Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Complex Postprandial Period as Framework in Low-Grade Inflammation
2.1. Three Metabolic States
2.2. Postprandial Period as an Endocrine, Metabolic and Inflammatory Frame-Work
3. Nutrients
3.1. Hyperglycemia and Hyperinsulinemia
3.2. Fatty Acids and TLR4 Activation
3.3. Amino-Acids
4. Endotoxemia and Nutrients
4.1. Postprandial Endotoxemia
4.2. Lipopolysaccharide Translocation
5. Other Players in Inflammation
5.1. Transcription Factor Nf-κB in Postprandial Inflammatory Signaling
5.2. Oxidative Stress and Reactive Oxygen Species Production
5.3. Complement Component Factor 3 Is Activated in the Postprandial State
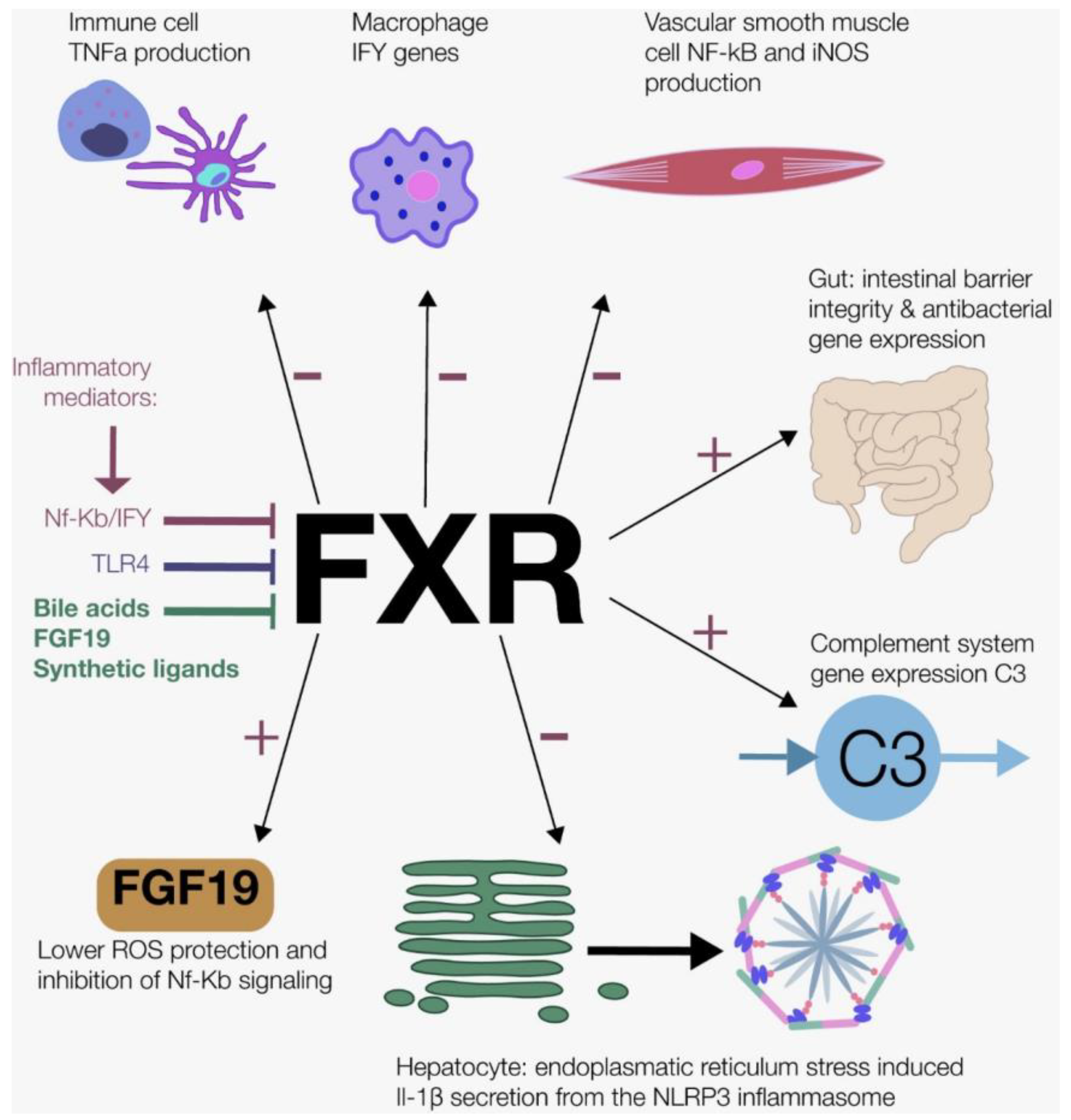
6. The Anti-Inflammatory Effects of Postprandial Bile Acid Signaling
6.1. Bile Acid Physiology
6.2. Farnesoid X Receptor and Fibroblast Growth Factor 19
6.3. Takeda G-Protein-Coupled Receptor 5
7. Acute Postprandial Inflammation: A Physiological Phenomenon?
8. Role of Nutrition in Chronic Low-Grade Inflammation Conditions Such as Rheumatoid Arthritis
8.1. General Interventions and the Gut
8.2. Omega-3 Fatty Acids
8.3. Fasting Paradigms
9. Conclusions, Implications and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Bozzetto, L.; Annuzzi, G.; Ragucci, M.; Di Donato, O.; Della Pepa, G.; Della Corte, G.; Griffo, E.; Anniballi, G.; Giacco, A.; Mancini, M.; et al. Insulin resistance, postprandial GLP-1 and adaptive immunity are the main predictors of NAFLD in a homogeneous population at high cardiovascular risk. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.W.; Koopman, F.A.; Visscher, J.P.M.; de Hair, M.J.; Gerlag, D.M.; Tak, P.P. Hormone, metabolic peptide, and nutrient levels in the earliest phases of rheumatoid arthritis—contribution of free fatty acids to an increased cardiovascular risk during very early disease. Clin. Rheumatol. 2017, 36, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Zilversmit, D.B. Atherogenesis: A postprandial phenomenon. Circulation 1979, 60, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Khan, K.M.; Cook, J.L.; Duronio, V. What is “inflammation”? Are we ready to move beyond Celsus? Br. J. Sports Med. 2004, 38, 248–249. [Google Scholar] [CrossRef]
- Ruiz-Núñez, B.; Pruimboom, L.; Dijck-Brouwer, D.A.J.; Muskiet, F.A.J. Lifestyle and nutritional imbalances associated with Western diseases: Causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J. Nutr. Biochem. 2013, 24, 1183–1201. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jönsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106, S5–S78. [Google Scholar] [CrossRef]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef]
- Wolowczuk, I.; Verwaerde, C.; Viltart, O.; Delanoye, A.; Delacre, M.; Pot, B.; Grangette, C. Feeding Our Immune System: Impact on Metabolism. Clin. Dev. Immunol. 2008, 2008, 1–19. [Google Scholar] [CrossRef]
- Dhindsa, S.; Tripathy, D.; Mohanty, P.; Ghanim, H.; Syed, T.; Aljada, A.; Dandona, P. Differential effects of glucose and alcohol on reactive oxygen species generation and intranuclear nuclear factor-κB in mononuclear cells. Metabolism 2004, 53, 330–334. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential Effects of Cream, Glucose, and Orange Juice on Inflammation, Endotoxin, and the Expression of Toll-Like Receptor-4 and Suppressor of Cytokine Signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Bakker, G.J.; Schnitzler, J.G.; Bekkering, S.; Clercq, N.C.; Koopen, A.M.; Hartstra, A.V.; Meessen, E.C.E.; Scheithauer, T.P.; Winkelmeijer, M.; Dallinga-Thie, G.M.; et al. Oral vancomycin treatment does not alter markers of postprandial inflammation in lean and obese subjects. Physiol. Rep. 2019, 7, e14199. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, S.; Samocha-Bonet, D.; Heilbronn, L.K.; Campbell, L.V. Inflammatory and Oxidative Stress Responses to High-Carbohydrate and High-Fat Meals in Healthy Humans. J. Nutr. Metab. