
METTL3 Regulates Osteoclast Biological Behaviors via iNOS/NO-Mediated Mitochondrial Dysfunction in Inflammatory Conditions

 and
and
Abstract
1. Introduction
2. Results
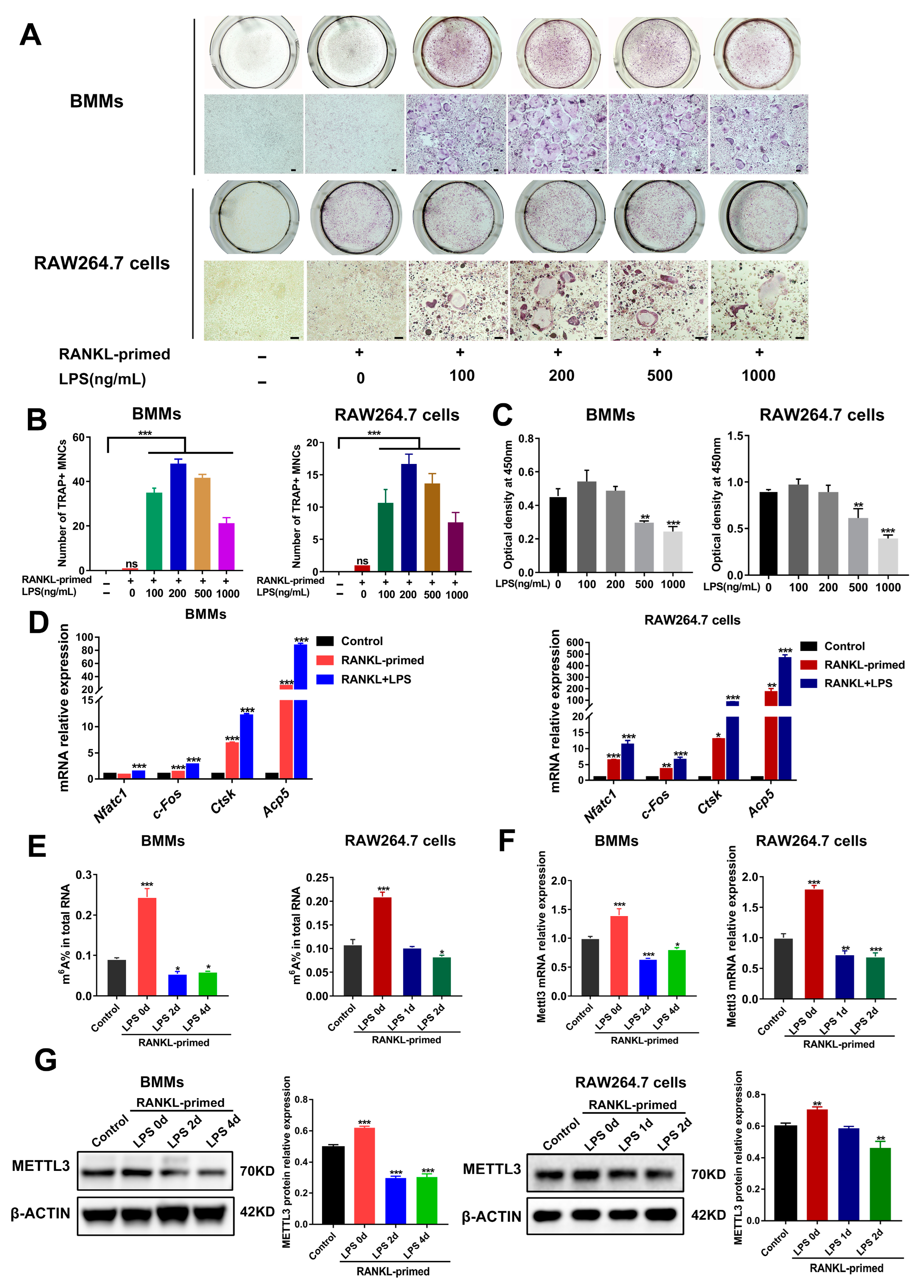
2.1. METTL3 Expression Is Downregulated during Inflammatory Osteoclastogenesis
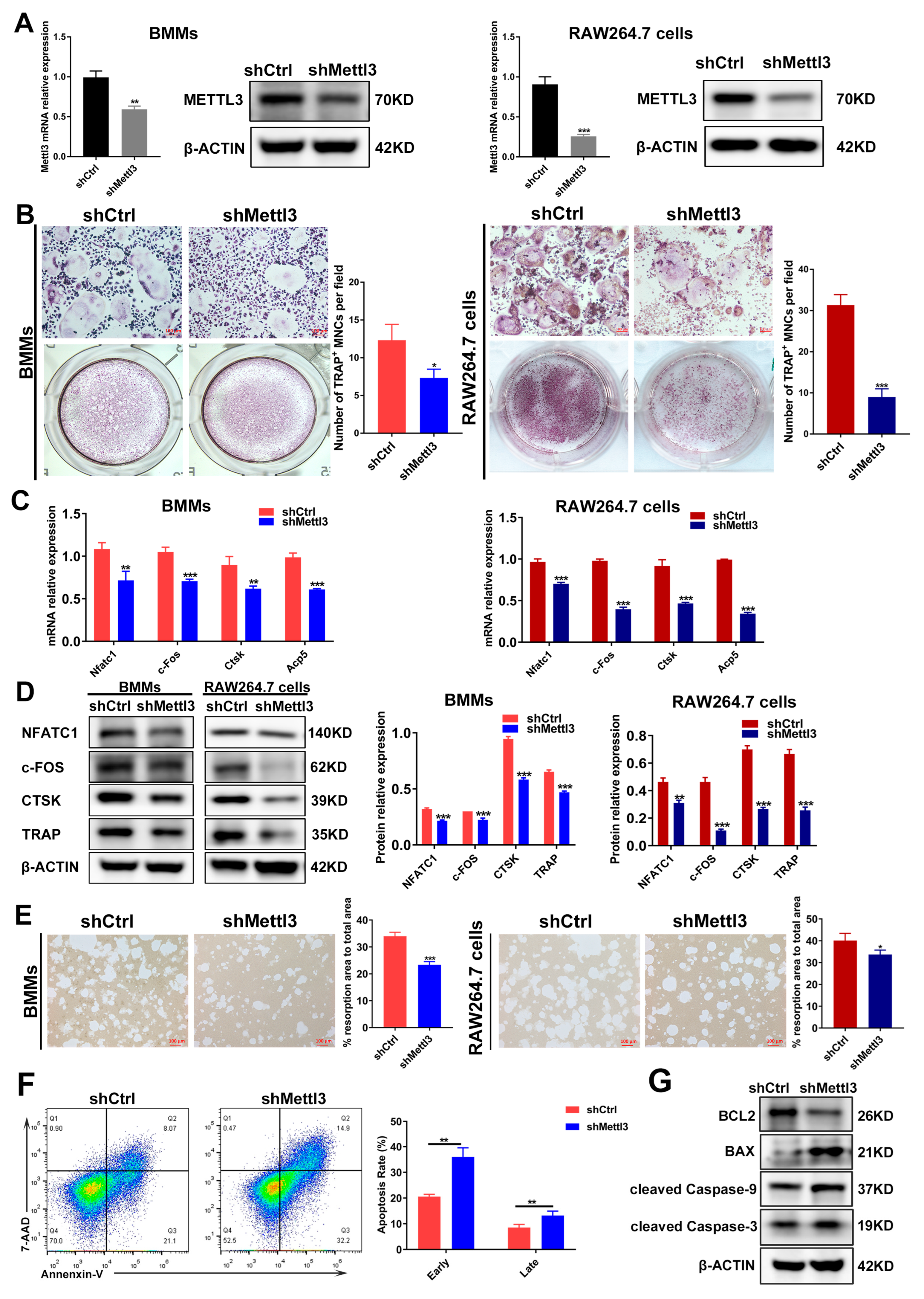
2.2. METTL3 Knockdown Regulates Osteoclast Differentiation, Bone Resorption Capacity and Apoptosis under Inflammatory Conditions
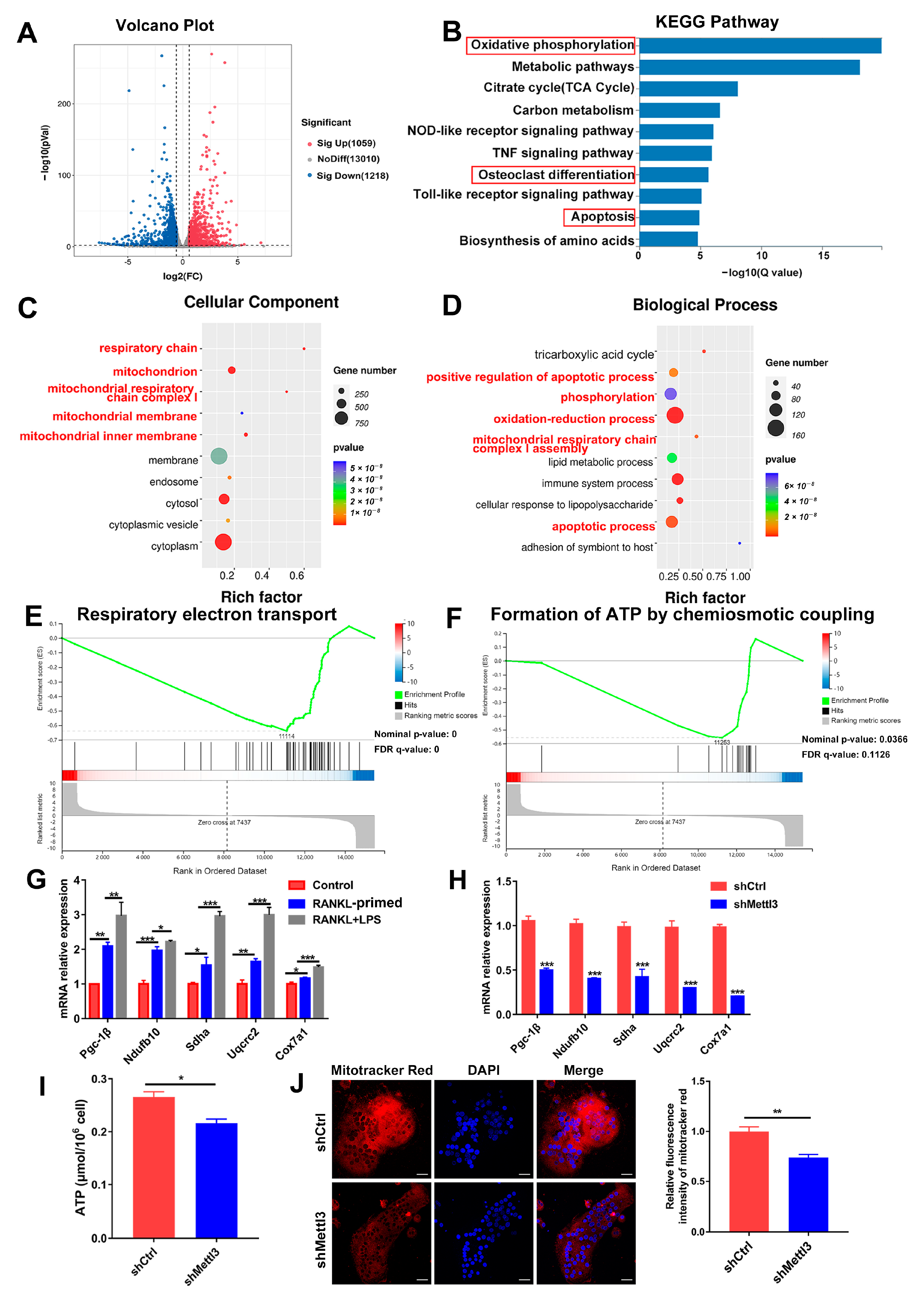
2.3. METTL3 Deficiency Triggers Mitochondrial Dysfunction during LPS-Induced Osteoclastogenesis
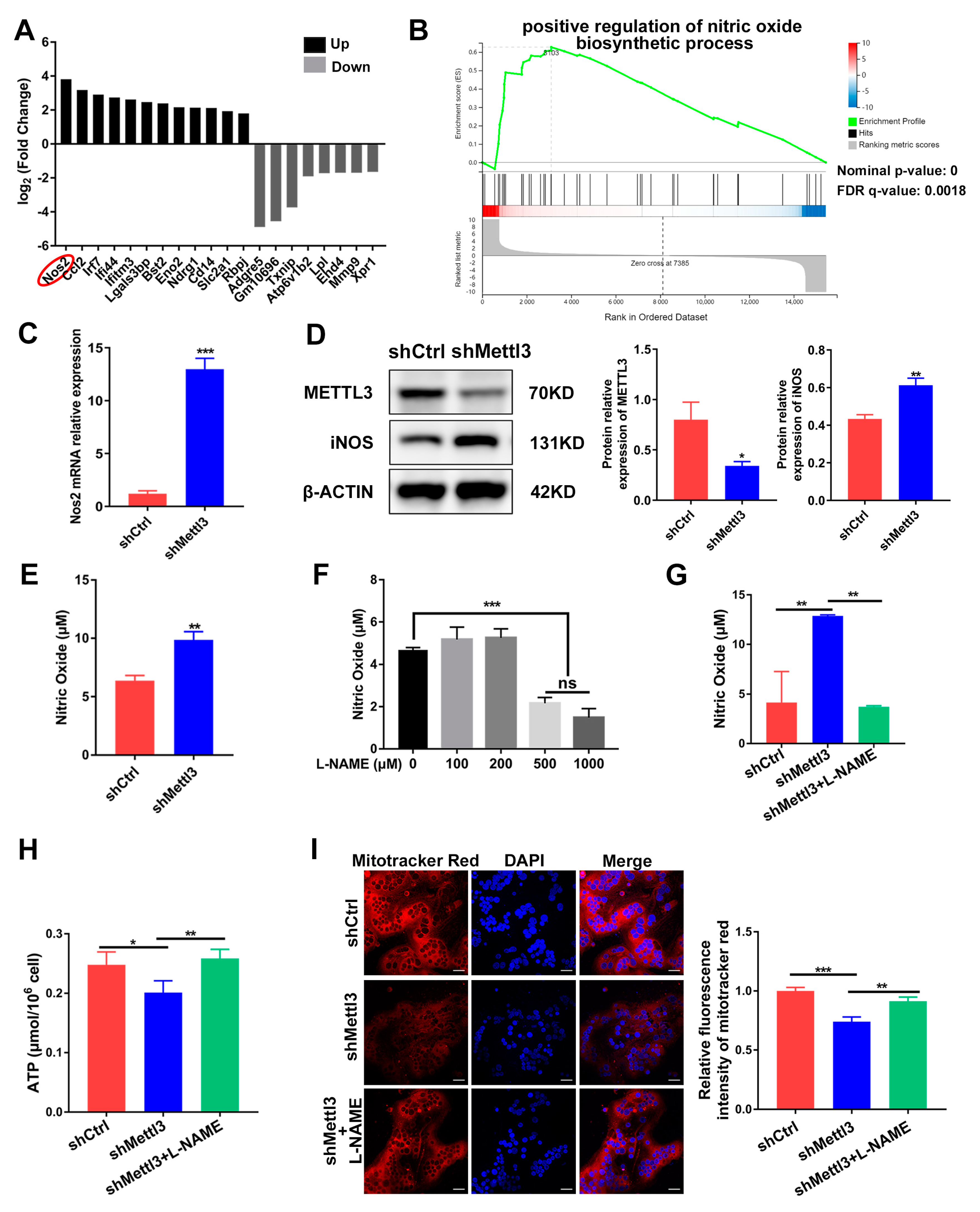
2.4. Mitochondrial Dysfunction in METTL3-Deficient Osteoclasts Is Mediated by iNOS/NO Signaling
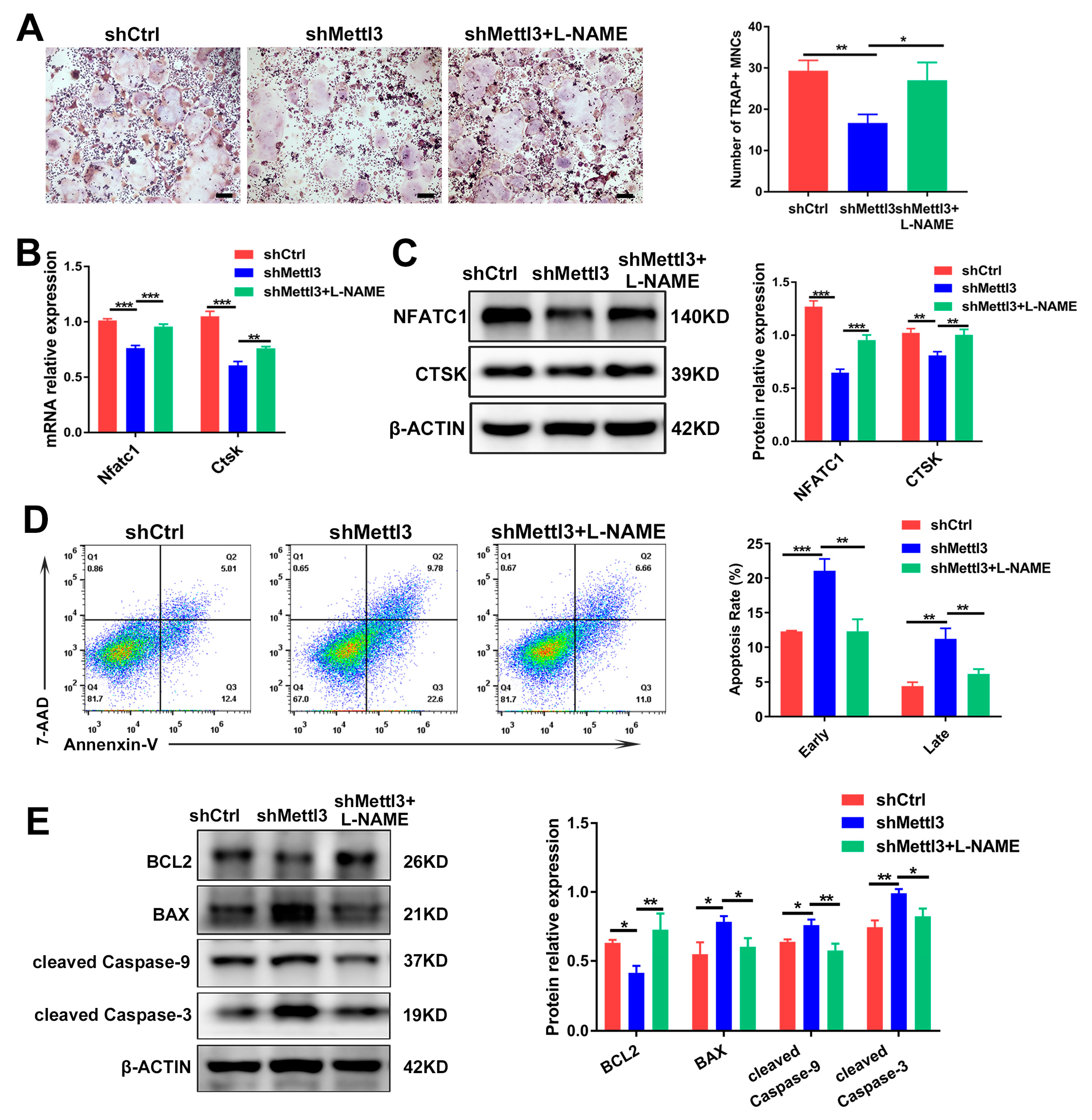
2.5. Inhibiting iNOS/NO Signaling Enhances Osteoclast Differentiation and Decreases Apoptosis in METTL3-Knockdown Cells
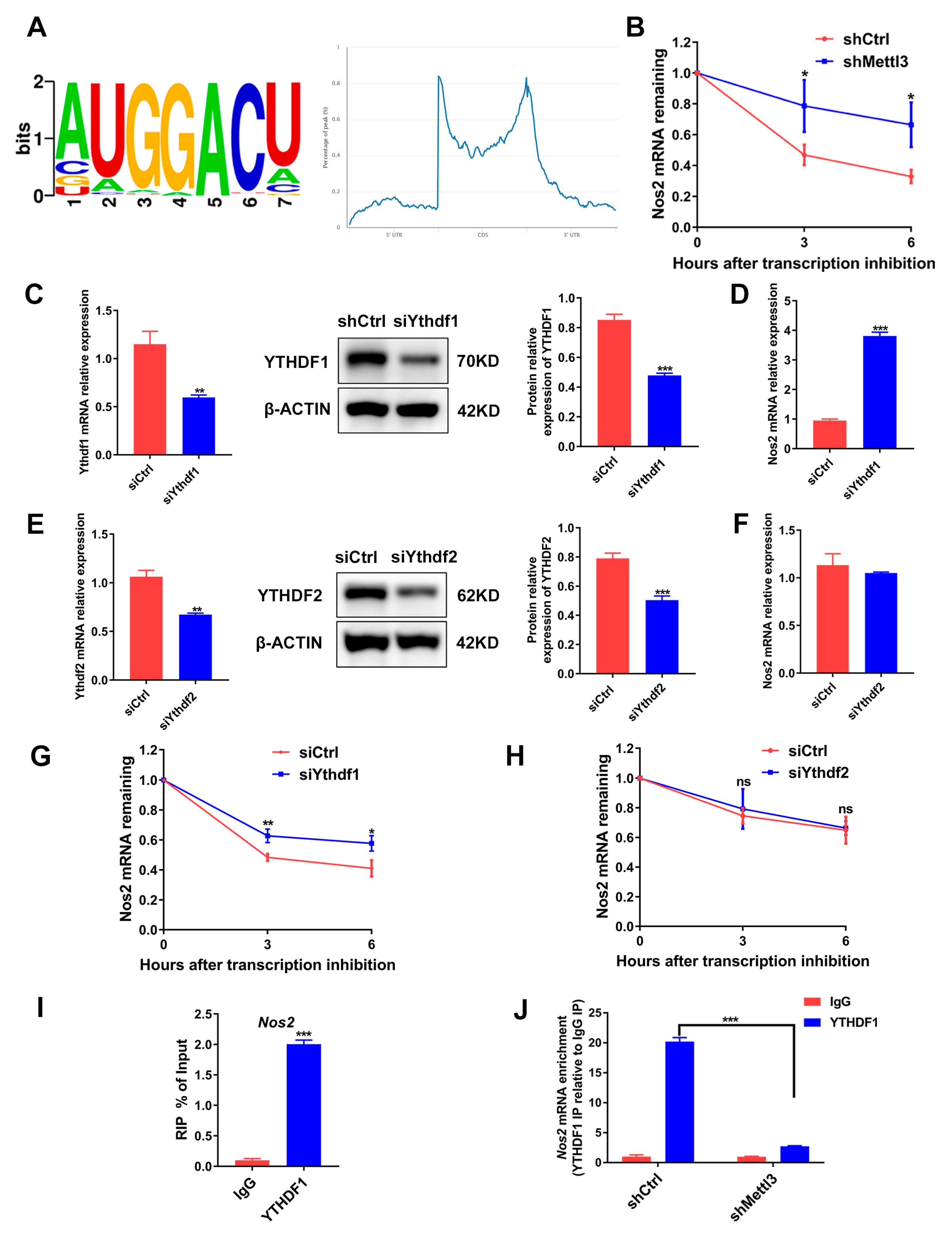
2.6. METTL3 Induces Nos2 mRNA Degradation in a YTHDF1-Dependent Manner
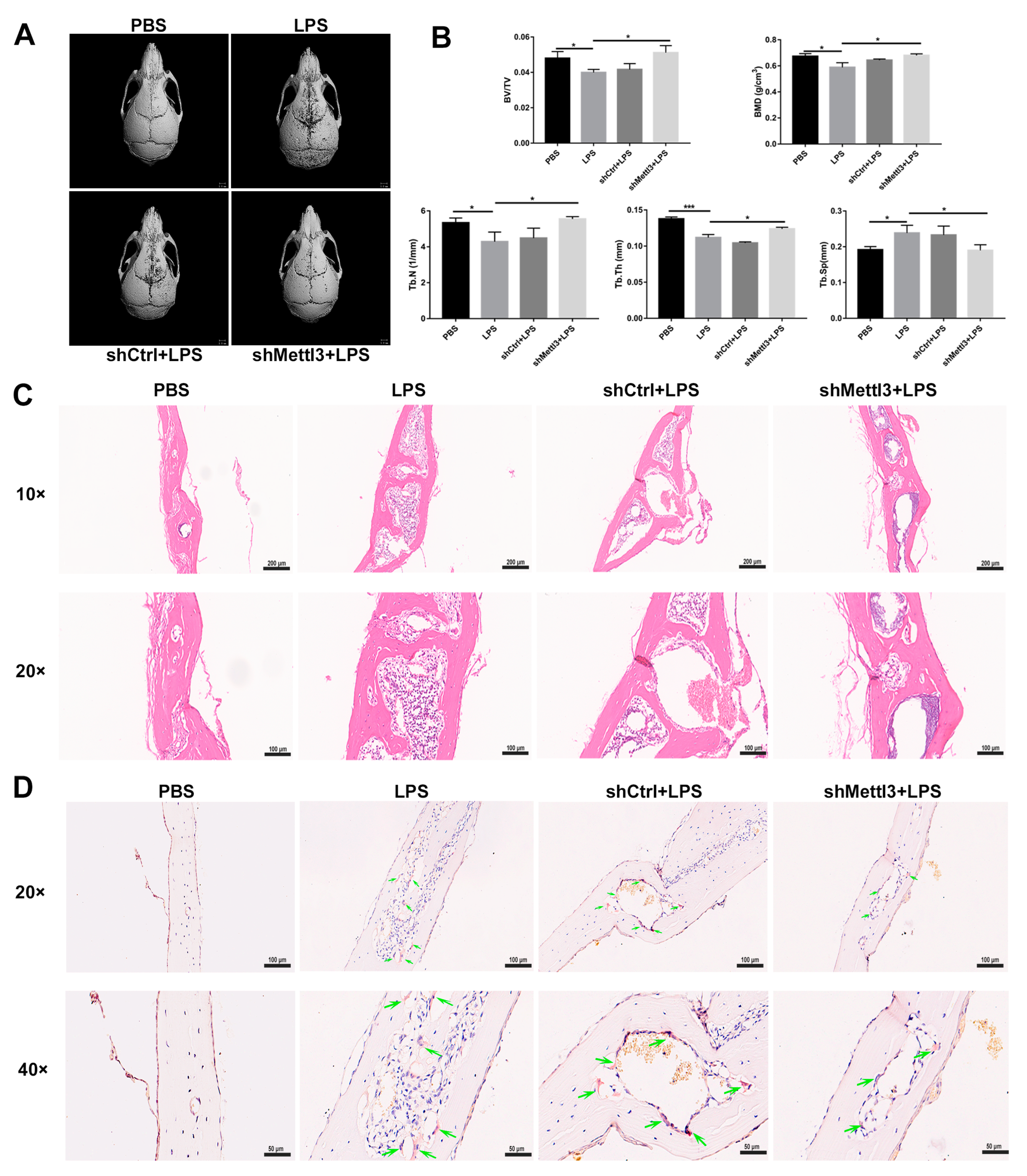
2.7. METTL3 Lentiviral Interference Suppresses Bone Destruction in an LPS-Induced Calvaria Osteolysis Murine Model
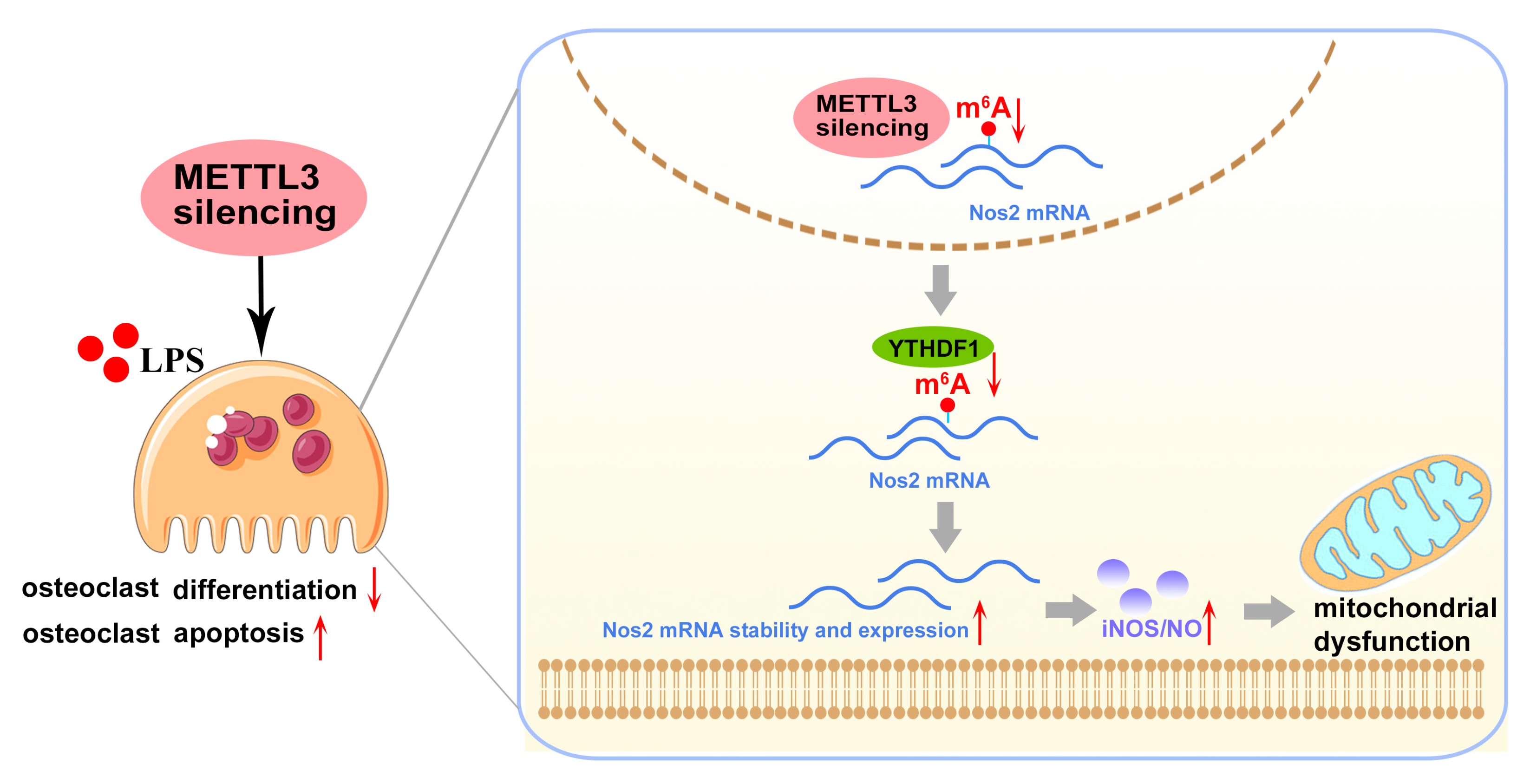
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.2. Cell Transfection
4.3. Cell Viability Assay
4.4. Tartrate-Resistant Acid Phosphatase (TRAP) Staining
4.5. Bone Resorption Assay
4.6. Flow Cytometry Assay of Apoptosis
4.7. Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
4.8. Quantitative Analysis of m6A Levels
4.9. RNA Sequencing
4.10. Western Blotting Analysis
4.11. Nitric Oxide Assay
4.12. ATP Level Detection
4.13. Mito-Tracker Red CMXRos Staining
4.14. RNA Stability Measurement
4.15. RNA-Binding Protein Immunoprecipitation (RIP)
4.16. Establishment and Analysis of LPS-Induced Osteolysis Murine Model
