Association of DNA Promoter Methylation and BRAF Mutation in Thyroid Cancer

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Genome-Wide Methylation Assay
2.3. BRAF and KRAS Mutation
3. Statistical Analysis
3.1. DNA Promoter Methylation in Thyroid Cancer
- DML (n = 51) common to BRAF mutation and wild type: These loci were differentially methylated in tumor tissue compared to healthy tissue, irrespective of BRAF mutation status, and the magnitude of the differential methylation was not different between BRAF mutant and wild-type tumors. Of these 51 DML, 34 were hypomethylated and 17 were hypermethylated in tumor tissue compared to corresponding normal thyroid tissue. An example of this group is shown in Figure 4A. The CCND1 gene was hypomethylated in both groups. Reasonable separation of tumor and healthy tissue in the PCA plot using these 51 common loci for all the patients is shown in Figure 4B.
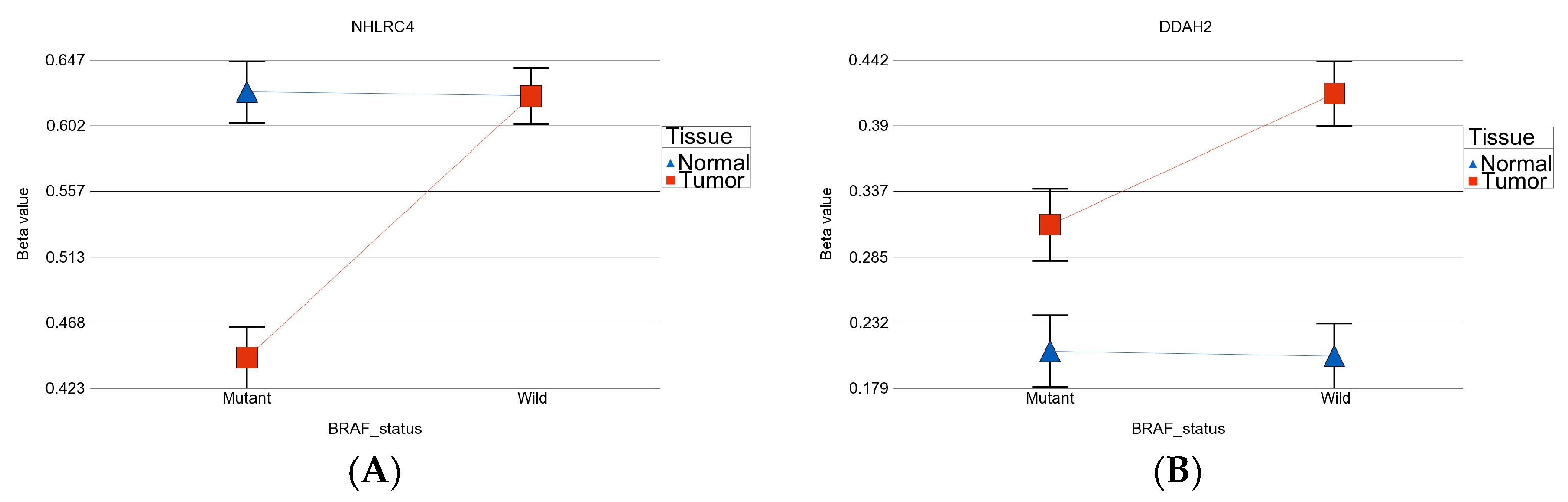
- BRAF mutation-specific DML (n = 35): These loci were significantly differentially methylated in tumor tissue only in the presence of BRAF mutation, and the magnitude of differential methylation was significantly greater than in wild type. Of these 35 DML, 29 were hypomethylated and 6 were hypermethylated in tumor tissue compared to corresponding normal thyroid tissue (for detail, see Supplementary Table S2). An example of this group is shown in Figure 4C. The TNFRSF1B gene (encoding TNFR2) was hypomethylated in the mutant group, suggesting potential overexpression of TNFR2 in BRAF mutant tumors. This is an important gene in terms of therapeutic targeting potential, because TNFR2 is rarely expressed in normal tissues. Reasonable separation of tumor and healthy tissue in the PCA plot, using these 35 BRAF mutation-specific loci for patients with BRAF mutation, is shown in Figure 4D.