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in Plasma Endotoxin Concentrations and the Expression of Toll-Like Receptors and Suppressor of Cytokine Signaling-3 in Mononuclear Cells After a High-Fat, High-Carbohydrate Meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Schaap, F.G.; Trauner, M.; Jansen, P.L.M. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2013, 11, 55–67. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Pols, T.; Eggink, H.; Soeters, M. TGR5 ligands as potential therapeutics in inflammatory diseases. Int. J. Interf. Cytokine Mediat. Res. 2014, 6, 27–38. [Google Scholar] [CrossRef]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef]
- Frayn, K.N.; Shadid, S.; Hamlani, R.; Humphrey, S.M.; Clark, M.L.; Fielding, B.A.; Boland, O.; Coppack, S.W.; Humphreys, S.M.; Field-Ing, B.A. Regulation of fatty acid movement in human adipose tissue in the postabsorptive-to-postprandial transition. Am. J. Physiol. Endocrinol. Metab. 1994, 266, E308–E317. [Google Scholar] [CrossRef]
- Klop, B.; Proctor, S.D.; Mamo, J.C.; Botham, K.M.; Cabezas, M.C. Understanding Postprandial Inflammation and Its Relationship to Lifestyle Behaviour and Metabolic Diseases. Int. J. Vasc. Med. 2012, 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Sottero, B.; Gargiulo, S.; Russo, I.; Barale, C.; Poli, G.; Cavalot, F. Postprandial Dysmetabolism and Oxidative Stress in Type 2 Diabetes: Pathogenetic Mechanisms and Therapeutic Strategies. Med. Res. Rev. 2015, 35, 968–1031. [Google Scholar] [CrossRef] [PubMed]
- Waseem, T.; Duxbury, M.; Ito, H.; Ashley, S.W.; Robinson, M.K. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 2008, 143, 334–342. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jun, H.S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediat. Inflamm. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Fantuzzi, G.; Faggioni, R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J. Leukoc. Biol. 2000, 68, 437–446. [Google Scholar]
- Dandona, P.; Aljada, A.; Mohanty, P.; Ghanim, H.; Hamouda, W.; Assian, E.; Ahmad, S. Insulin Inhibits Intranuclear Nuclear Factor κB and Stimulates IκB in Mononuclear Cells in Obese Subjects: Evidence for an Anti-inflammatory Effect? J. Clin. Endocrinol. Metab. 2001, 86, 3257–3265. [Google Scholar] [CrossRef]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef]
- Slag, M.F.; Ahmed, M.; Gannon, M.C.; Nuttall, F.Q. Meal stimulation of cortisol secretion: A protein induced effect. Metabolism 1981, 30, 1104–1108. [Google Scholar] [CrossRef]
- Stimson, R.H.; Mohd-Shukri, N.A.; Bolton, J.L.; Andrew, R.; Reynolds, R.M.; Walker, B.R. The Postprandial Rise in Plasma Cortisol in Men Is Mediated by Macronutrient-Specific Stimulation of Adrenal and Extra-Adrenal Cortisol Production. J. Clin. Endocrinol. Metab. 2014, 99, 160–168. [Google Scholar] [CrossRef]
- Chrousos, G.P. The Hypothalamic–Pituitary–Adrenal Axis and Immune-Mediated Inflammation. N. Engl. J. Med. 1995, 332, 1351–1363. [Google Scholar] [CrossRef]
- Vors, C.; Pineau, G.; Drai, J.; Meugnier, E.; Pesenti, S.; Laville, M.; Laugerette, F.; Malpuech-Brugère, C.; Vidal, H.; Michalski, M.C. Postprandial Endotoxemia Linked With Chylomicrons and Lipopolysaccharides Handling in Obese Versus Lean Men: A Lipid Dose-Effect Trial. J. Clin. Endocrinol. Metab. 2015, 100, 3427–3435. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Queipo-Ortuño, M.I.; Murri, M.; Boto-Ordoñez, M.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.J. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J. Lipid Res. 2012, 53, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory Cytokine Concentrations Are Acutely Increased by Hyperglycemia in Humans. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Harte, A.L.; Varma, M.C.; Tripathi, G.; McGee, K.C.; Al-Daghri, N.M.; Al-Attas, O.S.; Sabico, S.; O’Hare, J.P.; Ceriello, A.; Saravanan, P.; et al. High Fat Intake Leads to Acute Postprandial Exposure to Circulating Endotoxin in Type 2 Diabetic Subjects. Diabetes Care 2012, 35, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef]
