Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging


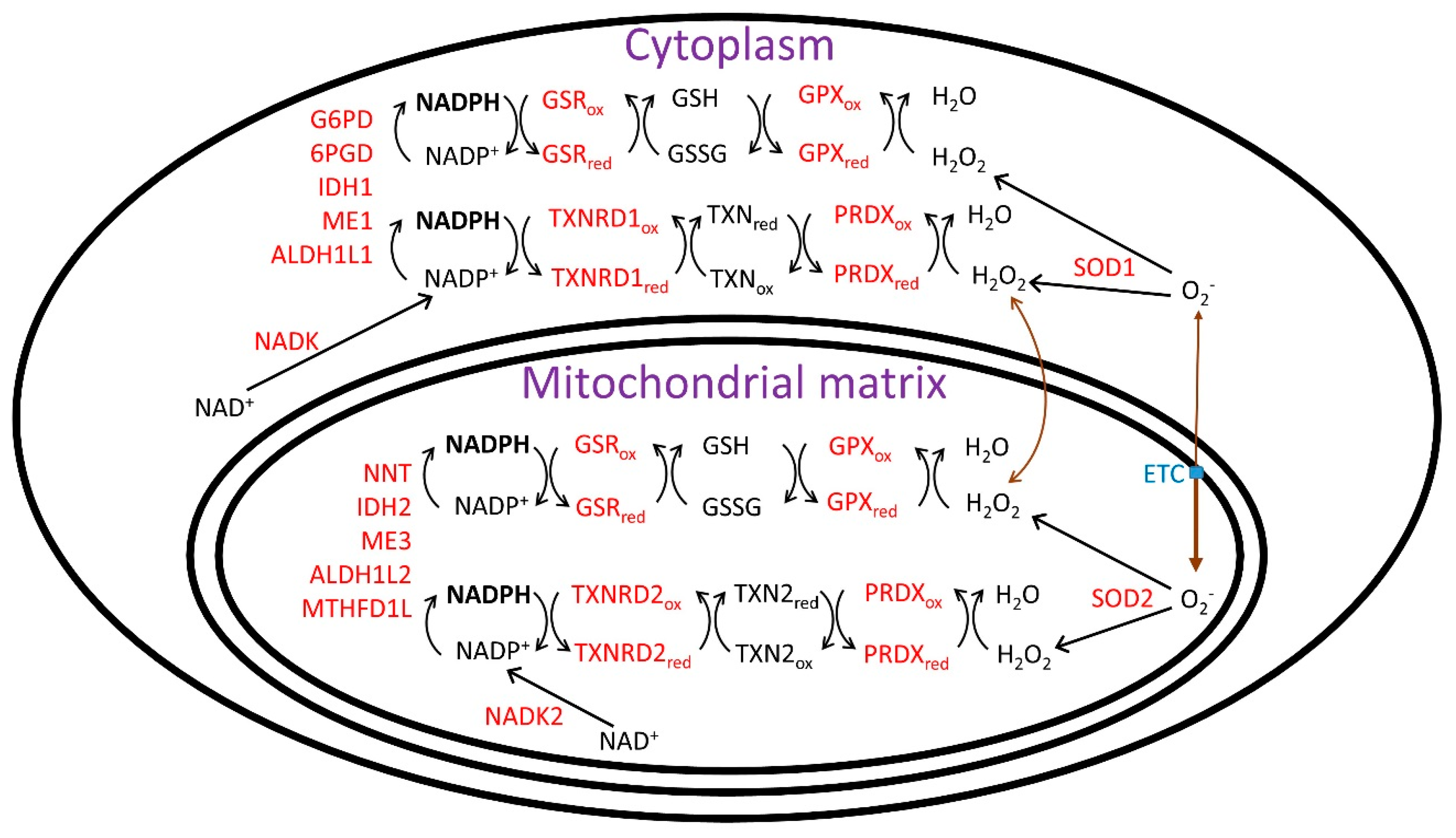{kind=link}
Abstract
1. NADPH and the Redox Theories of Aging
2. Loss of NAD+ as a Major Cause for Loss of NADPH With Aging
3. Aging is Associated with Redox Imbalance
4. Reductive Stress Can Influence the Rate of Aging
5. Could the Low Levels of Cytoplasmic Glutathione Peroxidase Activity in Long-Lived Species Function in the Preservation of [NADP+]/[NADPH] and GSSG/GSH with Aging to Promote Longevity?
6. The NADPH-Thioredoxin Reductase Redox System and Aging
7. The NADPH-Glutathione Reductase Redox System and Aging
8. Evidence for the Regulation of Longevity by the NADPH-Powered Redox Systems in Drosophila
9. Evidence for the Regulation of Longevity by the NADPH-Powered Redox Systems in Mammals
10. Evidence for the Regulation of Longevity by the NADPH-Powered Redox Systems in Budding Yeast
11. Evidence for the Regulation of Longevity by NADPH-Powered Redox Systems in C. Elegans Nematodes
12. Calorie Restriction May Extend Lifespan In Part through the NADPH-Driven Redox Systems
13. Regulation of Aging by NADPH and the Circadian Clock
14. NAD+ Kinases as Possible Regulators of NADPH Levels and Longevity
15. Pharmacological Modulation of Cellular Redox Status to Prevent Cellular Senescence and Delay Aging
16. Summary and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Aw, T.Y. Postnatal Changes in Pyridine Nucleotides in Rat Hepatocytes: Composition and O2 Dependence. Pediatr. Res. 1991, 30, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Levault, K.R.; Brewer, G.J. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 2014, 13, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar] [CrossRef]
- Baciou, L.; Masoud, R.; Souabni, H.; Serfaty, X.; Karimi, G.; Bizouarn, T.; Houee Levin, C. Phagocyte NADPH oxidase, oxidative stress and lipids: Anti- or pro ageing? Mech. Ageing Dev. 2018, 172, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sun, M.; Li, M.; Zhang, D.; Han, F.; Wu, J.C.; Fukunaga, K.; Chen, Z.; Qin, Z.H. Combination of NAD(+) and NADPH Offers Greater Neuroprotection in Ischemic Stroke Models by Relieving Metabolic Stress. Mol. Neurobiol. 2018, 55, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Doonan, R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Perez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.I.; Van Remmen, H.; Bokov, A.; Epstein, C.J.; Vijg, J.; Richardson, A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 2009, 8, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; McLaren, J.S.; Meyer, C.; Duncan, J.S.; Redman, P.; Collins, A.R.; Duthie, G.G.; Speakman, J.R. Life-long vitamin C supplementation in combination with cold exposure does not affect oxidative damage or lifespan in mice, but decreases expression of antioxidant protection genes. Mech. Ageing Dev. 2006, 127, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Ernst, I.M.; Pallauf, K.; Bendall, J.K.; Paulsen, L.; Nikolai, S.; Huebbe, P.; Roeder, T.; Rimbach, G. Vitamin E supplementation and lifespan in model organisms. Ageing Res. Rev. 2013, 12, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, D.; Cacho-Valadez, B.; Liu, J.L.; Wang, Y.; Yee, C.; Bernard, K.; Khaki, A.; Breton, L.; Hekimi, S. Antioxidants reveal an inverted U-shaped dose-response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans. Aging Cell 2017, 16, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Sohal, R.S. Does overexpression of Cu,Zn-SOD extend life span in Drosophila melanogaster? Exp. Gerontol. 2003, 38, 227–230. [Google Scholar] [CrossRef]
- Shibamura, A.; Ikeda, T.; Nishikawa, Y. A method for oral administration of hydrophilic substances to Caenorhabditis elegans: Effects of oral supplementation with antioxidants on the nematode lifespan. Mech. Ageing Dev. 2009, 130, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, H.N.; Gaetani, G.F. Catalase: A tetrameric enzyme with four tightly bound molecules of NADPH. Proc. Natl. Acad. Sci. USA 1984, 81, 4343–4347. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. (Lond. Engl. 1979) 2017, 131, 1669–1688. [Google Scholar] [CrossRef]
- Heidler, T.; Hartwig, K.; Daniel, H.; Wenzel, U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology 2010, 11, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Rate of generation of oxidative stress-related damage and animal longevity. Free Radic. Biol. Med. 2002, 33, 1167–1172. [Google Scholar] [CrossRef]
- Schindeldecker, M.; Stark, M.; Behl, C.; Moosmann, B. Differential cysteine depletion in respiratory chain complexes enables the distinction of longevity from aerobicity. Mech. Ageing Dev. 2011, 132, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R.; Barja, G.; Portero-Otin, M. Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: A homeoviscous-longevity adaptation? Ann. N. Y. Acad. Sci. 2002, 959, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B. Respiratory chain cysteine and methionine usage indicate a causal role for thiyl radicals in aging. Exp. Gerontol. 2011, 46, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal. 2014, 20, 1925–1953. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Pi, C.; Yang, Y.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; He, X. Nampt Expression Decreases Age-Related Senescence in Rat Bone Marrow Mesenchymal Stem Cells by Targeting Sirt1. PLoS ONE 2017, 12, e0170930. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Boslett, J.; Helal, M.; Chini, E.; Zweier, J.L. Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. J. Mol. Cell. Cardiol. 2018, 118, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Vu, C.Q.; Lu, P.J.; Chen, C.S.; Jacobson, M.K. 2′-Phospho-cyclic ADP-ribose, a calcium-mobilizing agent derived from NADP. J. Biol. Chem. 1996, 271, 4747–4754. [Google Scholar] [PubMed]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef]
- Ferrero, E.; Malavasi, F. A Natural History of the Human CD38 Gene. In Cyclic ADP-Ribose and NAADP: Structures, Metabolism and Functions; Lee, H.C., Ed.; Springer: Boston, MA, USA, 2002; pp. 65–79. [Google Scholar]
- Sur, M.; Dey, P.; Sarkar, A.; Bar, S.; Banerjee, D.; Bhat, S.; Mukherjee, P. Sarm1 induction and accompanying inflammatory response mediates age-dependent susceptibility to rotenone-induced neurotoxicity. Cell Death Discov. 2018, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Minnifield, N.; Sun, I.; Morre, D.J. NADP phosphatase as a marker in free-flow electrophoretic separations for cisternae of the Golgi apparatus midregion. Biochim. Biophys. Acta 1986, 881, 1–9. [Google Scholar] [CrossRef]
- Richter, C. NADP+ phosphatase: A novel mitochondrial enzyme. Biochem. Biophys. Res. Commun. 1987, 146, 253–257. [Google Scholar] [CrossRef]
