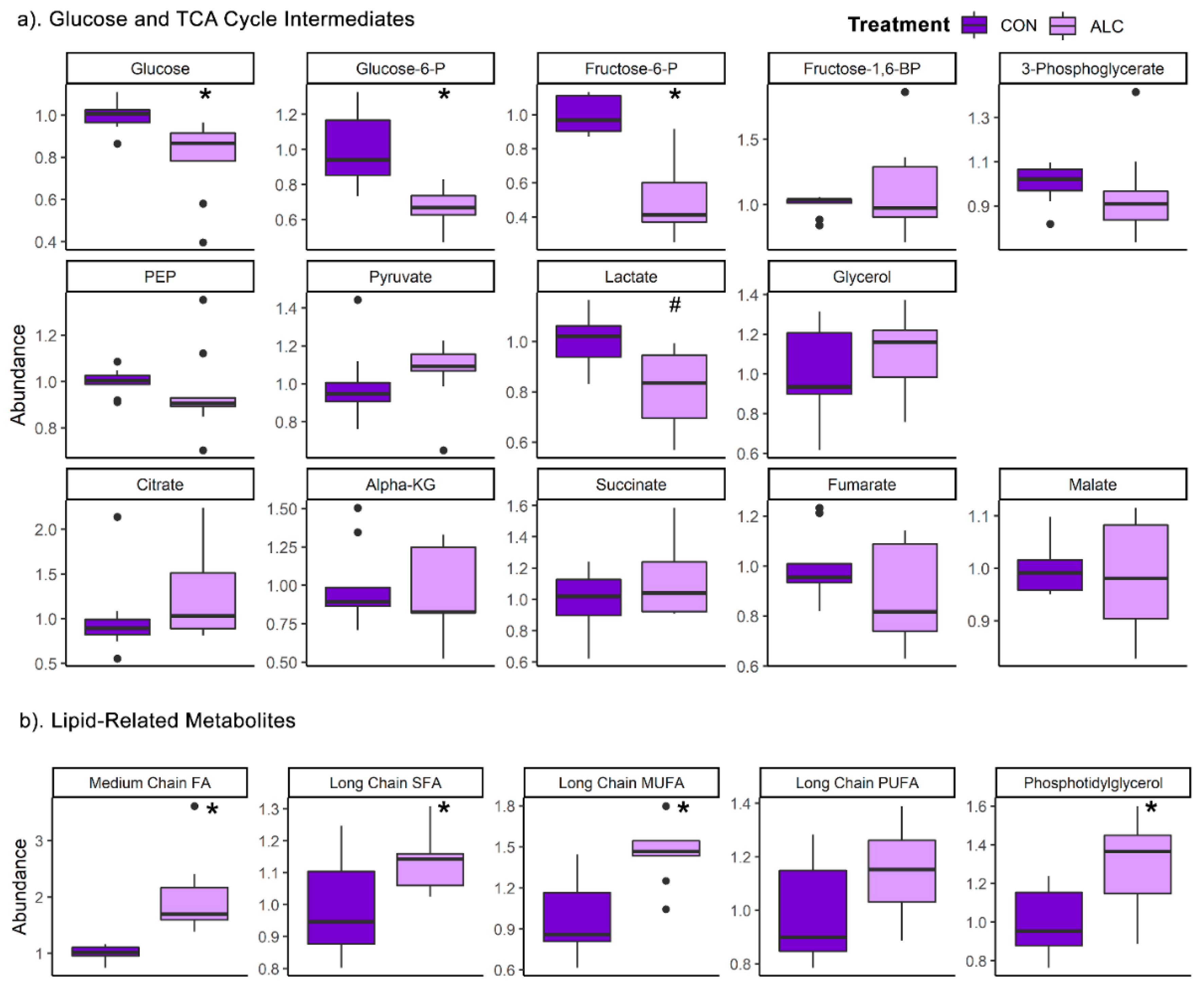
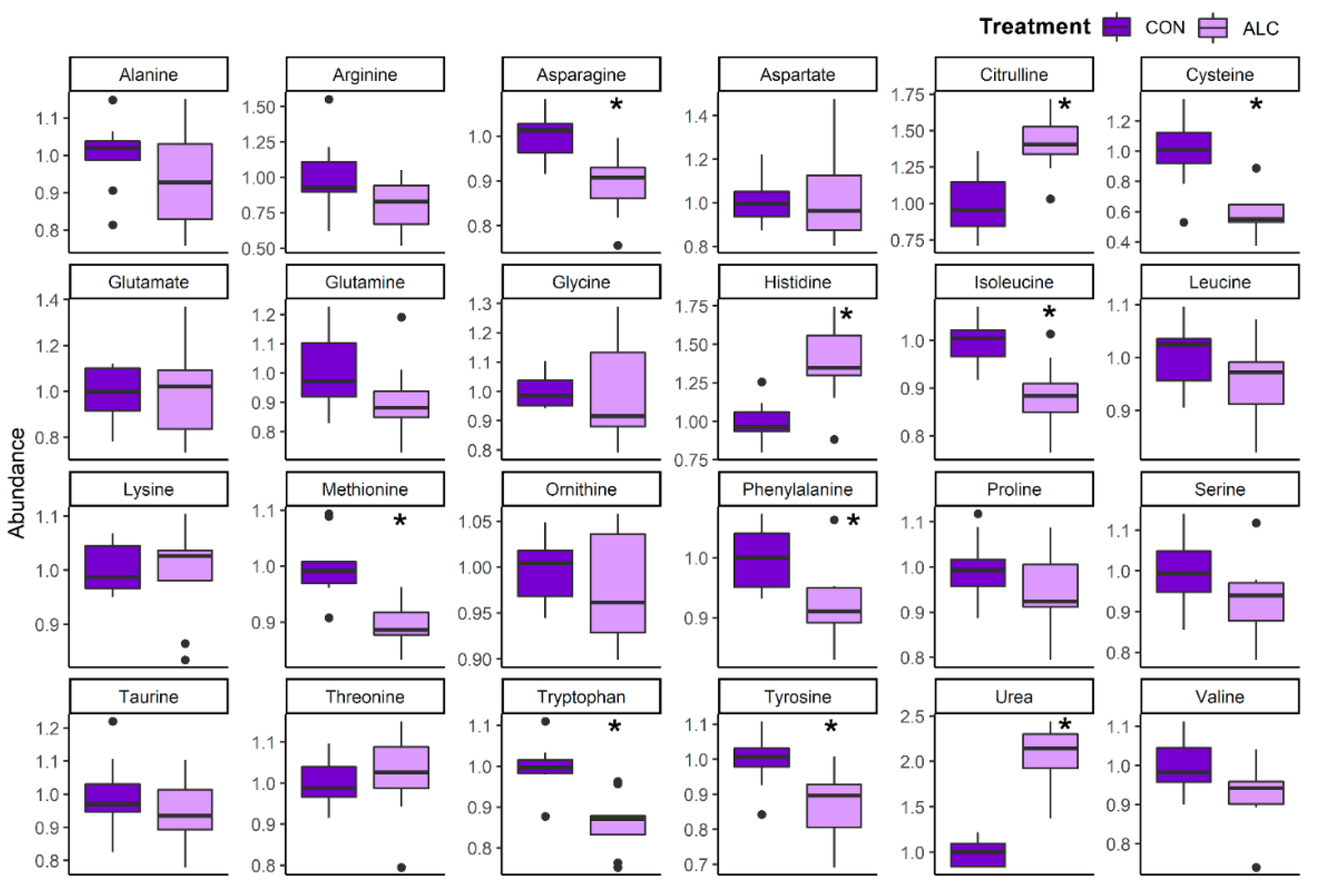
3.4. Maternal Metabolite Profile Is Consistent with the Known Impact of Alcohol on Hepatic Metabolism
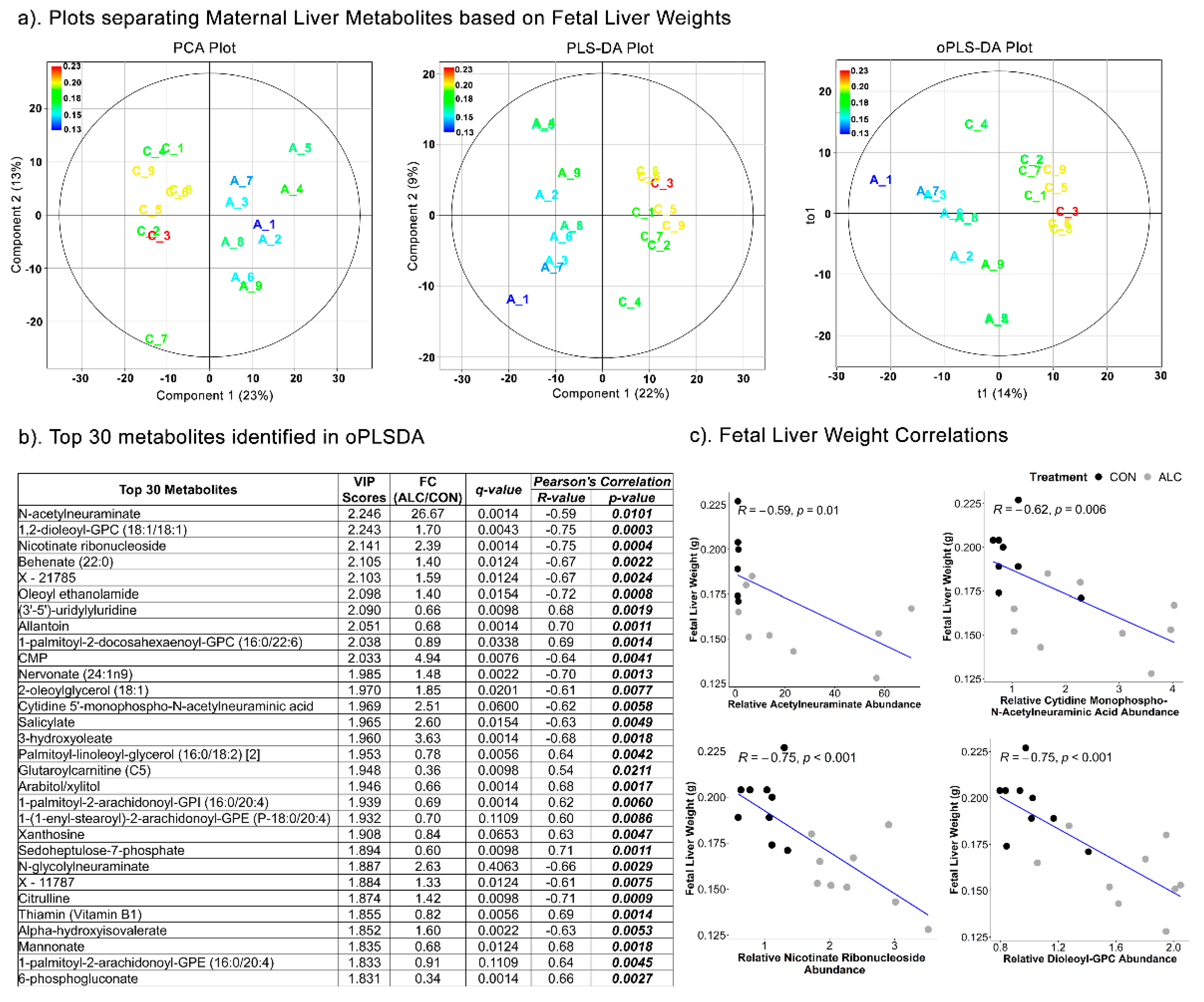
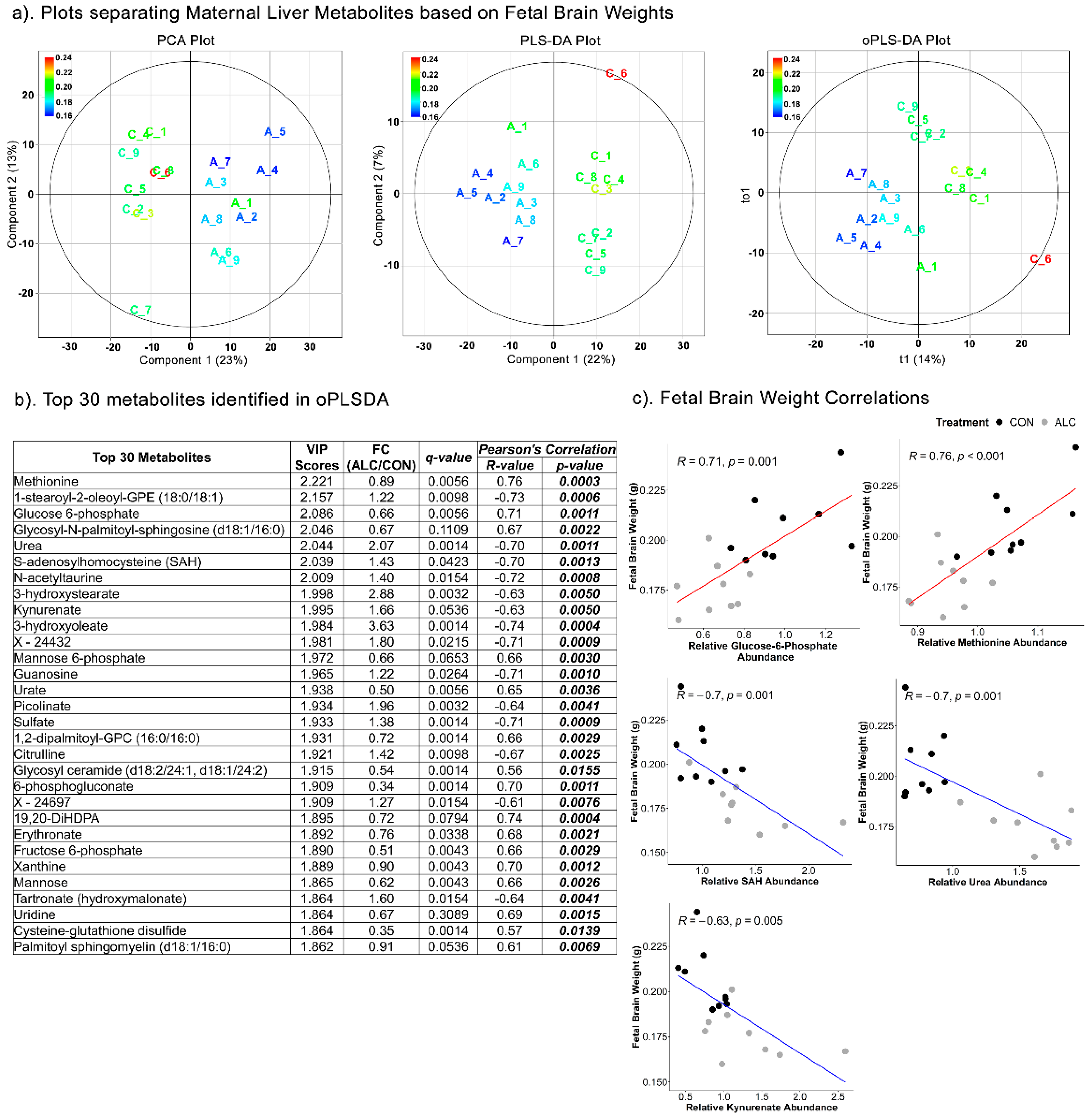
3.5. Maternal Hepatic Metabolites Are Predictive of Fetal Phenotypic Outcomes
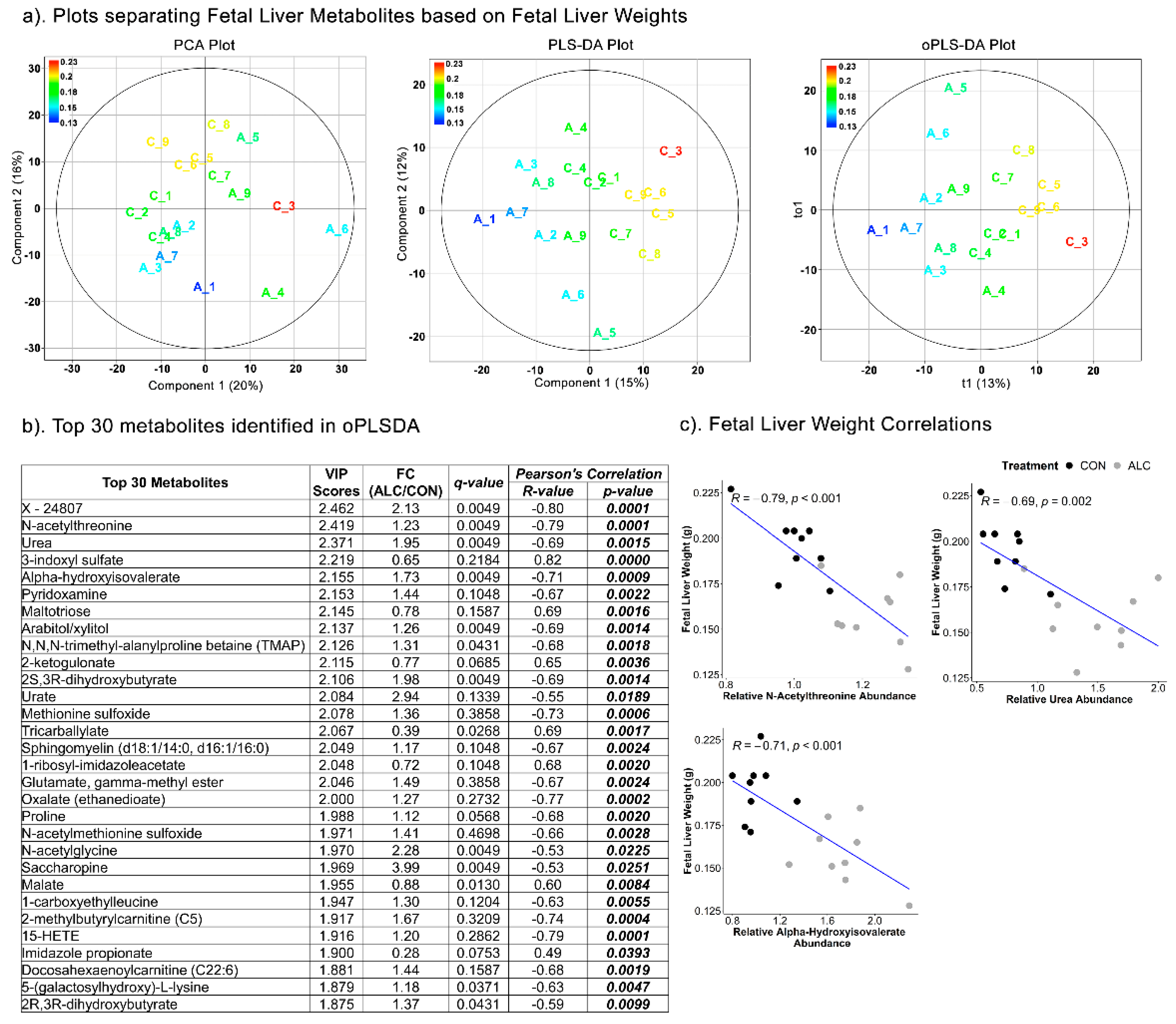
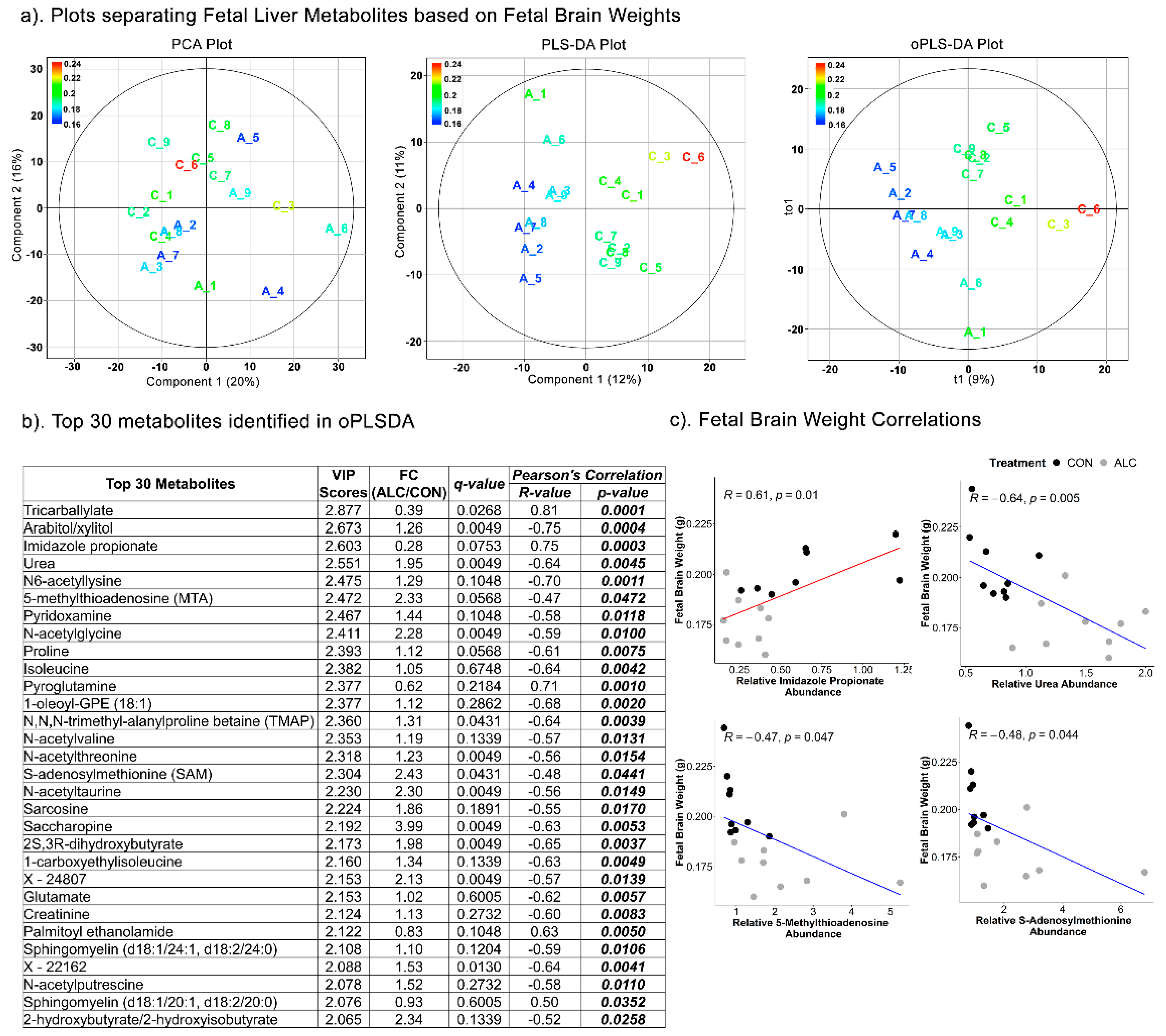
3.6. Fetal Hepatic Metabolites Predict Fetal Brain Weight but Not Fetal Body and Liver Weight
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, J.K.; Giesbrecht, H.E.; Eskin, M.N.; Aliani, M.; Suh, M. Nutrition implications for fetal alcohol spectrum disorder. Adv. Nutr. 2014, 5, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Borras-Novell, C.; Casanova, M.A.; Pascual Tutusaus, M.; Ferrero Martinez, S.; Gomez Roig, M.D.; Garcia-Algar, O. The Effects of Alcohol and Drugs of Abuse on Maternal Nutritional Profile during Pregnancy. Nutrients 2018, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.R.; Fink, B.A.; Fuglestad, A.J.; Eckerle, J.K.; Boys, C.J.; Sandness, K.E.; Radke, J.P.; Miller, N.C.; Lindgren, C.; Brearley, A.M.; et al. Four-year follow-up of a randomized controlled trial of choline for neurodevelopment in fetal alcohol spectrum disorder. J. Neurodev. Disord. 2020, 12, 9. [Google Scholar] [CrossRef]
- Thomas, J.D. Choline supplementation as an intervention for fetal alcohol spectrum disorders: A commentary. Alcohol. Clin. Exp. Res. 2021, 45, 2465–2467. [Google Scholar] [CrossRef]
- Helfrich, K.K.; Saini, N.; Kling, P.J.; Smith, S.M. Maternal iron nutriture as a critical modulator of fetal alcohol spectrum disorder risk in alcohol-exposed pregnancies. Biochem. Cell Biol. 2018, 96, 204–212. [Google Scholar] [CrossRef]
- Serrano, M.; Han, M.; Brinez, P.; Linask, K.K. Fetal alcohol syndrome: Cardiac birth defects in mice and prevention with folate. Am. J. Obstet. Gynecol. 2010, 203, 75.e7–75.e15. [Google Scholar] [CrossRef]
- Shi, Y.; Li, J.; Chen, C.; Gong, M.; Chen, Y.; Liu, Y.; Chen, J.; Li, T.; Song, W. 5-Mehtyltetrahydrofolate rescues alcohol-induced neural crest cell migration abnormalities. Mol. Brain 2014, 7, 67. [Google Scholar] [CrossRef]