- Turina, M.; Fry, D.E.; Polk, H.C. Acute hyperglycemia and the innate immune system: Clinical, cellular, and molecular aspects. Crit. Care Med. 2005, 33, 1624–1633. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Plomgaard, P.; Akerstrom, T.; Møller, K.; Schmitz, O.; Pedersen, B.K. Effect of short-term intralipid infusion on the immune response during low-dose endotoxemia in humans. Am. J. Physiol. Metab. 2008, 294, E371–E379. [Google Scholar] [CrossRef]
- Dandona, P.; Chaudhuri, A.; Mohanty, P.; Ghanim, H. Anti-inflammatory effects of insulin. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 511–517. [Google Scholar] [CrossRef]
- Hansen, T.K.; Thiel, S.; Wouters, P.J.; Christiansen, J.S.; van den Berghe, G. Intensive Insulin Therapy Exerts Antiinflammatory Effects in Critically Ill Patients and Counteracts the Adverse Effect of Low Mannose-Binding Lectin Levels. J. Clin. Endocrinol. Metab. 2003, 88, 1082–1088. [Google Scholar] [CrossRef]
- Devaraj, S.; Venugopal, S.K.; Singh, U.; Jialal, I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-α and -β. Diabetes 2005, 54, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.H.; Joosten, L.A.B.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Vogel, S.N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002, 4, 903–914. [Google Scholar] [CrossRef]
- Rogero, M.; Calder, P. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef]
- Lien, E.; Means, T.K.; Heine, H.; Yoshimura, A.; Kusumoto, S.; Fukase, K.; Fenton, M.J.; Oikawa, M.; Qureshi, N.; Monks, B.; et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Investig. 2000, 105, 497–504. [Google Scholar] [CrossRef]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The Bile Acid Sensor FXR Is Required for Immune-Regulatory Activities of TLR-9 in Intestinal Inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.K.; Yin, J.; Zhu, X.P.; Liu, G.; Li, N.Z.; Peng, Y.Y.; Yin, Y.Y. Glutamine on Intestinal Inflammation: A Mechanistic Perspective. Eur. J. Inflamm. 2013, 11, 315–326. [Google Scholar] [CrossRef]
- Uehara, K.; Takahashi, T.; Fujii, H.; Shimizu, H.; Omori, E.; Matsumi, M.; Yokoyama, M.; Morita, K.; Akagi, R.; Sassa, S. The lower intestinal tract-specific induction of heme oxygenase-1 by glutamine protects against endotoxemic intestinal injury. Crit. Care Med. 2005, 33, 381–390. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Z.; Lin, G.; Hu, S.; Wang, B.; Dai, Z.; Wu, G. Glycine Stimulates Protein Synthesis and Inhibits Oxidative Stress in Pig Small Intestinal Epithelial Cells. J. Nutr. 2014, 144, 1540–1548. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, X.; Wang, L.; Gao, K.; Jiang, Z. L-Arginine Inhibited Inflammatory Response and Oxidative Stress Induced by Lipopolysaccharide via Arginase-1 Signaling in IPEC-J2 Cells. Int. J. Mol. Sci. 2019, 20, 1800. [Google Scholar] [CrossRef]
- Ren, W.; Yin, J.; Wu, M.; Liu, G.; Yang, G.; Xion, Y.; Su, D.; Wu, L.; Li, T.; Chen, S.; et al. Serum Amino Acids Profile and the Beneficial Effects of L-Arginine or L-Glutamine Supplementation in Dextran Sulfate Sodium Colitis. PLoS ONE 2014, 9, e88335. [Google Scholar] [CrossRef]
- Perng, W.; Gillman, M.W.; Fleisch, A.F.; Michalek, R.D.; Watkins, S.M.; Isganaitis, E.; Patti, M.-E.; Oken, E. Metabolomic profiles and childhood obesity. Obesity 2014, 22, 2570–2578. [Google Scholar] [CrossRef]
- Newgard, C.B. Interplay between Lipids and Branched-Chain Amino Acids in Development of Insulin Resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef]