- Balan, V.; Miller, G.S.; Kaplun, L.; Balan, K.; Chong, Z.Z.; Li, F.; Kaplun, A.; VanBerkum, M.F.; Arking, R.; Freeman, D.C.; et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 2008, 283, 27810–27819. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, T.L.; Huang, L.; Lange, S.E.; Hanna-Rose, W. Nicotinamidase modulation of NAD+ biosynthesis and nicotinamide levels separately affect reproductive development and cell survival in C. elegans. Development 2009, 136, 3637–3646. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, M.R.; Wang, W.; Holleran, L.M.; Hanna-Rose, W. Uridine monophosphate synthetase enables eukaryotic de novo NAD+ biosynthesis from quinolinic acid. J. Biol. Chem. 2017, 292, 11147–11153. [Google Scholar] [CrossRef] [PubMed]
- Breda, C.; Sathyasaikumar, K.V.; Sograte Idrissi, S.; Notarangelo, F.M.; Estranero, J.G.; Moore, G.G.; Green, E.W.; Kyriacou, C.P.; Schwarcz, R.; Giorgini, F. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc. Natl. Acad. Sci. USA 2016, 113, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Copes, N.; Edwards, C.; Chaput, D.; Saifee, M.; Barjuca, I.; Nelson, D.; Paraggio, A.; Saad, P.; Lipps, D.; Stevens, S.M., Jr.; et al. Metabolome and proteome changes with aging in Caenorhabditis elegans. Exp. Gerontol. 2015, 72, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Arnold, L.; Orr, W.C. Effect of age on superoxide dismutase, catalase, glutathione reductase, inorganic peroxides, TBA-reactive material, GSH/GSSG, NADPH/NADP+ and NADH/NAD+ in Drosophila melanogaster. Mech. Ageing Dev. 1990, 56, 223–235. [Google Scholar] [CrossRef]
- Pollak, N.; Dolle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides—Small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Horikawa, M.; Nomura, T.; Sakamoto, K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology 2010, 11, 31–43. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Christensen, K.C.; Gazzaniga, F.; Pletnev, A.A.; Brenner, C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Boil. Chem. 2009, 284, 158–164. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Andziak, B.; Buffenstein, R. Disparate patterns of age-related changes in lipid peroxidation in long-lived naked mole-rats and shorter-lived mice. Aging Cell 2006, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969, 115, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Kwon, Y.; Son, T.; Cho, Y.S. Restoration of Mitochondrial NAD(+) Levels Delays Stem Cell Senescence and Facilitates Reprogramming of Aged Somatic Cells. Stem Cells 2016, 34, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Tischler, M.E.; Friedrichs, D.; Coll, K.; Williamson, J.R. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 1977, 184, 222–236. [Google Scholar] [CrossRef]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.C.; Ashraf, S.S.; Rokutan, K.; Johnston, R.B., Jr.; Thomas, J.A. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N.; et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Yap, L.P.; Garcia, J.V.; Han, D.; Cadenas, E. The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 2009, 61, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Canepa, L.; Ferraris, A.M.; Miglino, M.; Gaetani, G.F. Bound and unbound pyridine dinucleotides in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Biochim. Biophys. Acta 1991, 1074, 101–104. [Google Scholar] [CrossRef]
- Booty, L.M.; Gawel, J.M.; Cvetko, F.; Caldwell, S.T.; Hall, A.R.; Mulvey, J.F.; James, A.M.; Hinchy, E.C.; Prime, T.A.; Arndt, S.; et al. Selective Disruption of Mitochondrial Thiol Redox State in Cells and In Vivo. Cell Chem. Boil. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Mody, V.C., Jr.; Carlson, J.L.; Lynn, M.J.; Sternberg, P., Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD(+) Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.W.; Santos, S.R.; Morris, M.; Evans, M.; Alminana, D.; Guarente, L.; Marcotulli, E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: A randomized, double-blind, placebo-controlled study. Npj Aging Mech. Dis. 2017, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, J.; Sheng, R.; Li, M.; Wang, Y.; Han, R.; Han, F.; Chen, Z.; Qin, Z.H. Reduced Nicotinamide Adenine Dinucleotide Phosphate Inhibits MPTP-Induced Neuroinflammation and Neurotoxicity. Neuroscience 2018, 391, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C.; Halliwell, B. Mini-Review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, A.; Shi, M.; Chen, X.; Liu, R.; Li, T.; Zhang, C.; Zhang, Z.; Zhu, L.; Ju, Z.; et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018, 13, 2362–2386. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Gutscher, M.; Pauleau, A.L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.C.; Barata, A.G.; Grosshans, J.; Teleman, A.A.; Dick, T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Limphong, P.; Pieper, J.; Liu, Q.; Rodesch, C.K.; Christians, E.; Benjamin, I.J. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012, 26, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wadey, A.L.; Muyderman, H.; Kwek, P.T.; Sims, N.R. Mitochondrial glutathione uptake: Characterization in isolated brain mitochondria and astrocytes in culture. J. Neurochem. 2009, 109 (Suppl. 1), 101–108. [Google Scholar] [CrossRef] [PubMed]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 2015, 1847, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Benjamin, I.J. Reductive stress on life span extension in C. elegans. BMC Res. Notes 2008, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Boil. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, M.; Laun, P.; Dickinson, J.R.; Klocker, A.; Rinnerthaler, M.; Dawes, I.W.; Aung-Htut, M.T.; Breitenbach-Koller, L.; Caballero, A.; Nystrom, T.; et al. The role of mitochondria in the aging processes of yeast. Sub-Cell. Biochem. 2012, 57, 55–78. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Boil. 2015, 11, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Andziak, B.; Yang, T.; Buffenstein, R. The naked mole-rat response to oxidative stress: Just deal with it. Antioxid. Redox Signal. 2013, 19, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-nitrosoglutathione. Biochim. Biophys. Acta 2013, 1830, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Liang, H.; Ikeno, Y.; Qi, W.; Prolla, T.A.; Roberts, L.J., 2nd; Wolf, N.; Van Remmen, H.; Richardson, A. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Mockett, R.J.; Orr, W.C.; Rahmandar, J.J.; Sohal, B.H.; Sohal, R.S. Antioxidant status and stress resistance in long- and short-lived lines of Drosophila melanogaster. Exp. Gerontol. 2001, 36, 441–463. [Google Scholar] [CrossRef]
- Lopez-Torres, M.; Perez-Campo, R.; Rojas, C.; Cadenas, S.; Barja, G. Maximum life span in vertebrates: Relationship with liver antioxidant enzymes, glutathione system, ascorbate, urate, sensitivity to peroxidation, true malondialdehyde, in vivo H2O2, and basal and maximum aerobic capacity. Mech. Ageing Dev. 1993, 70, 177–199. [Google Scholar] [CrossRef]