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonciulea, A.; de Beur, S.J. The dynamic skeleton. Rev. Endocr. Metab. Disord. 2015, 16, 79–91. [Google Scholar] [CrossRef]
- Soysa, N.S.; Alles, N. Osteoclast function and bone-resorbing activity: An overview. Biochem. Biophys. Res. Commun. 2016, 476, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. [Google Scholar] [CrossRef] [PubMed]
- Nason, R.; Jung, J.Y.; Chole, R.A. Lipopolysaccharide-induced osteoclastogenesis from mononuclear precursors: A mechanism for osteolysis in chronic otitis. J. Assoc. Res. Otolaryngol. 2009, 10, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Wu, B.; Li, L.; Huang, Y.; Ma, J.; Min, J. Readers, writers and erasers of N(6)-methylated adenosine modification. Curr. Opin. Struct. Biol. 2017, 47, 67–76. [Google Scholar] [CrossRef]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G.M.; Zeller, M.; Anderson, K.G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A.F.; et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35, 109091. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Wang, J.; Yan, S.; Lu, H.; Wang, S.; Xu, D. METTL3 Attenuates LPS-Induced Inflammatory Response in Macrophages via NF-kappaB Signaling Pathway. Mediat. Inflamm. 2019, 2019, 3120391. [Google Scholar] [CrossRef]
- Shi, W.; Zheng, Y.; Luo, S.; Li, X.; Zhang, Y.; Meng, X.; Huang, C.; Li, J. METTL3 Promotes Activation and Inflammation of FLSs Through the NF-kappaB Signaling Pathway in Rheumatoid Arthritis. Front. Med. 2021, 8, 607585. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Xue, S.; Jiang, Y.; Lu, H.; Zhu, L.; Wang, C.; Ma, J. METTL3 involves the progression of osteoarthritis probably by affecting ECM degradation and regulating the inflammatory response. Life Sci. 2021, 278, 119528. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Avram, V.F.; Merce, A.P.; Hancu, I.M.; Batran, A.D.; Kennedy, G.; Rosca, M.G.; Muntean, D.M. Impairment of Mitochondrial Respiration in Metabolic Diseases: An Overview. Int. J. Mol. Sci. 2022, 23, 8852. [Google Scholar] [CrossRef]