- Mu, W.C.; VanHoosier, E.; Elks, C.; Grant, R. Long-Term Effects of Dietary Protein and Branched-Chain Amino Acids on Metabolism and Inflammation in Mice. Nutrients 2018, 10, 918. [Google Scholar] [CrossRef]
- Zhenyukh, O.; Civantos, E.; Ruiz-Ortega, M.; Sánchez, M.S.; Vázquez, C.; Peiró, C.; Egido, J.; Mas, S. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol. Med. 2017, 104, 165–177. [Google Scholar] [CrossRef]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kiers, D.; Leijte, G.P.; Gerretsen, J.; Zwaag, J.; Kox, M.; Pickkers, P. Comparison of different lots of endotoxin and evaluation of in vivo potency over time in the experimental human endotoxemia model. Innate Immun. 2019, 25, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Laugerette, F.; vors, C.; Géloën, A.; Chauvin, M.A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Netto Candido, T.L.; Alfenas, R.C.G.; Bressan, J. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr. Hosp. 2018, 35, 1432–1440. [Google Scholar] [CrossRef]
- Agwunobi, A.O.; Reid, C.; Maycock, P.; Little, R.A.; Carlson, G.L. Insulin Resistance and Substrate Utilization in Human Endotoxemia. J. Clin. Endocrinol. Metab. 2000, 85, 3770–3778. [Google Scholar] [CrossRef]
- Van der Crabben, S.N.; Blümer, R.M.E.; Stegenga, M.E.; Ackermans, M.T.; Endert, E.; Tanck, M.W.T.; Serlie, M.J.; van der Poll, T.; Sauerwein, H.P. Early endotoxemia increases peripheral and hepatic insulin sensitivity in healthy humans. J. Clin. Endocrinol. Metab. 2009, 94, 463–468. [Google Scholar] [CrossRef]
- Laugerette, F.; Alligier, M.; Bastard, J.P.; Drai, J.; Chanséaume, E.; Lambert-Porcheron, S.; Laville, M.; Morio, B.; Vidal, H.; Michalski, M.C. Overfeeding increases postprandial endotoxemia in men: Inflammatory outcome may depend on LPS transporters LBP and sCD14. Mol. Nutr. Food Res. 2014, 58, 1513–1518. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-3 Fatty Acids in Inflammation and Autoimmune Diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef]
- Simopoulos, A. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.F.; Costanzo, A.; Gareau, M.G.; Armando, A.M.; Quehenberger, O.; Jameson, J.M.; Olefsky, J.M. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE 2015, 10, e0122195. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Beller, A.; Rausch, S.; Strandmark, J.; Zänker, M.; Arbach, O.; Kruglov, A.; Berek, C. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity 2014, 40, 582–593. [Google Scholar] [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.E.; Vaillant, J.C.; Oppert, J.M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef]
- Wolff, N.S.; Hugenholtz, F.; Wiersinga, W.J. The emerging role of the microbiota in the ICU. Crit. Care 2018, 22, 78. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. NF-κB Signaling Pathway and Its Therapeutic Implications in Human Diseases. Int. Rev. Immunol. 2008, 27, 293–319. [Google Scholar] [CrossRef]
- Ehlers, K.; Brand, T.; Bangert, A.; Hauner, H.; Laumen, H. Postprandial activation of metabolic and inflammatory signalling pathways in human peripheral mononuclear cells. Br. J. Nutr. 2014, 111, 2167–2175. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, H.W.; Sternsdorf, T.; Liese, J.; Belohradsky, B.; Weber, C.; Wedel, A.; Schreck, R.; Bäuerle, P.; Ströbel, M. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J. Immunol. 1993, 151, 6986–6993. [Google Scholar]
- Barnes, P.J. Nuclear factor-kappa B. Int. J. Biochem. Cell Biol. 1997, 29, 867–870. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing Pieces in the NF-κB Puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Aljada, A.; Friedman, J.; Ghanim, H.; Mohanty, P.; Hofmeyer, D.; Chaudhuri, A.; Dandona, P. Glucose ingestion induces an increase in intranuclear nuclear factor κB, a fall in cellular inhibitor κB, and an increase in tumor necrosis factor α messenger RNA by mononuclear cells in healthy human subjects. Metabolism 2006, 55, 1177–1185. [Google Scholar] [CrossRef]