- Heinze, I.; Bens, M.; Calzia, E.; Holtze, S.; Dakhovnik, O.; Sahm, A.; Kirkpatrick, J.M.; Szafranski, K.; Romanov, N.; Sama, S.N.; et al. Species comparison of liver proteomes reveals links to naked mole-rat longevity and human aging. BMC Biol. 2018, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Munro, D.; Baldy, C.; Pamenter, M.E.; Treberg, J.R. The exceptional longevity of the naked mole-rat may be explained by mitochondrial antioxidant defenses. Aging Cell 2019, e12916. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.M.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Emanuele, E.; Lucia, A.; Galvez, B.G. ‘Adipaging’: Ageing and obesity share biological hallmarks related to a dysfunctional adipose tissue. J. Physiol. 2016, 594, 3187–3207. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Purkayastha, S.; Cai, D. Hypothalamic microinflammation: A common basis of metabolic syndrome and aging. Trends Neurosci. 2015, 38, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Abdrakhmanova, A.; Zwicker, K.; Kerscher, S.; Zickermann, V.; Brandt, U. Tight binding of NADPH to the 39-kDa subunit of complex I is not required for catalytic activity but stabilizes the multiprotein complex. Biochim. Biophys. Acta 2006, 1757, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Hoppel, C.L. Oxidative phosphorylation and aging. Ageing Res. Rev. 2006, 5, 402–433. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. Mitochondrial complex I: A central regulator of the aging process. Cell Cycle 2011, 10, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Flores, L.C.; Roman, M.G.; Cheng, C.; Dube, S.; Allen, C.; Valentine, J.M.; Hubbard, G.B.; Bai, Y.; Saunders, T.L.; et al. Thioredoxin overexpression in both the cytosol and mitochondria accelerates age-related disease and shortens lifespan in male C57BL/6 mice. GeroScience 2018, 40, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Flores, L.C.; Roman, M.G.; Cunningham, G.M.; Cheng, C.; Dube, S.; Allen, C.; Van Remmen, H.; Hubbard, G.B.; Saunders, T.L.; Ikeno, Y. Continuous overexpression of thioredoxin 1 enhances cancer development and does not extend maximum lifespan in male C57BL/6 mice. Pathobiol. Aging Age Relat. Dis. 2018, 8, 1533754. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.M.; Lehr, M.; Gendron, C.M.; Pletcher, S.D.; Miller, R.A. Mitochondrial thioredoxin reductase 2 is elevated in long-lived primate as well as rodent species and extends fly mean lifespan. Aging Cell 2017, 16, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Mockett, R.J.; Benes, J.J.; Sohal, R.S. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J. Biol. Chem. 2003, 278, 26418–26422. [Google Scholar] [CrossRef] [PubMed]
- Mockett, R.J.; Sohal, R.S.; Orr, W.C. Overexpression of glutathione reductase extends survival in transgenic Drosophila melanogaster under hyperoxia but not normoxia. FASEB J. 1999, 13, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Rojanathammanee, L.; Rakoczy, S.; Brown-Borg, H.M. Growth hormone alters the glutathione S-transferase and mitochondrial thioredoxin systems in long-living Ames dwarf mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 Removal in Brain Mitochondria via the Thioredoxin/Peroxiredoxin System. J. Boil. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.; Morgan, B.; Riemer, J. Mitochondrial Glutathione: Regulation and Functions. Antioxid. Redox Signal. 2017, 27, 1162–1177. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Mendiratta, S.; Hill, K.E.; Burk, R.F. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J. Biol. Chem. 1997, 272, 22607–22610. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Bjorkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arner, E.S.; Spyrou, G.; Bjornstedt, M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Nalvarte, I.; Damdimopoulos, A.E.; Spyrou, G. Human mitochondrial thioredoxin reductase reduces cytochrome c and confers resistance to complex III inhibition. Free Radic. Biol. Med. 2004, 36, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Nordberg, J.; Holmgren, A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem. Biophys. Res. Commun. 1996, 225, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bjornstedt, M.; Hamberg, M.; Kumar, S.; Xue, J.; Holmgren, A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J. Biol. Chem. 1995, 270, 11761–11764. [Google Scholar] [CrossRef] [PubMed]
- Jee, C.; Vanoaica, L.; Lee, J.; Park, B.J.; Ahnn, J. Thioredoxin is related to life span regulation and oxidative stress response in Caenorhabditis elegans. Genes Cells 2005, 10, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Vizuete, A.; Fierro Gonzalez, J.C.; Gahmon, G.; Burghoorn, J.; Navas, P.; Swoboda, P. Lifespan decrease in a Caenorhabditis elegans mutant lacking TRX-1, a thioredoxin expressed in ASJ sensory neurons. FEBS Lett. 2006, 580, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bandyopadhyay, J.; Hwaang, H.S.; Park, B.J.; Cho, J.H.; Lee, J.I.; Ahnn, J.; Lee, S.K. Two thioredoxin reductases, trxr-1 and trxr-2, have differential physiological roles in Caenorhabditis elegans. Mol. Cells 2012, 34, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Cacho-Valadez, B.; Munoz-Lobato, F.; Pedrajas, J.R.; Cabello, J.; Fierro-Gonzalez, J.C.; Navas, P.; Swoboda, P.; Link, C.D.; Miranda-Vizuete, A. The characterization of the Caenorhabditis elegans mitochondrial thioredoxin system uncovers an unexpected protective role of thioredoxin reductase 2 in beta-amyloid peptide toxicity. Antioxid. Redox Signal. 2012, 16, 1384–1400. [Google Scholar] [CrossRef] [PubMed]
- Arodin, L.; Miranda-Vizuete, A.; Swoboda, P.; Fernandes, A.P. Protective effects of the thioredoxin and glutaredoxin systems in dopamine-induced cell death. Free Radic. Biol. Med. 2014, 73, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Oláhová, M.; Veal, E.A. A peroxiredoxin, PRDX-2, is required for insulin secretion and insulin/IIS-dependent regulation of stress resistance and longevity. Aging Cell 2015, 14, 558–568. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.; Huebner, C.; Markowitz, M.; Taube, N.; Harvanek, Z.M.; Jakob, U.; Knoefler, D. Do developmental temperatures affect redox level and lifespan in C. elegans through upregulation of peroxiredoxin? Redox Boil. 2017, 14, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Thamsen, M.; Kumsta, C.; Li, F.; Jakob, U. Is overoxidation of peroxiredoxin physiologically significant? Antioxid. Redox Signal. 2011, 14, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Moskovitz, J. The Functions of the Mammalian Methionine Sulfoxide Reductase System and Related Diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, H.M.; Kim, S.; Avanesov, A.S.; Lee, A.; Chun, B.-H.; Vorbruggen, G.; Gladyshev, V.N. Expression of the methionine sulfoxide reductase lost during evolution extends Drosophila lifespan in a methionine-dependent manner. Sci. Rep. 2018, 8, 1010. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Gladyshev, V.N. Methionine sulfoxide reductases: Selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem. J. 2007, 407, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Minniti, A.N.; Cataldo, R.; Trigo, C.; Vasquez, L.; Mujica, P.; Leighton, F.; Inestrosa, N.C.; Aldunate, R. Methionine sulfoxide reductase A expression is regulated by the DAF-16/FOXO pathway in Caenorhabditis elegans. Aging Cell 2009, 8, 690–705. [Google Scholar] [CrossRef] [PubMed]