- Wang, T.; Mo, L.; Ou, J.; Fang, Q.; Wu, H.; Wu, Y.; Nandakumar, K.S. Proteus mirabilis Vesicles Induce Mitochondrial Apoptosis by Regulating miR96-5p/Abca1 to Inhibit Osteoclastogenesis and Bone Loss. Front. Immunol. 2022, 13, 833040. [Google Scholar] [CrossRef]
- Xu, Y.; Tan, H.; Liu, K.; Wen, C.; Pang, C.; Liu, H.; Xu, R.; Li, Q.; He, C.; Nandakumar, K.S.; et al. Targeted inhibition of ATP5B gene prevents bone erosion in collagen-induced arthritis by inhibiting osteoclastogenesis. Pharmacol. Res. 2021, 165, 105458. [Google Scholar] [CrossRef]
- Arnett, T.R.; Orriss, I.R. Metabolic properties of the osteoclast. Bone 2018, 115, 25–30. [Google Scholar] [CrossRef]
- Kwak, H.B.; Lee, B.K.; Oh, J.; Yeon, J.T.; Choi, S.W.; Cho, H.J.; Lee, M.S.; Kim, J.J.; Bae, J.M.; Kim, S.H.; et al. Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone 2010, 46, 724–731. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Yang, M.; Du, Y.; Wan, Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014, 20, 483–498. [Google Scholar] [CrossRef]
- Eggelbusch, M.; Shi, A.; Broeksma, B.C.; Vazquez-Cruz, M.; Soares, M.N.; de Wit, G.; Everts, B.; Jaspers, R.T.; Wust, R. The NLRP3 inflammasome contributes to inflammation-induced morphological and metabolic alterations in skeletal muscle. J. Cachexia Sarcopenia Muscle 2022, 13, 3048–3061. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Z.; Li, R.; Wu, Y.; Chi, M.; Gao, S.; Sun, X.; Meng, X.; Wang, B. Potential roles of N6-methyladenosine (m6A) in immune cells. J. Transl. Med. 2021, 19, 251. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, Q.; Meng, R.; Yi, B.; Xu, Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J. Cell. Mol. Med. 2018, 22, 2558–2568. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, R.; Kong, Y.; Feng, Z.; Xu, Q. METTL3 regulates LPS-induced inflammatory response via the NOD1 signaling pathway. Cell. Signal. 2022, 93, 110283. [Google Scholar] [CrossRef]
- Castora, F.J. Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 83–108. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef]
- Murakami, S.; Jaffrey, S.R. Hidden codes in mRNA: Control of gene expression by m6A. Mol. Cell 2022, 82, 2236–2251. [Google Scholar] [CrossRef]
- Zaccara, S.; Jaffrey, S.R. A Unified Model for the Function of YTHDF Proteins in Regulating m6A-Modified mRNA. Cell 2020, 181, 1582–1595. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, G.; Zhou, X.; Huang, L.; Meng, J.; He, B.; Qi, Y. alpha-Mangostin inhibits LPS-induced bone resorption by restricting osteoclastogenesis via NF-kappaB and MAPK signaling. Chin. Med. 2022, 17, 34. [Google Scholar] [CrossRef]
- Oliveira, T.C.; Gomes, M.S.; Gomes, A.C. The Crossroads between Infection and Bone Loss. Microorganisms 2020, 8, 1765. [Google Scholar] [CrossRef]
- Yan, G.; Guo, Y.; Guo, J.; Wang, Q.; Wang, C.; Wang, X. N-Acetylcysteine Attenuates Lipopolysaccharide-Induced Osteolysis by Restoring Bone Remodeling Balance via Reduction of Reactive Oxygen Species Formation During Osteoclastogenesis. Inflammation 2020, 43, 1279–1292. [Google Scholar] [CrossRef]
- Park, H.; Noh, A.L.; Kang, J.H.; Sim, J.S.; Lee, D.S.; Yim, M. Peroxiredoxin II negatively regulates lipopolysaccharide-induced osteoclast formation and bone loss via JNK and STAT3. Antioxid. Redox. Signal. 2015, 22, 63–77. [Google Scholar] [CrossRef]