- Dickinson, S.; Hancock, D.P.; Petocz, P.; Ceriello, A.; Brand-Miller, J. High–glycemic index carbohydrate increases nuclear factor-κB activation in mononuclear cells of young, lean healthy subjects. Am. J. Clin. Nutr. 2008, 87, 1188–1193. [Google Scholar]
- Bellido, C.; López-Miranda, J.; Blanco-Colio, L.M.; Pérez-Martínez, P.; Muriana, F.J.; Martín-Ventura, J.L.; Marín, C.; Gómez, P.; Fuentes, F.; Egido, J.; et al. Butter and walnuts, but not olive oil, elicit postprandial activation of nuclear transcription factor κB in peripheral blood mononuclear cells from healthy men. Am. J. Clin. Nutr. 2004, 80, 1487–1491. [Google Scholar] [CrossRef]
- Peairs, A.D.; Rankin, J.W.; Lee, Y.W. Effects of acute ingestion of different fats on oxidative stress and inflammation in overweight and obese adults. Nutr. J. 2011, 10, 122. [Google Scholar] [CrossRef]
- Moldovan, L.; Moldovan, N.I. Oxygen free radicals and redox biology of organelles. Histochem. Cell Biol. 2004, 122, 395–412. [Google Scholar] [CrossRef]
- Mohanty, P.; Hamouda, W.; Garg, R.; Aljada, A.; Ghanim, H.; Dandona, P. Glucose Challenge Stimulates Reactive Oxygen Species (ROS) Generation by Leucocytes. J. Clin. Endocrinol. Metab. 2000, 85, 2970–2973. [Google Scholar] [CrossRef]
- Mohanty, P.; Ghanim, H.; Hamouda, W.; Aljada, A.; Garg, R.; Dandona, P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am. J. Clin. Nutr. 2002, 75, 767–772. [Google Scholar] [CrossRef]
- Patel, C.; Ghanim, H.; Ravishankar, S.; Sia, C.L.; Viswanathan, P.; Mohanty, P.; Dandona, P. Prolonged Reactive Oxygen Species Generation and Nuclear Factor-κB Activation after a High-Fat, High-Carbohydrate Meal in the Obese. J. Clin. Endocrinol. Metab. 2007, 92, 4476–4479. [Google Scholar] [CrossRef]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes Are Oxidative Organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef]
- Bokisch, V.A.; Dierich, M.P.; Muller-Eberhard, H.J. Third component of complement (C3): Structural properties in relation to functions. Proc. Natl. Acad. Sci. 1975, 72, 1989–1993. [Google Scholar] [CrossRef]
- Scantlebury, T.; Maslowska, M.; Cianflone, K. Chylomicron-specific enhancement of acylation stimulating protein and precursor protein C3 production in differentiated human adipocytes. J. Biol. Chem. 1998, 273, 20903–20909. [Google Scholar] [CrossRef]
- Halkes, C.J.M.; van Dijk, H.; de Jaegere, P.P.T.; Plokker, H.W.M.; van der Helm, Y.; Erkelens, D.W.; Castro Cabezas, M. Postprandial Increase of Complement Component 3 in Normolipidemic Patients With Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1526–1530. [Google Scholar] [CrossRef]
- Murray, I.; Sniderman, A.D.; Cianflone, K. Mice lacking acylation stimulating protein (ASP) have delayed postprandial triglyceride clearance. J. Lipid Res. 1999, 40, 1671–1676. [Google Scholar]
- Van Oostrom, A.J.; van Dijk, H.; Verseyden, C.; Sniderman, A.D.; Cianflone, K.; Rabelink, T.J.; Castro Cabezas, M. Addition of glucose to an oral fat load reduces postprandial free fatty acids and prevents the postprandial increase in complement component 3. Am. J. Clin. Nutr. 2004, 79, 510–515. [Google Scholar] [CrossRef]
- Liu, Z.; Tang, Q.; Wen, J.; Tang, Y.; Huang, D.; Huang, Y.; Xie, J.; Luo, Y.; Liang, M.; Wu, C.; et al. Elevated serum complement factors 3 and 4 are strong inflammatory markers of the metabolic syndrome development: A longitudinal cohort study. Sci. Rep. 2016, 6, 18713. [Google Scholar] [CrossRef]
- Phillips, C.M.; Kesse-Guyot, E.; Ahluwalia, N.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. Dietary fat, abdominal obesity and smoking modulate the relationship between plasma complement component 3 concentrations and metabolic syndrome risk. Atherosclerosis 2012, 220, 513–519. [Google Scholar] [CrossRef]