- Patten, A.R.; Sickmann, H.M.; Dyer, R.A.; Innis, S.M.; Christie, B.R. Omega-3 fatty acids can reverse the long-term deficits in hippocampal synaptic plasticity caused by prenatal ethanol exposure. Neurosci. Lett. 2013, 551, 7–11. [Google Scholar] [CrossRef]
- Wellmann, K.A.; George, F.; Brnouti, F.; Mooney, S.M. Docosahexaenoic acid partially ameliorates deficits in social behavior and ultrasonic vocalizations caused by prenatal ethanol exposure. Behav. Brain Res. 2015, 286, 201–211. [Google Scholar] [CrossRef]
- Kusat Ol, K.; Kanbak, G.; Oglakci Ilhan, A.; Burukoglu, D.; Yucel, F. The investigation of the prenatal and postnatal alcohol exposure-induced neurodegeneration in rat brain: Protection by betaine and/or omega-3. Childs Nerv. Syst. 2016, 32, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Feltham, B.A.; Louis, X.L.; Eskin, M.N.A.; Suh, M. Docosahexaenoic Acid: Outlining the Therapeutic Nutrient Potential to Combat the Prenatal Alcohol-Induced Insults on Brain Development. Adv. Nutr. 2020, 11, 724–735. [Google Scholar] [CrossRef]
- Sandlers, Y. The future perspective: Metabolomics in laboratory medicine for inborn errors of metabolism. Transl. Res. 2017, 189, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Parfieniuk, E.; Zbucka-Kretowska, M.; Ciborowski, M.; Kretowski, A.; Barbas, C. Untargeted metabolomics: An overview of its usefulness and future potential in prenatal diagnosis. Exp. Rev. Proteom. 2018, 15, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Chabenne, A.; Moon, C.; Ojo, C.; Khogali, A.; Nepal, B.; Sharma, S. Biomarkers in fetal alcohol syndrome. Biomark. Genom. Med. 2014, 6, 12–22. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Aliani, M. Metabolomics and fetal alcohol spectrum disorder. Biochem. Cell Biol. 2018, 96, 198–203. [Google Scholar] [CrossRef]
- Campbell, I. Liver: Metabolic functions. Anaesth. Intensive Care Med. 2006, 7, 51–54. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Ferrarini, A.; Di Poto, C.; He, S.; Tu, C.; Varghese, R.S.; Kara Balla, A.; Jayatilake, M.; Li, Z.; Ghaffari, K.; Fan, Z.; et al. Metabolomic Analysis of Liver Tissues for Characterization of Hepatocellular Carcinoma. J. Proteome Res. 2019, 18, 3067–3076. [Google Scholar] [CrossRef]
- Zheng, H.; Cai, A.; Shu, Q.; Niu, Y.; Xu, P.; Li, C.; Lin, L.; Gao, H. Tissue-Specific Metabolomics Analysis Identifies the Liver as a Major Organ of Metabolic Disorders in Amyloid Precursor Protein/Presenilin 1 Mice of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 1218–1227. [Google Scholar] [CrossRef]