- BRAF wild-type-specific DML (n = 62): These loci are significantly differentially methylated in tumor tissue only in the absence of BRAF mutation, and the magnitude of differential methylation is significantly greater than in patients with BRAF mutation. Of these 62 DML, 20 were hypomethylated and 42 were hypermethylated in tumor tissue compared to corresponding normal thyroid tissue. An example of this group is shown in Figure 4E. The REC8 gene is hypermethylated in the BRAF wild-type group. Reasonable separation of tumor and healthy tissue in the PCA plot, using these 35 BRAF wild-type-specific loci for patients with wild-type BRAF, is shown in Figure 4F.
3.2. Validation of the DML in Independent Set of Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henke, L.E.; Pfeifer, J.D.; Baranski, T.J.; DeWees, T.; Grigsby, P.W. Long-term outcomes of follicular variant vs classic papillary thyroid carcinoma. Endocr. Connect. 2018, 7, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Bibbins-Domingo, K.; Grossman, D.C.; Curry, S.J.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Krist, A.H.; Kurth, A.E.; et al. Screening for Thyroid Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2017, 317, 1882–1887. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.R.; Nam, S.H.; Roh, J.L.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Thyroid stimulating hormone suppression and recurrence after thyroid lobectomy for papillary thyroid carcinoma. Endocrine 2022, 75, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Nasr, C.; Bischoff, L.; Busaidy, N.L.; Byrd, D.; Callender, G.; Dickson, P.; Duh, Q.Y.; Ehya, H.; Goldner, W.; et al. NCCN Guidelines Insights: Thyroid Carcinoma, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Marotta, V.; Chiofalo, M.G.; Di Gennaro, F.; Daponte, A.; Sandomenico, F.; Vallone, P.; Costigliola, L.; Botti, G.; Ionna, F.; Pezzullo, L. Kinase-inhibitors for iodine-refractory differentiated thyroid cancer: Still far from a structured therapeutic algorithm. Crit. Rev. Oncol. Hematol. 2021, 162, 103353. [Google Scholar] [CrossRef] [PubMed]
- Schmidbauer, B.; Menhart, K.; Hellwig, D.; Grosse, J. Differentiated Thyroid Cancer-Treatment: State of the Art. Int. J. Mol. Sci. 2017, 18, 1292. [Google Scholar] [CrossRef]
- Shonka, D.C., Jr.; Ho, A.; Chintakuntlawar, A.V.; Geiger, J.L.; Park, J.C.; Seetharamu, N.; Jasim, S.; Abdelhamid Ahmed, A.H.; Bible, K.C.; Brose, M.S.; et al. American Head and Neck Society Endocrine Surgery Section and International Thyroid Oncology Group consensus statement on mutational testing in thyroid cancer: Defining advanced thyroid cancer and its targeted treatment. Head Neck 2022, 44, 1277–1300. [Google Scholar] [CrossRef]
- Sipos, J.A.; Ringel, M.D. Molecular testing in thyroid cancer diagnosis and management. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 37, 101680. [Google Scholar] [CrossRef]