- Isaacs, P.E.; Ladas, S.; Forgacs, I.C.; Dowling, R.H.; Ellam, S.V.; Adrian, T.E.; Bloom, S.R. Comparison of effects of ingested medium and long-chain triglyceride on gallbladder volume and release of cholecystokinin and other gut peptides. Dig. Dis. Sci. 1987, 32, 481–486. [Google Scholar] [CrossRef]
- LaRusso, N.F.; Korman, M.G.; Hoffman, N.E.; Hofmann, A.F. Dynamics of the enterohepatic circulation of bile acids. Postprandial serum concentrations of conjugates of cholic acid in health, cholecystectomized patients, and patients with bile acid malabsorption. N. Engl. J. Med. 1974, 291, 689–692. [Google Scholar] [CrossRef]
- Eggink, H.M.; van Nierop, F.S.; Schooneman, M.G.; Boelen, A.; Kalsbeek, A.; Koehorst, M.; ten Have, G.A.M.; de Brauw, L.M.; Groen, A.K.; Romijn, J.A.; et al. Transhepatic bile acid kinetics in pigs and humans. Clin. Nutr. 2018, 37, 1406–1414. [Google Scholar] [CrossRef]
- Van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. lancet. Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid X receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar]
- Mazuy, C.; Helleboid, A.; Staels, B.; Lefebvre, P. Nuclear bile acid signaling through the farnesoid X receptor. Cell. Mol. Life Sci. 2015, 72, 1631–1650. [Google Scholar] [CrossRef]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical Profiling of Nuclear Receptor Expression Reveals a Hierarchical Transcriptional Network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef]
- Chiang, J.Y.; Kimmel, R.; Weinberger, C.; Stroup, D. Farnesoid X receptor responds to bile acids and represses cholesterol 7α-hydroxylase gene (CYP7A1) transcription. J. Biol. Chem. 2000, 275, 10918–10924. [Google Scholar] [CrossRef]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a Postprandial, Insulin-Independent Activator of Hepatic Protein and Glycogen Synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1α Pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Fiorucci, S. Reciprocal regulation of the bile acid-activated receptor FXR and the interferon-γ-STAT-1 pathway in macrophages. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 564–573. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Li, Y.T.Y.; Swales, K.E.; Thomas, G.J.; Warner, T.D.; Bishop-Bailey, D. Farnesoid X Receptor Ligands Inhibit Vascular Smooth Muscle Cell Inflammation and Migration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2606–2611. [Google Scholar] [CrossRef]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef]
- Li, J.; Pircher, P.C.; Schulman, I.G.; Westin, S.K. Regulation of complement C3 expression by the bile acid receptor FXR. J. Biol. Chem. 2005, 280, 7427–7434. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim. Biophys. Acta-Mol. Basis Dis. 2011, 1812, 851–858. [Google Scholar] [CrossRef]
- Uchiyama, K.; Naito, Y.; Takagi, T.; Mizushima, K.; Hayashi, N.; Handa, O.; Ishikawa, T.; Yagi, N.; Kokura, S.; Yoshikawa, T. FGF19 Protects Colonic Epithelial Cells against Hydrogen Peroxide. Digestion 2011, 83, 180–183. [Google Scholar] [CrossRef]
- Shimizu, M.; Morimoto, H.; Maruyama, R.; Inoue, J.; Sato, R. Selective Regulation of FGF19 and FGF21 Expression by Cellular and Nutritional Stress. J. Nutr. Sci. Vitaminol. 2015, 61, 154–160. [Google Scholar] [CrossRef][Green Version]
- Drafahl, K.A.; McAndrew, C.W.; Meyer, A.N.; Haas, M.; Donoghue, D.J. The Receptor Tyrosine Kinase FGFR4 Negatively Regulates NF-kappaB Signaling. PLoS ONE 2010, 5, e14412. [Google Scholar] [CrossRef]
- Morton, G.J.; Kaiyala, K.J.; Foster-Schubert, K.E.; Cummings, D.E.; Schwartz, M.W. Carbohydrate Feeding Dissociates the Postprandial FGF19 Response From Circulating Bile Acid Levels in Humans. J. Clin. Endocrinol. Metab. 2014, 99, E241–E245. [Google Scholar] [CrossRef]