- Bruce, L.; Singkornrat, D.; Wilson, K.; Hausman, W.; Robbins, K.; Huang, L.; Foss, K.; Binninger, D. In Vivo Effects of Methionine Sulfoxide Reductase Deficiency in Drosophila melanogaster. Antioxidants 2018, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Tang, X.D.; Chen, M.L.; Joiner, M.L.; Sun, G.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Iverson, L.; Wu, C.F.; et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 2748–2753. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kim, A.K.; Jung, S.A.; Kim, S.W.; Yu, K.; Lee, J.H. The Drosophila homolog of methionine sulfoxide reductase A extends lifespan and increases nuclear localization of FOXO. FEBS Lett. 2010, 584, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.B.; Kim, G.; Liu, C.; Wren, J.D.; Georgescu, C.; Richardson, A.; Levine, R.L. Effects of transgenic methionine sulfoxide reductase A (MsrA) expression on lifespan and age-dependent changes in metabolic function in mice. Redox Boil. 2016, 10, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, S.; Kim, G.; Levine, R.L. Drosophila methionine sulfoxide reductase A (MSRA) lacks methionine oxidase activity. Free Radic. Biol. Med. 2018, 131, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Cadenas, S.; Rojas, C.; Lopez-Torres, M.; Perez-Campo, R. A decrease of free radical production near critical targets as a cause of maximum longevity in animals. Comp. Biochem. Physiol. Part B 1994, 108, 501–512. [Google Scholar] [CrossRef]
- Kanzok, S.M.; Fechner, A.; Bauer, H.; Ulschmid, J.K.; Muller, H.M.; Botella-Munoz, J.; Schneuwly, S.; Schirmer, R.; Becker, K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 2001, 291, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Mora-Lorca, J.A.; Saenz-Narciso, B.; Gaffney, C.J.; Naranjo-Galindo, F.J.; Pedrajas, J.R.; Guerrero-Gomez, D.; Dobrzynska, A.; Askjaer, P.; Szewczyk, N.J.; Cabello, J.; et al. Glutathione reductase gsr-1 is an essential gene required for Caenorhabditis elegans early embryonic development. Free Radic. Biol. Med. 2016, 96, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Luersen, K.; Stegehake, D.; Daniel, J.; Drescher, M.; Ajonina, I.; Ajonina, C.; Hertel, P.; Woltersdorf, C.; Liebau, E. The glutathione reductase GSR-1 determines stress tolerance and longevity in Caenorhabditis elegans. PLoS ONE 2013, 8, e60731. [Google Scholar] [CrossRef] [PubMed]
- Stenvall, J.; Fierro-Gonzalez, J.C.; Swoboda, P.; Saamarthy, K.; Cheng, Q.; Cacho-Valadez, B.; Arner, E.S.; Persson, O.P.; Miranda-Vizuete, A.; Tuck, S. Selenoprotein TRXR-1 and GSR-1 are essential for removal of old cuticle during molting in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2011, 108, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Gomez, D.; Mora-Lorca, J.A.; Saenz-Narciso, B.; Naranjo-Galindo, F.J.; Munoz-Lobato, F.; Parrado-Fernandez, C.; Goikolea, J.; Cedazo-Minguez, A.; Link, C.D.; Neri, C.; et al. Loss of glutathione redox homeostasis impairs proteostasis by inhibiting autophagy-dependent protein degradation. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Luchak, J.M.; Prabhudesai, L.; Sohal, R.S.; Radyuk, S.N.; Orr, W.C. Modulating longevity in Drosophila by over- and underexpression of glutamate-cysteine ligase. Ann. N. Y. Acad. Sci. 2007, 1119, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Radyuk, S.N.; Prabhudesai, L.; Toroser, D.; Benes, J.J.; Luchak, J.M.; Mockett, R.J.; Rebrin, I.; Hubbard, J.G.; Sohal, R.S. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J. Biol. Chem. 2005, 280, 37331–37338. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, X.; Cui, X.; Li, J.; Wang, Z. Recombinant buckwheat glutaredoxin intake increases lifespan and stress resistance via hsf-1 upregulation in Caenorhabditis elegans. Exp. Gerontol. 2018, 104, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Andziak, B.; O’Connor, T.P.; Qi, W.; DeWaal, E.M.; Pierce, A.; Chaudhuri, A.R.; Van Remmen, H.; Buffenstein, R. High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell 2006, 5, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.; Go, Y.M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Rebrin, I.; Sohal, R.S. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp. Gerontol. 2004, 39, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Legan, S.K.; Rebrin, I.; Mockett, R.J.; Radyuk, S.N.; Klichko, V.I.; Sohal, R.S.; Orr, W.C. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 2008, 283, 32492–32499. [Google Scholar] [CrossRef] [PubMed]
- Luckinbill, L.S.; Riha, V.; Rhine, S.; Grudzien, T.A. The role of glucose-6-phosphate dehydrogenase in the evolution of longevity in Drosophila melanogaster. Heredity 1990, 65 Pt 1, 29–38. [Google Scholar] [CrossRef]
- Wang, C.-T.; Chen, Y.-C.; Wang, Y.-Y.; Huang, M.-H.; Yen, T.-L.; Li, H.; Liang, C.-J.; Sang, T.-K.; Ciou, S.-C.; Yuh, C.-H.; et al. Reduced neuronal expression of ribose-5-phosphate isomerase enhances tolerance to oxidative stress, extends lifespan, and attenuates polyglutamine toxicity in Drosophila. Aging Cell 2012, 11, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Farkas, R.; Danis, P.; Medved’ova, L.; Mechler, B.M.; Knopp, J. Regulation of cytosolic malate dehydrogenase by juvenile hormone in Drosophila melanogaster. Cell Biochem. Biophys. 2002, 37, 37–52. [Google Scholar] [CrossRef]
- Kim, G.H.; Lee, Y.E.; Lee, G.H.; Cho, Y.H.; Lee, Y.N.; Jang, Y.; Paik, D.; Park, J.J. Overexpression of malic enzyme in the larval stage extends Drosophila lifespan. Biochem. Biophys. Res. Commun. 2015, 456, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.; Jang, Y.G.; Lee, Y.E.; Lee, Y.N.; Yamamoto, R.; Gee, H.Y.; Yoo, S.; Bae, E.; Min, K.J.; Tatar, M.; et al. Misexpression screen delineates novel genes controlling Drosophila lifespan. Mech. Ageing Dev. 2012, 133, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Merritt, T.J.S.; Kuczynski, C.; Sezgin, E.; Zhu, C.-T.; Kumagai, S.; Eanes, W.F. Quantifying interactions within the NADP(H) enzyme network in Drosophila melanogaster. Genetics 2009, 182, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.S.; Golic, K.G. A targeted gene knockout in Drosophila. Genetics 2001, 157, 1307–1312. [Google Scholar] [PubMed]
- Tremblay, G.B.; Sohi, S.S.; Retnakaran, A.; MacKenzie, R.E. NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase is targeted to the cytoplasm in insect cell lines. FEBS Lett. 1995, 368, 177–182. [Google Scholar] [CrossRef]