- Deng, P.; Barney, J.; Petriello, M.C.; Morris, A.J.; Wahlang, B.; Hennig, B. Hepatic metabolomics reveals that liver injury increases PCB 126-induced oxidative stress and metabolic dysfunction. Chemosphere 2019, 217, 140–149. [Google Scholar] [CrossRef]
- Shi, X.; Wahlang, B.; Wei, X.; Yin, X.; Falkner, K.C.; Prough, R.A.; Kim, S.H.; Mueller, E.G.; McClain, C.J.; Cave, M.; et al. Metabolomic analysis of the effects of polychlorinated biphenyls in nonalcoholic fatty liver disease. J. Proteome Res. 2012, 11, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. ALCOHOL: Its metabolism and interaction with nutrients. Annu. Rev. Nutr. 2000, 20, 395–430. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Mandrekar, P. Focus on: Alcohol and the liver. Alcohol. Res. Health 2010, 33, 87–96. [Google Scholar] [PubMed]
- Singal, A.K.; Charlton, M.R. Nutrition in alcoholic liver disease. Clin. Liver. Dis. 2012, 16, 805–826. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. 2017, 38, 147–161. [Google Scholar]
- Cioarca-Nedelcu, R.; Atanasiu, V.; Stoian, I. Alcoholic liver disease-from steatosis to cirrhosis—A biochemistry approach. J. Med. Life 2021, 14, 594–599. [Google Scholar] [CrossRef]
- Pohl, K.; Moodley, P.; Dhanda, A.D. Alcohol’s Impact on the Gut and Liver. Nutrients 2021, 13, 3170. [Google Scholar] [CrossRef]
- Virdee, M.S.; Saini, N.; Kay, C.D.; Neilson, A.P.; Kwan, S.T.C.; Helfrich, K.K.; Mooney, S.M.; Smith, S.M. An enriched biosignature of gut microbiota-dependent metabolites characterizes maternal plasma in a mouse model of fetal alcohol spectrum disorder. Sci. Rep. 2021, 11, 248. [Google Scholar] [CrossRef]
- Mooney, S.M.; Pjetri, E.; Friday, W.B.; Smith, S.M. Growth and behavioral differences in a C57BL/6J mouse model of prenatal alcohol exposure. Alcohol 2021, 97, 51–57. [Google Scholar] [CrossRef]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef]
- Saini, N.; Virdee, M.; Helfrich, K.K.; Kwan, S.T.C.; Smith, S.M. Global metabolomic profiling reveals hepatic biosignatures that reflect the unique metabolic needs of late-term mother and fetus. Metabolomics 2021, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Zorn, A.M. Liver development. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2008. [Google Scholar]