- Loo, E.; Khalili, P.; Beuhler, K.; Siddiqi, I.; Vasef, M.A. BRAF V600E Mutation across Multiple Tumor Types: Correlation between DNA-based Sequencing and Mutation-specific Immunohistochemistry. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 709–713. [Google Scholar] [CrossRef]
- Chen, D.; Su, X.; Zhu, L.; Jia, H.; Han, B.; Chen, H.; Liang, Q.; Hu, C.; Yang, H.; Liu, L.; et al. Papillary thyroid cancer organoids harboring BRAF(V600E) mutation reveal potentially beneficial effects of BRAF inhibitor-based combination therapies. J. Transl. Med. 2023, 21, 9. [Google Scholar] [CrossRef]
- da Silva, T.N.; Rodrigues, R.; Saramago, A.; Pires, C.; Rito, M.; Horta, M.; Martins, C.; Leite, V.; Cavaco, B.M. Target therapy for BRAF mutated anaplastic thyroid cancer: A clinical and molecular study. Eur. J. Endocrinol. 2023, 188, lvac011. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zheng, X.; Zou, X.; Ye, Z.; Liu, J.; Lei, J.; Li, Z. Genetic and clinical profiles of 160 papillary thyroid cancers with lateral neck lymph node metastasis. Front. Oncol. 2022, 12, 1057532. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, S.J.; Ha, S.Y.; Xu, Z.; Han, Y.; Jee, H.G.; Cho, S.W.; Park, Y.J.; Lee, K.E. BRAFV600E Mutation Enhances Estrogen-Induced Metastatic Potential of Thyroid Cancer by Regulating the Expression of Estrogen Receptors. Endocrinol. Metab. 2022, 37, 879–890. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, V.; Canberk, S.; Deftereos, G. Molecular testing in fine-needle aspiration of thyroid nodules. Diagn. Cytopathol. 2023, 51, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef]
- Reis, J.S.; Costa, I.; Costa, M.; Valente, A.C.; Freitas, M.B.; Almeida, C.L.; Gonçalves, M.; Teixeira, C.; Ribeiro, M.; Caeiro, C.; et al. Metastatic BRAF V600E-Mutated Thyroid Carcinoma: Molecular/Genetic Profiling Brings a New Therapeutic Option. Case Rep. Oncol. 2022, 15, 960–966. [Google Scholar] [CrossRef]
- Silver, J.A.; Bogatchenko, M.; Pusztaszeri, M.; Forest, V.I.; Hier, M.P.; Yang, J.W.; Tamilia, M.; Payne, R.J. BRAF V600E mutation is associated with aggressive features in papillary thyroid carcinomas ≤1.5 cm. J. Otolaryngol. Head Neck Surg. 2021, 50, 63. [Google Scholar] [CrossRef]
- Turchini, J.; Sioson, L.; Clarkson, A.; Sheen, A.; Delbridge, L.; Glover, A.; Sywak, M.; Sidhu, S.; Gill, A.J. The Presence of Typical “BRAFV600E-Like” Atypia in Papillary Thyroid Carcinoma is Highly Specific for the Presence of the BRAFV600E Mutation. Endocr. Pathol. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Zhang, M.; Gu, J.; Wang, W.; Wang, K.; Zheng, L.; Feng, J.; Shang, J. Combined expression of the BRAF(V600E) mutation and PD-L1 in early papillary thyroid carcinoma and its relationship with clinicopathological features and recurrence-a retrospective cohort study. Gland. Surg. 2022, 11, 1908–1923. [Google Scholar] [CrossRef]