- Wang, S.; Lv, W.; Li, T.; Zhang, S.; Wang, H.; Li, X.; Wang, L.; Ma, D.; Zang, Y.; Shen, J.; et al. Dynamic regulation and functions of mRNA m6A modification. Cancer Cell Int. 2022, 22, 48. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tian, Y.; Zhang, Q.; Ren, D.; Zhang, Q.; Yan, X.; Wang, L.; He, Z.; Zhang, W.; Zhang, T.; et al. Novel Insights Into the Role of N6-Methyladenosine RNA Modification in Bone Pathophysiology. Stem Cells Dev. 2021, 30, 17–28. [Google Scholar] [CrossRef]
- Mi, B.; Xiong, Y.; Yan, C.; Chen, L.; Xue, H.; Panayi, A.C.; Hu, L.; Hu, Y.; Zhou, W.; Cao, F.; et al. Methyltransferase-like 3-mediated N6-methyladenosine modification of miR-7212-5p drives osteoblast differentiation and fracture healing. J. Cell. Mol. Med. 2020, 24, 6385–6396. [Google Scholar] [CrossRef]
- Li, D.; Cai, L.; Meng, R.; Feng, Z.; Xu, Q. METTL3 Modulates Osteoclast Differentiation and Function by Controlling RNA Stability and Nuclear Export. Int. J. Mol. Sci. 2020, 21, 1660. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Kang, J.Y.; Kang, N.; Yang, Y.M.; Hong, J.H.; Shin, D.M. The Role of Ca (2+)-NFATc1 Signaling and Its Modulation on Osteoclastogenesis. Int. J. Mol. Sci. 2020, 21, 3646. [Google Scholar] [CrossRef]
- Vikramdeo, K.S.; Sudan, S.K.; Singh, A.P.; Singh, S.; Dasgupta, S. Mitochondrial respiratory complexes: Significance in human mitochondrial disorders and cancers. J. Cell. Physiol. 2022, 237, 4049–4078. [Google Scholar] [CrossRef]
- Ghosh-Choudhary, S.K.; Liu, J.; Finkel, T. The role of mitochondria in cellular senescence. FASEB J. 2021, 35, e21991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Figueiredo-Pereira, C.; Oudot, C.; Vieira, H.L.; Brenner, C. Mitochondrion: A Common Organelle for Distinct Cell Deaths? Int. Rev. Cell Mol. Biol. 2017, 331, 245–287. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Sboarina, M.; Porporato, P.E.; Zini, N.; Sonveaux, P.; Di Pompo, G.; Baldini, N.; Avnet, S. Energy metabolism in osteoclast formation and activity. Int. J. Biochem. Cell Biol. 2016, 79, 168–180. [Google Scholar] [CrossRef]