- Yu, S.; Jang, Y.; Paik, D.; Lee, E.; Park, J.J. Nmdmc overexpression extends Drosophila lifespan and reduces levels of mitochondrial reactive oxygen species. Biochem. Biophys. Res. Commun. 2015, 465, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, G.L.; de Oliveira, A.K. Citric acid cycle: A mainstream metabolic pathway influencing life span in Drosophila melanogaster? Exp. Gerontol. 1996, 31, 705–715. [Google Scholar] [CrossRef]
- Arking, R.; Burde, V.; Graves, K.; Hari, R.; Feldman, E.; Zeevi, A.; Soliman, S.; Saraiya, A.; Buck, S.; Vettraino, J.; et al. Forward and reverse selection for longevity in Drosophila is characterized by alteration of antioxidant gene expression and oxidative damage patterns. Exp. Gerontol. 2000, 35, 167–185. [Google Scholar] [CrossRef]
- Alonso, J.; Rodriguez, J.M.; Baena-Lopez, L.A.; Santaren, J.F. Characterization of the Drosophila melanogaster mitochondrial proteome. J. Proteome Res. 2005, 4, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Iijima-Ando, K.; Iijima, K.; Lee, W.J.; Lee, J.H.; Yu, K.; Lee, D.S. JNK/FOXO-mediated neuronal expression of fly homologue of peroxiredoxin II reduces oxidative stress and extends life span. J. Biol. Chem. 2009, 284, 29454–29461. [Google Scholar] [CrossRef] [PubMed]
- Radyuk, S.N.; Michalak, K.; Klichko, V.I.; Benes, J.; Rebrin, I.; Sohal, R.S.; Orr, W.C. Peroxiredoxin 5 confers protection against oxidative stress and apoptosis and also promotes longevity in Drosophila. Biochem. J. 2009, 419, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Odnokoz, O.; Nakatsuka, K.; Klichko, V.I.; Nguyen, J.; Solis, L.C.; Ostling, K.; Badinloo, M.; Orr, W.C.; Radyuk, S.N. Mitochondrial peroxiredoxins are essential in regulating the relationship between Drosophila immunity and aging. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Radyuk, S.N.; Sohal, R.S. Involvement of redox state in the aging of Drosophila melanogaster. Antioxid. Redox Signal. 2013, 19, 788–803. [Google Scholar] [CrossRef] [PubMed]
- Radyuk, S.N.; Rebrin, I.; Klichko, V.I.; Sohal, B.H.; Michalak, K.; Benes, J.; Sohal, R.S.; Orr, W.C. Mitochondrial peroxiredoxins are critical for the maintenance of redox state and the survival of adult Drosophila. Free Radic. Biol. Med. 2010, 49, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- Klichko, V.I.; Orr, W.C.; Radyuk, S.N. The role of peroxiredoxin 4 in inflammatory response and aging. Biochim. Biophys. Acta 2016, 1862, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Nóbrega-Pereira, S. NADPH: New oxygen for the ROS theory of aging. Oncotarget 2016, 7, 50814–50815. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Nobrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Vina, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef] [PubMed]
- Dietzmann, K. Histochemical demonstration of enzyme activity changes in the regio postcentralis of the mouse brain under the effect of the aging process. J. Hirnforsch. 1981, 22, 405–408. [Google Scholar] [PubMed]
- Kaliman, P.A.; Konovalova, E.O. Age-dependent characteristics of the regulation of cytoplasmic NADP+-dehydrogenases in the liver of rats on different diets. Ukrainskii Biokhimicheskii Zhurnal (1978) 1985, 57, 38–42. [Google Scholar]
- Lemeshko, V.V.; Nikitchenko Iu, V.; Kaliman, P.A. Enzymes of the antioxidant system of rat liver during aging. Ukrainskii Biokhimicheskii Zhurnal (1978) 1983, 55, 523–528. [Google Scholar]
- Maurya, P.K.; Kumar, P.; Chandra, P. Age-dependent detection of erythrocytes glucose-6-phosphate dehydrogenase and its correlation with oxidative stress. Arch. Physiol. Biochem. 2016, 122, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gartler, S.M.; Hornung, S.K.; Motulsky, A.G. Effect of chronologic age on induction of cystathionine synthase, uroporphyrinogen I synthase, and glucose-6-phosphate dehydrogenase activities in lymphocytes. Proc. Natl. Acad. Sci. USA 1981, 78, 1916–1919. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.; Ayala, A.; Gordillo, E.; Revilla, E.; Santa Maria, C. Relationship between enzymatic activity loss and post-translational protein modification in aging. Arch. Gerontol. Geriatr. 1991, 12, 187–197. [Google Scholar] [CrossRef]
- Kil, I.S.; Lee, Y.S.; Bae, Y.S.; Huh, T.L.; Park, J.W. Modulation of NADP(+)-dependent isocitrate dehydrogenase in aging. Redox Rep. 2004, 9, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Blair, A.R.; Morahan, G.; Andrikopoulos, S. The deletion variant of nicotinamide nucleotide transhydrogenase (Nnt) does not affect insulin secretion or glucose tolerance. Endocrinology 2010, 151, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Worthington, D.J.; Rosemeyer, M.A. Glutathione reductase from human erythrocytes. Catalytic properties and aggregation. Eur. J. Biochem. 1976, 67, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Koziel, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Durr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Goy, C.; Czypiorski, P.; Altschmied, J.; Jakob, S.; Rabanter, L.L.; Brewer, A.C.; Ale-Agha, N.; Dyballa-Rukes, N.; Shah, A.M.; Haendeler, J. The imbalanced redox status in senescent endothelial cells is due to dysregulated Thioredoxin-1 and NADPH oxidase 4. Exp. Gerontol. 2014, 56, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Responses to reductive stress in the cardiovascular system. Free Radic. Boil. Med. 2017, 109, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Rezende, F.; Schürmann, C.; Schütz, S.; Harenkamp, S.; Herrmann, E.; Seimetz, M.; Weißmann, N.; Schröder, K. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox Boil. 2016, 11, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Hirschhauser, C.; Bornbaum, J.; Reis, A.; Bohme, S.; Kaludercic, N.; Menabo, R.; Di Lisa, F.; Boengler, K.; Shah, A.M.; Schulz, R.; et al. NOX4 in Mitochondria: Yeast Two-Hybrid-Based Interaction with Complex I Without Relevance for Basal Reactive Oxygen Species? Antioxid. Redox Signal. 2015, 23, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, H.; Kawai, S.; Murata, K. Two sources of mitochondrial NADPH in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 7553–7560. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Liu, W.; Zhou, C.Z. Crystal structure of Saccharomyces cerevisiae 6-phosphogluconate dehydrogenase Gnd1. BMC Struct. Boil. 2007, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Minard, K.I.; McAlister-Henn, L. Sources of NADPH in yeast vary with carbon source. J. Biol. Chem. 2005, 280, 39890–39896. [Google Scholar] [CrossRef] [PubMed]
- Garay, E.; Campos, S.E.; Gonzalez de la Cruz, J.; Gaspar, A.P.; Jinich, A.; Deluna, A. High-resolution profiling of stationary-phase survival reveals yeast longevity factors and their genetic interactions. PLoS Genet. 2014, 10, e1004168. [Google Scholar] [CrossRef] [PubMed]