- Gong, T.; Zhang, C.; Ni, X.; Li, X.; Li, J.; Liu, M.; Zhan, D.; Xia, X.; Song, L.; Zhou, Q.; et al. A time-resolved multi-omic atlas of the developing mouse liver. Genome Res. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.N.; Shashidhar, A.; Ashok, C. In utero fuel homeostasis: Lessons for a clinician. Indian J. Endocrinol. Metab. 2013, 17, 60–68. [Google Scholar] [CrossRef]
- Gruppuso, P.A.; Brautigan, D.L. Induction of hepatic glycogenesis in the fetal rat. Am. J. Physiol. 1989, 256, E49–E54. [Google Scholar] [CrossRef] [PubMed]
- Blakley, P.M.; Scott, W.J., Jr. Determination of the proximate teratogen of the mouse fetal alcohol syndrome. 2. Pharmacokinetics of the placental transfer of ethanol and acetaldehyde. Toxicol. Appl. Pharmacol. 1984, 72, 364–371. [Google Scholar] [CrossRef]
- Zorzano, A.; Herrera, E. Disposition of ethanol and acetaldehyde in late pregnant rats and their fetuses. Pediatr. Res. 1989, 25, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Burd, L.; Blair, J.; Dropps, K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J. Perinatol. 2012, 32, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.W.; Smith, G.N.; Patrick, J.; Richardson, B.; Brien, J.F. Activity of alcohol dehydrogenase and aldehyde dehydrogenase in maternal liver, fetal liver and placenta of the near-term pregnant ewe. Dev. Pharmacol. Ther. 1989, 12, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Nakhoul, M.R.; Seif, K.E.; Haddad, N.; Haddad, G.E. Fetal Alcohol Exposure: The Common Toll. J. Alcohol. Drug Depend. 2017, 5, 1000257. [Google Scholar] [CrossRef]
- Kwan, S.T.C.; Presswood, B.H.; Helfrich, K.K.; Baulch, J.W.; Mooney, S.M.; Smith, S.M. An interaction between fetal sex and placental weight and efficiency predicts intrauterine growth in response to maternal protein insufficiency and gestational exposure window in a mouse model of FASD. Biol. Sex. Differ. 2020, 11, 40. [Google Scholar] [CrossRef]
- Krebs, H.A.; Freedland, R.A.; Hems, R.; Stubbs, M. Inhibition of hepatic gluconeogenesis by ethanol. Biochem. J. 1969, 112, 117–124. [Google Scholar] [CrossRef]
- Lumeng, L.; Davis, E.J. Mechanism of ethanol suppression of gluconeogenesis. Inhibition of phosphoenolpyruvate synthesis from glutamate and alpha-ketaglutarate. J. Biol. Chem. 1970, 245, 3179–3185. [Google Scholar] [CrossRef]