- Yi, J.W.; Ha, S.Y.; Jee, H.G.; Kim, K.; Kim, S.J.; Chai, Y.J.; Choi, J.Y.; Lee, K.E. Induction of the BRAFV600E Mutation in Thyroid Cells Leads to Frequent Hypermethylation. Clin. Exp. Otorhinolaryngol. 2022, 15, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Tufano, R.P.; Teixeira, G.V.; Bishop, J.; Carson, K.A.; Xing, M. BRAF mutation in papillary thyroid cancer and its value in tailoring initial treatment: A systematic review and meta-analysis. Medicine 2012, 91, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–470, discussion 470–461. [Google Scholar] [CrossRef] [PubMed]
- Segev, D.L.; Clark, D.P.; Zeiger, M.A.; Umbricht, C. Beyond the suspicious thyroid fine needle aspirate. A review. Acta Cytol. 2003, 47, 709–722. [Google Scholar] [CrossRef]
- Acuña-Ruiz, A.; Carrasco-López, C.; Santisteban, P. Genomic and epigenomic profile of thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 37, 101656. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Zeng, Y.; Ma, W.; Zhang, W.; Liu, Y.; Guo, F.; Ruan, X.; Chi, J.; Zheng, X.; Gao, M. Characterization of the CpG island methylator phenotype subclass in papillary thyroid carcinoma. Front. Endocrinol. 2022, 13, 1008301. [Google Scholar] [CrossRef]
- Klimaitė, R.; Kazokaitė, M.; Kondrotienė, A.; Daukšienė, D.; Sabaliauskaitė, R.; Žukauskaitė, K.; Žilaitienė, B.; Jarmalaitė, S.; Daukša, A. The Role of TSHR, PTEN and RASSF1A Promoters’ Methylation Status for Non-Invasive Detection of Papillary Thyroid Carcinoma. J. Clin. Med. 2022, 11, 4917. [Google Scholar] [CrossRef]
- Rodríguez-Rodero, S.; Morales-Sánchez, P.; Tejedor, J.R.; Coca-Pelaz, A.; Mangas, C.; Peñarroya, A.; Fernández-Vega, I.; Fernández-Fernández, L.; Álvarez-López, C.M.; Fernández, A.F.; et al. Classification of follicular-patterned thyroid lesions using a minimal set of epigenetic biomarkers. Eur. J. Endocrinol. 2022, 187, 335–347. [Google Scholar] [CrossRef]
- White, M.G.; Nagar, S.; Aschebrook-Kilfoy, B.; Jasmine, F.; Kibriya, M.G.; Ahsan, H.; Angelos, P.; Kaplan, E.L.; Grogan, R.H. Epigenetic Alterations and Canonical Pathway Disruption in Papillary Thyroid Cancer: A Genome-wide Methylation Analysis. Ann. Surg. Oncol. 2016, 23, 2302–2309. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, P.; Ying, T.; Wang, Y.; Wang, X.; Shu, L.; Mo, Z.; Chen, Z.; Wang, X.; Wang, W.; et al. Integrative Analysis of DNA Methylation and Gene Expression Identified Follicular Thyroid Cancer-Specific Diagnostic Biomarkers. Front. Endocrinol. 2021, 12, 736068. [Google Scholar] [CrossRef]
- Ying, T.; Wang, X.; Yao, Y.; Yuan, J.; Chen, S.; Wen, L.; Chen, Z.; Wang, X.; Luo, C.; Sheng, J.; et al. Integrative Methylome and Transcriptome Characterization Identifies SERINC2 as a Tumor-Driven Gene for Papillary Thyroid Carcinoma. Cancers 2022, 15, 243. [Google Scholar] [CrossRef] [PubMed]