- Laschober, G.T.; Ruli, D.; Hofer, E.; Muck, C.; Carmona-Gutierrez, D.; Ring, J.; Hutter, E.; Ruckenstuhl, C.; Micutkova, L.; Brunauer, R.; et al. Identification of evolutionarily conserved genetic regulators of cellular aging. Aging Cell 2010, 9, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, I.; Stamerra, G.; Vai, M. Altered Expression of Mitochondrial NAD(+) Carriers Influences Yeast Chronological Lifespan by Modulating Cytosolic and Mitochondrial Metabolism. Front. Genet. 2018, 9, 676. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Tienson, H.; Lindemann, A.; Vitvitsky, V.; Reichmann, D.; Banerjee, R.; Jakob, U. Time line of redox events in aging postmitotic cells. eLife 2013, 2, e00306. [Google Scholar] [CrossRef] [PubMed]
- Kwolek-Mirek, M.; Maslanka, R.; Molon, M. Disorders in NADPH generation via pentose phosphate pathway influence the reproductive potential of the Saccharomyces cerevisiae yeast due to changes in redox status. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Picazo, C.; Matallana, E.; Aranda, A. Yeast thioredoxin reductase Trr1p controls TORC1-regulated processes. Sci. Rep. 2018, 8, 16500. [Google Scholar] [CrossRef] [PubMed]
- Matecic, M.; Smith, D.L.; Pan, X.; Maqani, N.; Bekiranov, S.; Boeke, J.D.; Smith, J.S. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 2010, 6, e1000921. [Google Scholar] [CrossRef] [PubMed]
- Musa, M.; Peric, M.; Bou Dib, P.; Sobocanec, S.; Saric, A.; Lovric, A.; Rudan, M.; Nikolic, A.; Milosevic, I.; Vlahovicek, K.; et al. Heat-induced longevity in budding yeast requires respiratory metabolism and glutathione recycling. Aging 2018, 10, 2407–2427. [Google Scholar] [CrossRef] [PubMed]
- Managbanag, J.R.; Witten, T.M.; Bonchev, D.; Fox, L.A.; Tsuchiya, M.; Kennedy, B.K.; Kaeberlein, M. Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS ONE 2008, 3, e3802. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Tsuchiya, M.; Fox, L.A.; Dang, N.; Hu, D.; Kerr, E.O.; Johnston, E.D.; Tchao, B.N.; Pak, D.N.; Welton, K.L.; et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008, 18, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.M.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Yu, H.; Liu, Y.C.; Chen, T.L.; Stern, A.; Lo, S.J.; Chiu, D.T. IDH-1 deficiency induces growth defects and metabolic alterations in GSPD-1-deficient Caenorhabditis elegans. J. Mol. Med. 2019, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Larsen, P.L. DAF-16-dependent and independent expression targets of DAF-2 insulin receptor-like pathway in Caenorhabditis elegans include FKBPs. J. Mol. Biol. 2001, 314, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, G.; Xie, F.; Petyuk, V.A.; Smolders, A.; Brewer, H.M.; Camp, D.G., 2nd; Smith, R.D.; Braeckman, B.P. LC-MS Proteomics Analysis of the Insulin/IGF-1-Deficient Caenorhabditis elegans daf-2(e1370) Mutant Reveals Extensive Restructuring of Intermediary Metabolism. J. Proteome Res. 2014, 13, 1938–1956. [Google Scholar] [CrossRef] [PubMed]
- Martell, J.; Seo, Y.; Bak, D.W.; Kingsley, S.F.; Tissenbaum, H.A.; Weerapana, E. Global Cysteine-Reactivity Profiling during Impaired Insulin/IGF-1 Signaling in C. elegans Identifies Uncharacterized Mediators of Longevity. Cell Chem. Boil. 2016, 23, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Pinkston-Gosse, J.; Kenyon, C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nat. Genet. 2007, 39, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Judy, M.; Lee, S.J.; Kenyon, C. Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: Roles for GATA factors and lipid gene regulators. Cell Metab. 2013, 17, 85–100. [Google Scholar] [CrossRef]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Erkut, C.; Gade, V.R.; Laxman, S.; Kurzchalia, T.V. The glyoxylate shunt is essential for desiccation tolerance in C. elegans and budding yeast. eLife 2016, 5, e13614. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Park, D.; Riddle, D.L. Increased longevity of some C. elegans mitochondrial mutants explained by activation of an alternative energy-producing pathway. Mech. Ageing Dev. 2011, 132, 515–518. [Google Scholar] [CrossRef]
- Zuryn, S.; Kuang, J.; Tuck, A.; Ebert, P.R. Mitochondrial dysfunction in Caenorhabditis elegans causes metabolic restructuring, but this is not linked to longevity. Mech. Ageing Dev. 2010, 131, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.; Dong, Y.; Shindo, M.; Liu, W.; Odell, I.; Ruvkun, G.; Lee, S.S. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005, 19, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sun, H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 2007, 6, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.L.; Yang, H.C.; Hung, C.Y.; Ou, M.H.; Pan, Y.Y.; Cheng, M.L.; Stern, A.; Lo, S.J.; Chiu, D.T. Impaired embryonic development in glucose-6-phosphate dehydrogenase-deficient Caenorhabditis elegans due to abnormal redox homeostasis induced activation of calcium-independent phospholipase and alteration of glycerophospholipid metabolism. Cell Death Dis. 2017, 8, e2545. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Kwon, J.J.; Chen, C.; Russell, J.; Acosta, K.; Burnaevskiy, N.; Crane, M.M.; Bitto, A.; Vander Wende, H.; Simko, M.; et al. Transaldolase inhibition impairs mitochondrial respiration and induces a starvation-like longevity response in Caenorhabditis elegans. PLoS Genet. 2017, 13, e1006695. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Robida-Stubbs, S.; Tullet, J.M.; Rual, J.F.; Vidal, M.; Blackwell, T.K. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010, 6, e1001048. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Kaeberlein, M. The mitochondrial unfolded protein response and increased longevity: Cause, consequence, or correlation? Exp. Gerontol. 2014, 56, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; DeJonge, H.; Blackwell, T.K.; et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [PubMed]
- Rzezniczak, T.Z.; Merritt, T.J. Interactions of NADP-reducing enzymes across varying environmental conditions: A model of biological complexity. G3 Genes Genomes Genet. 2012, 2, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Kim, M.J.; Ding, D.; Han, C.; Park, H.J.; Meneses, Z.; Tanokura, M.; Linser, P.; Salvi, R.; Someya, S. G6pd Deficiency Does Not Affect the Cytosolic Glutathione or Thioredoxin Antioxidant Defense in Mouse Cochlea. J. Neurosci. 2017, 37, 5770–5781. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.; Gover, S.; Lam, V.M.; Adams, M.J. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency. Structure (Lond. Engl. 1993) 2000, 8, 293–303. [Google Scholar]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995, 14, 5209–5215. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; Tsitsipatis, D.; Hausig, F.; Kreuzer, K.; Erler, K.; Stein, V.; Ristow, M.; Steinbrenner, H.; Klotz, L.O. Non-linear impact of glutathione depletion on C. elegans life span and stress resistance. Redox Boil. 2017, 11, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Inoue, H.; Ookuma, S.; Satoh, T.; Kano, K.; Honjoh, S.; Hisamoto, N.; Matsumoto, K.; Nishida, E. The ERK-MAPK pathway regulates longevity through SKN-1 and insulin-like signaling in Caenorhabditis elegans. J. Biol. Chem. 2010, 285, 30274–30281. [Google Scholar] [CrossRef] [PubMed]