- Mokuda, O.; Tanaka, H.; Hayashi, T.; Ooka, H.; Okazaki, R.; Sakamoto, Y. Ethanol stimulates glycogenolysis and inhibits both glycogenesis via gluconeogenesis and from exogenous glucose in perfused rat liver. Ann. Nutr. Metab. 2004, 48, 276–280. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Hales, C.N. Role of fetal and infant growth in programming metabolism in later life. Biol. Rev. Camb. Philos. Soc. 1997, 72, 329–348. [Google Scholar] [CrossRef]
- Kwon, E.J.; Kim, Y.J. What is fetal programming? A lifetime health is under the control of in utero health. Obstet. Gynecol. Sci. 2017, 60, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Ali Shah, S.M.; Munir, N.; Daniyal, M.; Tahir, I.M.; Mahmood, Z.; Irshad, M.; Akhlaq, M.; Sultana, S.; Zainab, R. Hexose monophosphate shunt, the role of its metabolites and associated disorders: A review. J. Cell Physiol. 2019, 234, 14473–14482. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Aziz, H.; Mohiuddin, S.S. Biochemistry, Hexose Monophosphate Pathway. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Badawy, A.A.B. A Review of the Effects of Alcohol on Carbohydrate Metabolism. Alcohol Alcohol. 1977, 12, 120–136. [Google Scholar] [CrossRef]
- Tran, M.; Yang, Z.; Liangpunsakul, S.; Wang, L. Metabolomics Analysis Revealed Distinct Cyclic Changes of Metabolites Altered by Chronic Ethanol-Plus-Binge and Shp Deficiency. Alcohol. Clin. Exp. Res. 2016, 40, 2548–2556. [Google Scholar] [CrossRef]
- Ma, T.; Li, Y.; Zhu, Y.; Jiang, S.; Cheng, C.; Peng, Z.; Xu, L. Differential Metabolic Pathways and Metabolites in a C57BL/6J Mouse Model of Alcoholic Liver Disease. Med. Sci. Monit. 2020, 26, e924602. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, M.W.; Sevin, D.C.; Klee, M.L.; Dieter, S.; Sauer, U.; Sommer, W.H. The neurometabolic fingerprint of excessive alcohol drinking. Neuropsychopharmacology 2015, 40, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.W.; Carter, R.C.; Molteno, C.D.; Stanton, M.E.; Herbert, J.S.; Lindinger, N.M.; Lewis, C.E.; Dodge, N.C.; Hoyme, H.E.; Zeisel, S.H.; et al. Efficacy of Maternal Choline Supplementation During Pregnancy in Mitigating Adverse Effects of Prenatal Alcohol Exposure on Growth and Cognitive Function: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 2018, 42, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Kaminen-Ahola, N. Fetal alcohol spectrum disorders: Genetic and epigenetic mechanisms. Prenat. Diagn. 2020, 40, 1185–1192. [Google Scholar] [CrossRef]