- Sonne, D.P.; Samuel van Nierop, F.; Kulik, W.; Soeters, M.R.; Vilsbøll, T.; Knop, F.K. Postprandial Plasma Concentrations of Individual Bile Acids and FGF-19 in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3002–3009. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, M.S.; Choung, J.S.; Kim, S.S.; Oh, H.H.; Choi, C.S.; Ha, S.Y.; Kang, Y.; Kim, Y.; Jun, H.S. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 2012, 55, 2456–2468. [Google Scholar] [CrossRef]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Masuda, M.; Kohno, S.; Levi, M.; Shiozaki, Y.; Keenan, A.L.; Miyazaki, M. Simultaneous inhibition of FXR and TGR5 exacerbates atherosclerotic formation. J. Lipid Res. 2018, 59, 1709–1713. [Google Scholar] [CrossRef]
- Jadhav, K.; Xu, Y.; Xu, Y.; Li, Y.; Xu, J.; Zhu, Y.; Adorini, L.; Lee, Y.K.; Kasumov, T.; Yin, L.; et al. Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol. Metab. 2018, 9, 131–140. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Mustafi, R.; Cerda, S.; Chumsangsri, A.; Xia, Y.R.; Li, Y.C.; Bissonnette, M. Lithocholic acid down-regulation of NF-κB activity through vitamin D receptor in colonic cancer cells. J. Steroid Biochem. Mol. Biol. 2008, 111, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grün, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-κB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef]
- Kopf, J.C.; Suhr, M.J.; Clarke, J.; Eyun, S.; Riethoven, J.J.M.; Ramer-Tait, A.E.; Rose, D.J. Role of whole grains versus fruits and vegetables in reducing subclinical inflammation and promoting gastrointestinal health in individuals affected by overweight and obesity: A randomized controlled trial. Nutr. J. 2018, 17, 72. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Khanna, S.; Jaiswal, K.S.; Gupta, B. Managing Rheumatoid Arthritis with Dietary Interventions. Front. Nutr. 2017, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The Mediterranean Diets: What Is So Special about the Diet of Greece? The Scientific Evidence. J. Nutr. 2001, 131, 3065S–3073S. [Google Scholar] [CrossRef] [PubMed]
- Skoldstam, L.; Hagfors, L.; Johansson, G. An experimental study of a Mediterranean diet intervention for patients with rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 208–214. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The Importance of the Omega-6/Omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Fortin, P.R.; Lew, R.A.; Liang, M.H.; Wright, E.A.; Beckett, L.A.; Chalmers, T.C.; Sperling, R.I. Validation of a meta-analysis: The effects of fish oil in rheumatoid arthritis. J. Clin. Epidemiol. 1995, 48, 1379–1390. [Google Scholar] [CrossRef]
- Gioxari, A.; Kaliora, A.C.; Marantidou, F.; Panagiotakos, D.P. Intake of ω-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: A systematic review and meta-analysis. Nutrition 2018, 45, 114–124. [Google Scholar] [CrossRef]
- Goldberg, R.J.; Katz, J. A meta-analysis of the analgesic effects of omega-3 polyunsaturated fatty acid supplementation for inflammatory joint pain. Pain 2007, 129, 210–223. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C.; Song, G.G. Omega-3 Polyunsaturated Fatty Acids and the Treatment of Rheumatoid Arthritis: A Meta-analysis. Arch. Med. Res. 2012, 43, 356–362. [Google Scholar] [CrossRef]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef]
- Voon, P.T.; Ng, T.K.W.; Lee, V.K.M.; Nesaretnam, K. Diets high in palmitic acid (16:0), lauric and myristic acids (12:0 + 14:0), or oleic acid (18:1) do not alter postprandial or fasting plasma homocysteine and inflammatory markers in healthy Malaysian adults. Am. J. Clin. Nutr. 2011, 94, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Ott, B.; Skurk, T.; Hastreiter, L.; Lagkouvardos, I.; Fischer, S.; Büttner, J.; Kellerer, T.; Clavel, T.; Rychlik, M.; Haller, D.; et al. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci. Rep. 2017, 7, 11955. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmi de Toledo, F.; Grundler, F.; Bergouignan, A.; Drinda, S.; Michalsen, A. Safety, health improvement and well-being during a 4 to 21-day fasting period in an observational study including 1422 subjects. PLoS ONE 2019, 14, e0209353. [Google Scholar] [CrossRef]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef]