- Przybysz, A.J.; Choe, K.P.; Roberts, L.J.; Strange, K. Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech. Ageing Dev. 2009, 130, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; Crawford, D.K.; Kenyon, C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 2002, 298, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-β-hydroxybutyrate extends lifespan in C. elegans. Aging (Albany Ny) 2014, 6, 621–644. [Google Scholar]
- Schwartz, A.G.; Pashko, L.L. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res. Rev. 2004, 3, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Vogelauer, M.; Krall, A.S.; McBrian, M.A.; Li, J.Y.; Kurdistani, S.K. Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. J. Biol. Chem. 2012, 287, 32006–32016. [Google Scholar] [CrossRef] [PubMed]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC inhibitors: A new promising drug class in anti-aging research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Arkblad, E.L.; Tuck, S.; Pestov, N.B.; Dmitriev, R.I.; Kostina, M.B.; Stenvall, J.; Tranberg, M.; Rydstrom, J. A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic. Biol. Med. 2005, 38, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Bowman, M.J.; Winn, M.E.; Senchuk, M.M.; Van Raamsdonk, J.M. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic. Biol. Med. 2017, 108, 362–373. [Google Scholar] [CrossRef] [PubMed]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Choe, K.P. Characterization of skn-1/wdr-23 phenotypes in Caenorhabditis elegans; pleiotrophy, aging, glutathione, and interactions with other longevity pathways. Mech. Ageing Dev. 2015, 149, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Roman, I.; Barja, G. Regulation of longevity and oxidative stress by nutritional interventions: Role of methionine restriction. Exp. Gerontol. 2013, 48, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Hine, C.; Mitchell, J.R. Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp. Gerontol. 2015, 68, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Prigge, J.R.; Talago, E.A.; Arner, E.S.; Schmidt, E.E. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat. Commun. 2015, 6, 6479. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Brewer, G.J. External cys/cySS redox state modification controls the intracellular redox state and neurodegeneration via Akt in aging and Alzheimer’s disease mouse model neurons. J. Alzheimers Dis. 2014, 42, 313–324. [Google Scholar] [CrossRef] [PubMed]
- McIsaac, R.S.; Lewis, K.N.; Gibney, P.A.; Buffenstein, R. From yeast to human: Exploring the comparative biology of methionine restriction in extending eukaryotic life span. Ann. N. Y. Acad. Sci. 2016, 1363, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Uthus, E.O.; Brown-Borg, H.M. Methionine flux to transsulfuration is enhanced in the long living Ames dwarf mouse. Mech. Ageing Dev. 2006, 127, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Slekar, K.H.; Kosman, D.J.; Culotta, V.C. The Yeast Copper/Zinc Superoxide Dismutase and the Pentose Phosphate Pathway Play Overlapping Roles in Oxidative Stress Protection. J. Boil. Chem. 1996, 271, 28831–28836. [Google Scholar] [CrossRef]
- Campbell, K.; Vowinckel, J.; Keller, M.A.; Ralser, M. Methionine Metabolism Alters Oxidative Stress Resistance via the Pentose Phosphate Pathway. Antioxid. Redox Signal. 2016, 24, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kaya, A.; Ma, S.; Kim, G.; Gerashchenko, M.V.; Yim, S.H.; Hu, Z.; Harshman, L.G.; Gladyshev, V.N. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat. Commun. 2014, 5, 3592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Johnson, F.B. Methionine restriction activates the retrograde response and confers both stress tolerance and lifespan extension to yeast, mouse and human cells. PLoS ONE 2014, 9, e97729. [Google Scholar] [CrossRef] [PubMed]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef] [PubMed]
- Wipf, B.; Leisinger, T. Compartmentation of arginine biosynthesis in Saccharomyces cerevisiae. Fems Microbiol. Lett. 1977, 2, 239–242. [Google Scholar] [CrossRef]
- Zabriskie, T.M.; Jackson, M.D. Lysine biosynthesis and metabolism in fungi. Nat. Prod. Rep. 2000, 17, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. Calorie restriction: Is AMPK a key sensor and effector? Physiol. 2011, 26, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Iio, W.; Tokutake, Y.; Koike, H.; Matsukawa, N.; Tsukahara, T.; Chohnan, S.; Toyoda, A. Effects of chronic mild food restriction on behavior and the hypothalamic malonyl-CoA signaling pathway. Anim. Sci. J. 2015, 86, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.E.; Shi, Y.; Van Remmen, H. The effects of dietary restriction on oxidative stress in rodents. Free Radic. Boil. Med. 2014, 66, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Lyssiotis, C.A.; Zhang, Y.; Ying, H.; Asara, J.M.; Cantley, L.C.; Paik, J.H. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013, 32, 2589–2602. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.G.; Kim, H.J.; Chung, S.W.; Jung, K.J.; Shim, K.H.; Yu, B.P.; Yodoi, J.; Chung, H.Y. Modulation of glutathione and thioredoxin systems by calorie restriction during the aging process. Exp. Gerontol. 2003, 38, 539–548. [Google Scholar] [CrossRef]
- Rohrbach, S.; Gruenler, S.; Teschner, M.; Holtz, J. The thioredoxin system in aging muscle: Key role of mitochondrial thioredoxin reductase in the protective effects of caloric restriction? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R927–R935. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Distelmaier, K.; Lanza, I.R.; Irving, B.A.; Robinson, M.M.; Konopka, A.R.; Shulman, G.I.; Nair, K.S. Mechanism by Which Caloric Restriction Improves Insulin Sensitivity in Sedentary Obese Adults. Diabetes 2016, 65, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Molin, M.; Yang, J.; Hanzen, S.; Toledano, M.B.; Labarre, J.; Nystrom, T. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol. Cell 2011, 43, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Fierro-Gonzalez, J.C.; Gonzalez-Barrios, M.; Miranda-Vizuete, A.; Swoboda, P. The thioredoxin TRX-1 regulates adult lifespan extension induced by dietary restriction in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2011, 406, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Menger, K.E.; James, A.M.; Cocheme, H.M.; Harbour, M.E.; Chouchani, E.T.; Ding, S.; Fearnley, I.M.; Partridge, L.; Murphy, M.P. Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell Rep. 2015, 11, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e5. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef] [PubMed]