- Kharbanda, K.K. Alcoholic liver disease and methionine metabolism. Semin. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Choline: Critical role during fetal development and dietary requirements in adults. Annu. Rev. Nutr. 2006, 26, 229–250. [Google Scholar] [CrossRef] [PubMed]
- Kwan, S.T.C.; Ricketts, D.K.; Presswood, B.H.; Smith, S.M.; Mooney, S.M. Prenatal choline supplementation during mouse pregnancy has differential effects in alcohol-exposed fetal organs. Alcohol. Clin. Exp. Res. 2021, 45, 2471–2484. [Google Scholar] [CrossRef]
- Steane, S.E.; Fielding, A.M.; Kent, N.L.; Andersen, I.; Browne, D.J.; Tejo, E.N.; Gardebjer, E.M.; Kalisch-Smith, J.I.; Sullivan, M.A.; Moritz, K.M.; et al. Maternal choline supplementation in a rat model of periconceptional alcohol exposure: Impacts on the fetus and placenta. Alcohol. Clin. Exp. Res. 2021, 45, 2130–2146. [Google Scholar] [CrossRef]
- Palmer, J.A.; Poenitzsch, A.M.; Smith, S.M.; Conard, K.R.; West, P.R.; Cezar, G.G. Metabolic biomarkers of prenatal alcohol exposure in human embryonic stem cell-derived neural lineages. Alcohol. Clin. Exp. Res. 2012, 36, 1314–1324. [Google Scholar] [CrossRef]
- Sillanaukee, P.; Ponnio, M.; Seppa, K. Sialic acid: New potential marker of alcohol abuse. Alcohol. Clin. Exp. Res. 1999, 23, 1039–1043. [Google Scholar] [CrossRef]
- Romppanen, J.; Punnonen, K.; Anttila, P.; Jakobsson, T.; Blake, J.; Niemela, O. Serum sialic acid as a marker of alcohol consumption: Effect of liver disease and heavy drinking. Alcohol. Clin. Exp. Res. 2002, 26, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Chrostek, L.; Cylwik, B.; Krawiec, A.; Korcz, W.; Szmitkowski, M. Relationship between serum sialic acid and sialylated glycoproteins in alcoholics. Alcohol Alcohol. 2007, 42, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Sriharan, M.; Reichelt, A.J.; Opperman, M.L.; Duncan, B.B.; Mengue, S.S.; Crook, M.A.; Schmidt, M.I. Total sialic acid and associated elements of the metabolic syndrome in women with and without previous gestational diabetes. Diabetes Care 2002, 25, 1331–1335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Findik, R.B.; Yilmaz, F.M.; Yilmaz, G.; Yilmaz, H.; Karakaya, J. Sialic acid levels in the blood in pregnant women with impaired glucose tolerance test. Arch. Gynecol. Obstet. 