- Cope, A.P.; Schulze-Koops, H.; Aringer, M. The central role of T cells in rheumatoid arthritis. Clin. Exp. Rheumatol. 2007, 25, S4. [Google Scholar]
- Tripathy, A.; Khanna, S.; Padhan, P.; Smita, S.; Raghav, S.; Gupta, B. Direct recognition of LPS drive TLR4 expressing CD8+ T cell activation in patients with rheumatoid arthritis. Sci. Rep. 2017, 7, 933. [Google Scholar] [CrossRef]
- Fraser, D.A.; Thoen, J.; Reseland, J.E.; Førre, Ø.; Kjeldsen-Kragh, J. Decreased CD4+ Lymphocyte Activation and Increased Interleukin-4 Production in Peripheral Blood of Rheumatoid Arthritis Patients After Acute Starvation. Clin. Rheumatol. 1999, 18, 394–401. [Google Scholar] [CrossRef]
- Woodward, E.A.; Prêle, C.M.; Nicholson, S.E.; Kolesnik, T.B.; Hart, P.H. The anti-inflammatory effects of interleukin-4 are not mediated by suppressor of cytokine signalling-1 (SOCS1). Immunology 2010, 131, 118–127. [Google Scholar] [CrossRef]
- Hafström, I.; Ringertz, B.; Gyllenhammar, H.; Palmblad, J.; Harms-Ringdahl, M. Effects of fasting on disease activity, neutrophil function, fatty acid composition, and leukotriene biosynthesis in patients with rheumatoid arthritis. Arthritis Rheum. 1988, 31, 585–592. [Google Scholar] [CrossRef]
- Kjeldsen-Kragh, J.; Haugen, M.; Borchgrevink, C.F.; Laerum, E.; Eek, M.; Mowinkel, P.; Hovi, K.; Førre, O. Controlled trial of fasting and one-year vegetarian diet in rheumatoid arthritis. The Lancet 1991, 338, 899–902. [Google Scholar] [CrossRef]
- Komaki, G.; Kanazawa, F.; Sogawa, H.; Mine, K.; Tamai, H.; Okamura, S.; Kubo, C. Alterations in lymphocyte subsets and pituitary-adrenal gland-related hormones during fasting. Am. J. Clin. Nutr. 1997, 66, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Shegarfi, H.; Naddafi, F.; Mirshafiey, A. Natural Killer Cells and Their Role in Rheumatoid Arthritis: Friend or Foe? Sci. World, J. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, J.E.; Senturia, J.B. A comparison of bone marrow leukocytes in hibernating and nonhibernating woodchucks and ground squirrels. Cryobiology 1972, 9, 257–261. [Google Scholar] [CrossRef]
- Xu, D.L.; Wang, D.H. Fasting suppresses T cell-mediated immunity in female Mongolian gerbils (Meriones unguiculatus). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2010, 155, 25–33. [Google Scholar] [CrossRef]
- Sköldstam, L.; Larsson, L.; Lindström, F.D. Effects Of Fasting and Lactovegetarian Diet on Rheumatoid Arthritis. Scand. J. Rheumatol. 1979, 8, 249–255. [Google Scholar] [CrossRef]
- Willoughby, D.; Hewlings, S.; Kalman, D. Body Composition Changes in Weight Loss: Strategies and Supplementation for Maintaining Lean Body Mass, a Brief Review. Nutrients 2018, 10, 1876. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meessen, E.C.E.; Warmbrunn, M.V.; Nieuwdorp, M.; Soeters, M.R. Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review. Nutrients 2019, 11, 3000. https://doi.org/10.3390/nu11123000
Meessen ECE, Warmbrunn MV, Nieuwdorp M, Soeters MR. Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review. Nutrients. 2019; 11(12):3000. https://doi.org/10.3390/nu11123000
Chicago/Turabian StyleMeessen, Emma C.E., Moritz V. Warmbrunn, Max Nieuwdorp, and Maarten R. Soeters. 2019. "Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review" Nutrients 11, no. 12: 3000. https://doi.org/10.3390/nu11123000
APA StyleMeessen, E. C. E., Warmbrunn, M. V., Nieuwdorp, M., & Soeters, M. R. (2019). Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review. Nutrients, 11(12), 3000. https://doi.org/10.3390/nu11123000