- Park-Min, K.H. Metabolic reprogramming in osteoclasts. Semin. Immunopathol. 2019, 41, 565–572. [Google Scholar] [CrossRef]
- Da, W.; Tao, L.; Zhu, Y. The Role of Osteoclast Energy Metabolism in the Occurrence and Development of Osteoporosis. Front. Endocrinol. 2021, 12, 675385. [Google Scholar] [CrossRef]
- Caballano-Infantes, E.; Terron-Bautista, J.; Beltran-Povea, A.; Cahuana, G.M.; Soria, B.; Nabil, H.; Bedoya, F.J.; Tejedo, J.R. Regulation of mitochondrial function and endoplasmic reticulum stress by nitric oxide in pluripotent stem cells. World J. Stem Cells 2017, 9, 26–36. [Google Scholar] [CrossRef]
- Lan, M.; Tang, X.; Zhang, J.; Yao, Z. Insights in pathogenesis of multiple sclerosis: Nitric oxide may induce mitochondrial dysfunction of oligodendrocytes. Rev. Neurosci. 2018, 29, 39–53. [Google Scholar] [CrossRef]
- Ghasemi, M.; Mayasi, Y.; Hannoun, A.; Eslami, S.M.; Carandang, R. Nitric Oxide and Mitochondrial Function in Neurological Diseases. Neuroscience 2018, 376, 48–71. [Google Scholar] [CrossRef]
- Burke, A.S.; MacMillan-Crow, L.A.; Hinson, J.A. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem. Res. Toxicol. 2010, 23, 1286–1292. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, Y.; Shen, H.; Xie, W. m6A-binding proteins: The emerging crucial performers in epigenetics. J. Hematol. Oncol. 