2012, 286, 913–916. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top Pathways | Total Compounds | Hits | Raw p | FDR | Metabolites Identified |
|---|---|---|---|---|---|
| Purine metabolism | 66 | 6 | 4.01 × 10−10 | 1.80 × 10−8 | Xanthine, urate, allantoin, guanosine, sulfate, urea |
| Pentose phosphate pathway | 22 | 6 | 8.11 × 10−10 | 1.83 × 10−8 | Glucose 6-phosphate, sedoheptulose 7-phosphate, fructose 6-phosphate, 6-phosphogluconate, gluconic acid, glycerate |
| Pyrimidine metabolism | 39 | 4 | 2.21 × 10−8 | 3.31 × 10−7 | 3-ureidopropionate, cytidine, CMP, thymine |
| Nicotinate and nicotinamide metabolism | 15 | 3 | 4.40 × 10−7 | 3.96 × 10−6 | Quinolinate, nicotinate D-ribonucleoside, nicotinamide-beta-riboside |
| Glycolysis/gluconeogenesis | 26 | 3 | 3.12 × 10−6 | 2.00 × 10−5 | Thiamin diphosphate, fructose 6-phosphate, glucose 6-phosphate |
| Fatty acid biosynthesis | 47 | 3 | 7.81 × 10−6 | 4.39 × 10−5 | Tetradecanoic acid, dodecanoic acid, decanoic acid |
| Metabolite Name | FC (ALC/CON) | p-Value | q-Value |
|---|---|---|---|
| N-Acetylated Amino-acid Products | |||
| N-Acetylalanine | 0.82 | 0.0142 | 0.0536 |
| N-Acetylasparagine | 1.49 | 0.0188 | 0.0653 |
| N-Acetylaspartate (NAA) | 0.93 | 0.7304 | 0.8229 |
| N-Acetylglutamate | 1.92 | 0.0056 | 0.0264 |
| N-Acetylglutamine | 1.24 | 0.3401 | 0.4994 |
| N-Acetylglycine | 1.35 | 0.7304 | 0.8229 |
| N-Acetylserine | 0.93 | 0.2224 | 0.3773 |
| N-Acetylthreonine | 1.29 | 0.0106 | 0.0423 |
| N-Acetylhistidine | 1.49 | 0.0106 | 0.0423 |
| N-Acetylleucine | 1.02 | 0.9314 | 0.9512 |
| N-Acetylvaline | 0.98 | 0.8633 | 0.9140 |
| N-Acetylcysteine | 0.99 | 0.7962 | 0.8632 |
| N-Acetylmethionine | 0.85 | 0.0503 | 0.1334 |
| N-Acetyltaurine | 1.40 | 0.0028 | 0.0154 |
| N-Acetylphenylalanine | 1.15 | 0.1135 | 0.2443 |
| Essential Amino-Acid Catabolites | |||
| 2-hydroxy-3-methylvalerate | 1.43 | 0.0315 | 0.0961 |
| 3-hydroxyisobutyrate | 1.39 | 0.0625 | 0.1582 |
| 4-methyl-2-oxopentanoate | 0.53 | 0.2868 | 0.4505 |
| α-hydroxyisovalerate | 1.60 | 0.0001 | 0.0022 |