- De Nobrega, A.K.; Lyons, L.C. Aging and the clock: Perspective from flies to humans. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Froy, O. Circadian rhythms, aging, and life span in mammals. Physiology 2011, 26, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Mauvoisin, D.; Wang, J.; Jouffe, C.; Martin, E.; Atger, F.; Waridel, P.; Quadroni, M.; Gachon, F.; Naef, F. Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc. Natl. Acad. Sci. USA 2014, 111, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Mendez, I.; Vazquez-Martinez, O.; Hernandez-Munoz, R.; Valente-Godinez, H.; Diaz-Munoz, M. Redox regulation and pro-oxidant reactions in the physiology of circadian systems. Biochimie 2016, 124, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J. Boil. Chem. 2018, 293, 7508–7516. [Google Scholar] [CrossRef] [PubMed]
- Putker, M.; Crosby, P.; Feeney, K.A.; Hoyle, N.P.; Costa, A.S.H.; Gaude, E.; Frezza, C.; O’Neill, J.S. Mammalian Circadian Period, But Not Phase and Amplitude, Is Robust Against Redox and Metabolic Perturbations. Antioxid. Redox Signal. 2018, 28, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; Valekunja, U.K.; Feeney, K.A.; Wulund, L.; Milev, N.B.; Stangherlin, A.; Ansel-Bollepalli, L.; Velagapudi, V.; O’Neill, J.S.; Reddy, A.B. The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab. 2016, 24, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Cracan, V.; Titov, D.V.; Shen, H.; Grabarek, Z.; Mootha, V.K. A genetically encoded tool for manipulation of NADP(+)/NADPH in living cells. Nat. Chem. Boil. 2017, 13, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Sandbichler, A.M.; Jansen, B.; Peer, B.A.; Paulitsch, M.; Pelster, B.; Egg, M. Metabolic Plasticity Enables Circadian Adaptation to Acute Hypoxia in Zebrafish Cells. Cell. Physiol. Biochem. 2018, 46, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Reick, M.; Wu, L.C.; McKnight, S.L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 2001, 293, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Tajima, F.; Ishijima, S.; Sagami, I. Changes in pH and NADPH regulate the DNA binding activity of neuronal PAS domain protein 2, a mammalian circadian transcription factor. Biochemistry 2015, 54, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Goya, M.E.; Romanowski, A.; Caldart, C.S.; Benard, C.Y.; Golombek, D.A. Circadian rhythms identified in Caenorhabditis elegans by in vivo long-term monitoring of a bioluminescent reporter. Proc. Natl. Acad. Sci. USA 2016, 113, E7837–E7845. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; Milev, N.B.; Valekunja, U.K.; Ch, R.; Ray, S.; Silva Dos Santos, M.; Nagy, A.D.; Antrobus, R.; MacRae, J.I.; Reddy, A.B. Metabolic oscillations on the circadian time scale in Drosophila cells lacking clock genes. Mol. Syst. Biol. 2018, 14, e8376. [Google Scholar] [CrossRef] [PubMed]
- Causton, H.C.; Feeney, K.A.; Ziegler, C.A.; O’Neill, J.S. Metabolic Cycles in Yeast Share Features Conserved among Circadian Rhythms. Curr. Biol. 2015, 25, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Lerner, F.; Niere, M.; Ludwig, A.; Ziegler, M. Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun. 2001, 288, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Love, N.R.; Pollak, N.; Dolle, C.; Niere, M.; Chen, Y.; Oliveri, P.; Amaya, E.; Patel, S.; Ziegler, M. NAD kinase controls animal NADP biosynthesis and is modulated via evolutionarily divergent calmodulin-dependent mechanisms. Proc. Natl. Acad. Sci. USA 2015, 112, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Shianna, K.V.; Marchuk, D.A.; Strand, M.K. Genomic characterization of POS5, the Saccharomyces cerevisiae mitochondrial NADH kinase. Mitochondrion 2006, 6, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Kawai, S.; Murata, K. Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun. 2012, 3, 1248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R. MNADK, a novel liver-enriched mitochondrion-localized NAD kinase. Boil. Open 2013, 2, 432–438. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, R.; Sainsard-Chanet, A. Deletion of the mitochondrial NADH kinase increases mitochondrial DNA stability and life span in the filamentous fungus Podospora anserina. Exp. Gerontol. 2010, 45, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.M.; O’Donovan, M.; Sun, L.; Choi, J.Y.; Ren, M.; Cao, K. Anti-Aging Potentials of Methylene Blue for Human Skin Longevity. Sci. Rep. 2017, 7, 2475. [Google Scholar] [CrossRef] [PubMed]
- Gura, T. Hope in Alzheimer’s fight emerges from unexpected places. Nat. Med. 2008, 14, 894. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N.; Starkov, A.A. Methylene blue does not bypass Complex III antimycin block in mouse brain mitochondria. FEBS Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Kang, S.G.; Yoon, S.I.; Ko, H.J.; Kim, P.H.; Hong, E.J.; An, B.S.; Lee, E.; Lee, G.S. Methylene blue inhibits NLRP3, NLRC4, AIM2, and non-canonical inflammasome activation. Sci. Rep. 2017, 7, 12409. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wu, J.; Xu, L.; Wang, J.; Chen, Z.; Yang, R. Regulation of HSF1 protein stabilization: An updated review. Eur. J. Pharm. 2018, 822, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Copes, N. High-Throughput Screening of Age-Related Changes in Caenorhabditis elegans; University of South Florida: Tampa, FL, USA, 2015. [Google Scholar]
- Harrison, D.E.; Strong, R.; Allison, D.B.; Ames, B.N.; Astle, C.M.; Atamna, H.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nadon, N.L.; et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 2014, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Massie, H.R.; Aiello, V.R.; Williams, T.R. Influence of photosensitizers and light on the life span of Drosophila. Mech. Ageing Dev. 1993, 68, 175–182. [Google Scholar] [CrossRef]
- Vatolin, S.; Radivoyevitch, T.; Maciejewski, J.P. New drugs for pharmacological extension of replicative life span in normal and progeroid cells. Npj Aging Mech. Dis. 2019, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, A.; Lanasa, M.; Garcia, J.; Mora, H.; Fan, F.; Martin-Montalvo, A.; Di Francesco, A.; Calvo-Rubio, M.; Salvador-Pascual, A.; Aon, M.A.; et al. Overexpression of CYB5R3 and NQO1, two NAD(+) -producing enzymes, mimics aspects of caloric restriction. Aging Cell 2018, 17, e12767. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradshaw, P.C. Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients 2019, 11, 504. https://doi.org/10.3390/nu11030504
Bradshaw PC. Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients. 2019; 11(3):504. https://doi.org/10.3390/nu11030504
Chicago/Turabian StyleBradshaw, Patrick C. 2019. "Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging" Nutrients 11, no. 3: 504. https://doi.org/10.3390/nu11030504
APA StyleBradshaw, P. C. (2019). Cytoplasmic and Mitochondrial NADPH-Coupled Redox Systems in the Regulation of Aging. Nutrients, 11(3), 504. https://doi.org/10.3390/nu11030504