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, X.; Xia, M.; Zhong, J. The roles and mechanisms of the m6A reader protein YTHDF1 in tumor biology and human diseases. Mol. Ther. Nucleic Acids 2021, 26, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Lu, A.Q. The biological function of m6A reader YTHDF2 and its role in human disease. Cancer Cell Int. 2021, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, K.; Dong, X.; Xu, Y.; Sun, Q.; Wang, H.; Chen, Z.; Liu, C.; Liu, R.; Yang, Z.; et al. YTHDF1 promotes mRNA degradation via YTHDF1-AGO2 interaction and phase separation. Cell Prolif. 2022, 55, e13157. [Google Scholar] [CrossRef]
- Huang, H.; Jiang, W.; Hong, K.; Cai, J.; He, Y.; Ma, X.; Wu, P.; Lang, J.; Ma, Y.; Huang, C.; et al. Protocatechualdehyde inhibits receptor activator of nuclear factor kappa-B ligand-induced osteoclastogenesis and attenuates lipopolysaccharide-induced inflammatory osteolysis. Phytother. Res. 2021, 35, 3821–3835. [Google Scholar] [CrossRef]
- Guangtao, F.; Zhenkang, W.; Zhantao, D.; Mengyuan, L.; Qingtian, L.; Yuanchen, M.; Yuanfeng, C.; Qiujian, Z. Icariin Alleviates Wear Particle-Induced Periprosthetic Osteolysis via Down-Regulation of the Estrogen Receptor alpha-mediated NF-kappaB Signaling Pathway in Macrophages. Front. Pharm. 2021, 12, 746391. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| shRNA | Target Sequences |
|---|---|
| shMettl3 | GCACCCGCAAGATTGAGTTAT |
| shCtrl | TTCTCCGAACGTGTCACGT |
| siRNA | Target Sequences (5′-3′) |
|---|---|
| siYthdf1 | CCCGUAUCUCACUACCUAUTT AUAGGUAGUGAGAUACGGGTT |
| siYthdf2 | CCAUGAUUGAUGGACAGUCAGCUUU AAAGCUGACUGUCCAUCAAUCAUGG |
| siCtrl | Stealth RNAiTM siRNA Negative Control LO GC (12935-200) |
| Gene | Forward Primer (5′-3′) | Forward Primer (5′-3′) |
|---|---|---|
| Mettl3 | CTTTCTACCCCATCTTGAGTG | CCAACCTTCCGTAGTGATAGTC |
| Ctsk | CACCCAGTGGGAGCTATGGAA | GCCTCCAGGTTATGGGCAGA |
| Acp5 | ACCTTGGCAACGTCTCTGCAC | GTCCAGCATAAAGATGGCCACA |
| Nfatc1 | CCCGTCACATTCTGGTCCAT | CAAGTAACCGTGTAGCTGCACAA |