| β-hydroxyisovalerate | 0.98 | 0.8633 | 0.9140 |
| β-hydroxyisovaleroylcarnitine | 1.10 | 0.0400 | 0.1109 |
| Isovalerylcarnitine (C5) | 1.40 | 0.1903 | 0.3429 |
| Isovalerylglycine | 1.46 | 0.2581 | 0.4063 |
| Pipecolate | 1.67 | 0.0000 | 0.0014 |
| Picolinate | 1.96 | 0.0002 | 0.0032 |
| Fatty-Acyl Carnitines | |||
| Carnitine | 0.94 | 0.5457 | 0.6749 |
| Adipoylcarnitine (C6-DC) | 0.36 | 0.0062 | 0.0287 |
| 3-Methyladipoylcarnitine (C7-DC) | 0.66 | 0.0000 | 0.0014 |
| (R)-3-hydroxybutyrylcarnitine | 0.53 | 0.0106 | 0.0423 |
| (S)-3-hydroxybutyrylcarnitine | 0.51 | 0.0078 | 0.0338 |
| 3-hydroxyoleoylcarnitine | 1.98 | 0.0079 | 0.0343 |
| Acetylcarnitine (C2) | 0.87 | 0.2581 | 0.4063 |
| Propionylcarnitine (C3) | 0.67 | 0.1575 | 0.3089 |
| Malonylcarnitine | 0.65 | 0.0171 | 0.0637 |
| Isobutyrylcarnitine (C4) | 0.69 | 0.0625 | 0.1582 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saini, N.; Virdee, M.S.; Helfrich, K.K.; Kwan, S.T.C.; Mooney, S.M.; Smith, S.M. Untargeted Metabolome Analysis Reveals Reductions in Maternal Hepatic Glucose and Amino Acid Content That Correlate with Fetal Organ Weights in a Mouse Model of Fetal Alcohol Spectrum Disorders. Nutrients 2022, 14, 1096. https://doi.org/10.3390/nu14051096
Saini N, Virdee MS, Helfrich KK, Kwan STC, Mooney SM, Smith SM. Untargeted Metabolome Analysis Reveals Reductions in Maternal Hepatic Glucose and Amino Acid Content That Correlate with Fetal Organ Weights in a Mouse Model of Fetal Alcohol Spectrum Disorders. Nutrients. 2022; 14(5):1096. https://doi.org/10.3390/nu14051096
Chicago/Turabian StyleSaini, Nipun, Manjot S. Virdee, Kaylee K. Helfrich, Sze Ting Cecilia Kwan, Sandra M. Mooney, and Susan M. Smith. 2022. "Untargeted Metabolome Analysis Reveals Reductions in Maternal Hepatic Glucose and Amino Acid Content That Correlate with Fetal Organ Weights in a Mouse Model of Fetal Alcohol Spectrum Disorders" Nutrients 14, no. 5: 1096. https://doi.org/10.3390/nu14051096
APA StyleSaini, N., Virdee, M. S., Helfrich, K. K., Kwan, S. T. C., Mooney, S. M., & Smith, S. M. (2022). Untargeted Metabolome Analysis Reveals Reductions in Maternal Hepatic Glucose and Amino Acid Content That Correlate with Fetal Organ Weights in a Mouse Model of Fetal Alcohol Spectrum Disorders. Nutrients, 14(5), 1096. https://doi.org/10.3390/nu14051096