| c-fos | CGGCATCATCTAGGCCCAG | TCTGCTGCATAGAAGGAACCG |
| Pgc-1β | CTTGGCTGCGCTTACGAAGA | GAAAGCTCGTCCACGTCAGAC |
| Ndufb10 | GATTCTTGGGACAAGGATGTGT | CCTTCGTCAAGTAGGTGATGGG |
| Sdha | AATTTGCCATTTACCGATGGGA | AGCATCCAACACCATAGGTCC |
| Uqcrc2 | AAAGTTGCCCCGAAGGTTAAA | GAGCATAGTTTTCCAGAGAAGCA |
| Cox7a1 | ACAATGACCTCCCAGTACACT | CCAAGCAGTATAAGCAGTAGGC |
| Nos2 | ACATCGACCCGTCCACAGTAT | CAGAGGGGTAGGCTTGTCTC |
| Ythdf1 | ACAGTCCAATCCGAGTAACAGT | GGTAGTGAGATACGGGATGGGA |
| Ythdf2 | AGGCGGGTTCTGGATCTACT | ACCCGGCCATGTTTCAGATT |
| β-Actin | CATACCCAAGAAGGAAGGCTGG | GCTATGTTGCTCTAGACTTCGAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; He, J.; Fang, C.; Zhang, Y.; He, M.; Zhang, Z.; Hou, J.; Xu, Q. METTL3 Regulates Osteoclast Biological Behaviors via iNOS/NO-Mediated Mitochondrial Dysfunction in Inflammatory Conditions. Int. J. Mol. Sci. 2023, 24, 1403. https://doi.org/10.3390/ijms24021403
Li D, He J, Fang C, Zhang Y, He M, Zhang Z, Hou J, Xu Q. METTL3 Regulates Osteoclast Biological Behaviors via iNOS/NO-Mediated Mitochondrial Dysfunction in Inflammatory Conditions. International Journal of Molecular Sciences. 2023; 24(2):1403. https://doi.org/10.3390/ijms24021403
Chicago/Turabian StyleLi, Di, Jinlin He, Caihong Fang, Yiwen Zhang, Mingli He, Zhanqi Zhang, Jinsong Hou, and Qiong Xu. 2023. "METTL3 Regulates Osteoclast Biological Behaviors via iNOS/NO-Mediated Mitochondrial Dysfunction in Inflammatory Conditions" International Journal of Molecular Sciences 24, no. 2: 1403. https://doi.org/10.3390/ijms24021403
APA StyleLi, D., He, J., Fang, C., Zhang, Y., He, M., Zhang, Z., Hou, J., & Xu, Q. (2023). METTL3 Regulates Osteoclast Biological Behaviors via iNOS/NO-Mediated Mitochondrial Dysfunction in Inflammatory Conditions. International Journal of Molecular Sciences, 24(2), 1403. https://doi.org/10.3390/ijms24021403