- Zarkesh, M.; Arab, N.; Abooshahab, R.; Heydarzadeh, S.; Sheikholeslami, S.; Nozhat, Z.; Salehi Jahromi, M.; Fanaei, S.A.; Hedayati, M. CpG island status as an epigenetic alteration for NIS promoter in thyroid neoplasms; a cross-sectional study with a systematic review. Cancer Cell Int. 2022, 22, 310. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Dralle, H.; Machens, A.; Basa, J.; Fatourechi, V.; Franceschi, S.; Hay, I.D.; Nikiforov, Y.E.; Pacini, F.; Pasieka, J.L.; Sherman, S.I. Follicular cell-derived thyroid cancer. Nat. Rev. Dis. Prim. 2015, 1, 15077. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Kim, H.J.; Kim, Y.H.; Park, S.H.; Chwae, Y.J.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. B-RafV600E inhibits sodium iodide symporter expression via regulation of DNA methyltransferase 1. Exp. Mol. Med. 2014, 46, e120. [Google Scholar] [CrossRef]
- Galrão, A.L.; Camargo, R.Y.; Friguglietti, C.U.; Moraes, L.; Cerutti, J.M.; Serrano-Nascimento, C.; Suzuki, M.F.; Medeiros-Neto, G.; Rubio, I.G. Hypermethylation of a New Distal Sodium/Iodide Symporter (NIS) enhancer (NDE) is associated with reduced NIS expression in thyroid tumors. J. Clin. Endocrinol. Metab. 2014, 99, E944–E952. [Google Scholar] [CrossRef]
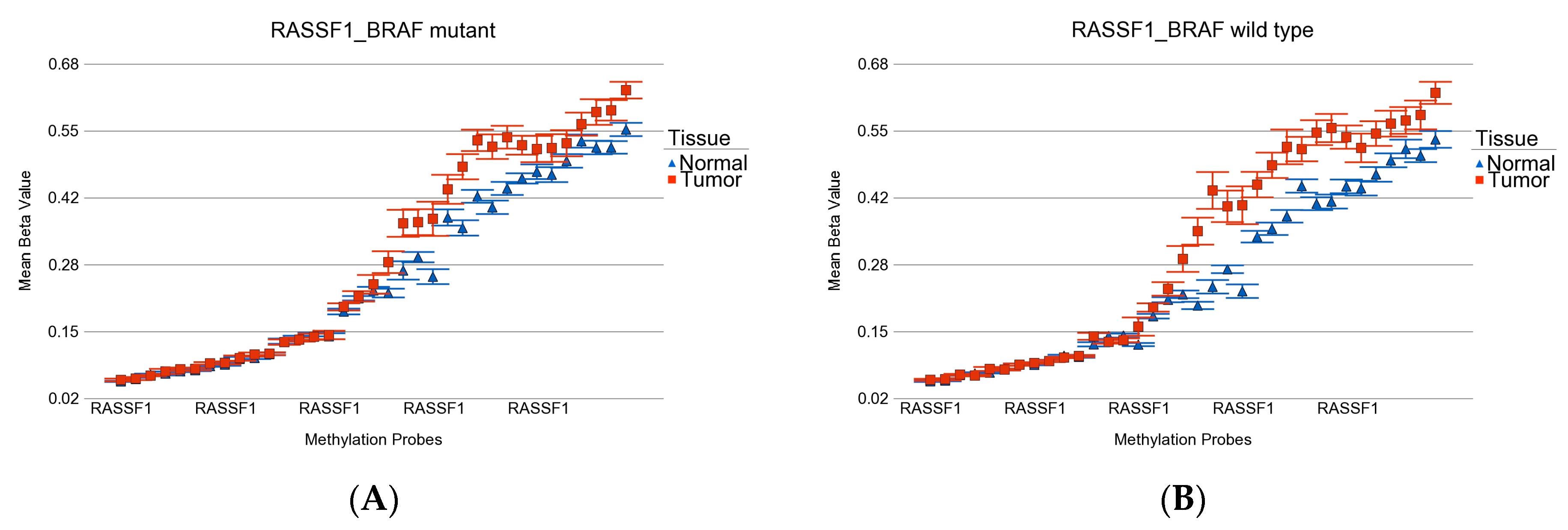
- Schagdarsurengin, U.; Gimm, O.; Hoang-Vu, C.; Dralle, H.; Pfeifer, G.P.; Dammann, R. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res. 2002, 62, 3698–3701. [Google Scholar]
- Hoque, M.O.; Rosenbaum, E.; Westra, W.H.; Xing, M.; Ladenson, P.; Zeiger, M.A.; Sidransky, D.; Umbricht, C.B. Quantitative assessment of promoter methylation profiles in thyroid neoplasms. J. Clin. Endocrinol. Metab. 2005, 90, 4011–4018. [Google Scholar] [CrossRef]
- Nakamura, N.; Carney, J.A.; Jin, L.; Kajita, S.; Pallares, J.; Zhang, H.; Qian, X.; Sebo, T.J.; Erickson, L.A.; Lloyd, R.V. RASSF1A and NORE1A methylation and BRAFV600E mutations in thyroid tumors. Lab. Investig. 2005, 85, 1065–1075. [Google Scholar] [CrossRef]
- Schagdarsurengin, U.; Gimm, O.; Dralle, H.; Hoang-Vu, C.; Dammann, R. CpG island methylation of tumor-related promoters occurs preferentially in undifferentiated carcinoma. Thyroid 2006, 16, 633–642. [Google Scholar] [CrossRef]
- Galrão, A.L.; Sodré, A.K.; Camargo, R.Y.; Friguglietti, C.U.; Kulcsar, M.A.; Lima, E.U.; Medeiros-Neto, G.; Rubio, I.G. Methylation levels of sodium-iodide symporter (NIS) promoter in benign and malignant thyroid tumors with reduced NIS expression. Endocrine 2013, 43, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.K.; Chen, K.M.; Merritt, J.; Chitale, D.; Divine, G.; Worsham, M.J. Methylation markers differentiate thyroid cancer from benign nodules. J. Endocrinol. Investig. 2018, 41, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kunstman, J.W.; Korah, R.; Healy, J.M.; Prasad, M.; Carling, T. Quantitative assessment of RASSF1A methylation as a putative molecular marker in papillary thyroid carcinoma. Surgery 2013, 154, 1255–1261, discussion 1261–1252. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.C.; Juhlin, C.C.; Healy, J.M.; Prasad, M.L.; Korah, R.; Carling, T. Frequent silencing of RASSF1A via promoter methylation in follicular thyroid hyperplasia: A potential early epigenetic susceptibility event in thyroid carcinogenesis. JAMA Surg. 2014, 149, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodero, S.; Fernández, A.F.; Fernández-Morera, J.L.; Castro-Santos, P.; Bayon, G.F.; Ferrero, C.; Urdinguio, R.G.; Gonzalez-Marquez, R.; Suarez, C.; Fernández-Vega, I.; et al. DNA methylation signatures identify biologically distinct thyroid cancer subtypes. J. Clin. Endocrinol. Metab. 2013, 98, 2811–2821. [Google Scholar] [CrossRef]
- Mancikova, V.; Buj, R.; Castelblanco, E.; Inglada-Pérez, L.; Diez, A.; de Cubas, A.A.; Curras-Freixes, M.; Maravall, F.X.; Mauricio, D.; Matias-Guiu, X.; et al. DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int. J. Cancer 2014, 135, 598–610. [Google Scholar] [CrossRef]
- Ellis, R.J.; Wang, Y.; Stevenson, H.S.; Boufraqech, M.; Patel, D.; Nilubol, N.; Davis, S.; Edelman, D.C.; Merino, M.J.; He, M.; et al. Genome-wide methylation patterns in papillary thyroid cancer are distinct based on histological subtype and tumor genotype. J. Clin. Endocrinol. Metab. 2014, 99, E329–E337. [Google Scholar] [CrossRef]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Bisarro Dos Reis, M.; Barros-Filho, M.C.; Marchi, F.A.; Beltrami, C.M.; Kuasne, H.; Pinto, C.A.L.; Ambatipudi, S.; Herceg, Z.; Kowalski, L.P.; Rogatto, S.R. Prognostic Classifier Based on Genome-Wide DNA Methylation Profiling in Well-Differentiated Thyroid Tumors. J. Clin. Endocrinol. Metab. 2017, 102, 4089–4099. [Google Scholar] [CrossRef]
- Affinito, O.; Salerno, P.; D’Alessio, A.; Cuomo, M.; Florio, E.; Carlomagno, F.; Proietti, A.; Giannini, R.; Basolo, F.; Chiariotti, L.; et al. Association between DNA methylation profile and malignancy in follicular-patterned thyroid neoplasms. Endocr. Relat. Cancer 2019, 26, 451–462. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Chen, M.; Sun, L.; Han, J.; Elena, V.K.; Qiao, H. CXCL12 methylation-mediated epigenetic regulation of gene expression in papillary thyroid carcinoma. Sci. Rep. 2017, 7, 44033. [Google Scholar] [CrossRef] [PubMed]
- Yim, J.H.; Choi, A.H.; Li, A.X.; Qin, H.; Chang, S.; Tong, S.T.; Chu, P.; Kim, B.W.; Schmolze, D.; Lew, R.; et al. Identification of Tissue-Specific DNA Methylation Signatures for Thyroid Nodule Diagnostics. Clin. Cancer Res. 2019, 25, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.M.; Dos Reis, M.B.; Barros-Filho, M.C.; Marchi, F.A.; Kuasne, H.; Pinto, C.A.L.; Ambatipudi, S.; Herceg, Z.; Kowalski, L.P.; Rogatto, S.R. Integrated data analysis reveals potential drivers and pathways disrupted by DNA methylation in papillary thyroid carcinomas. Clin. Epigenetics 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Pérez-González, B.; Jordà, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bosquet, J.; Calcei, J.; Wei, J.S.; Garcia-Closas, M.; Sherman, M.E.; Hewitt, S.; Vockley, J.; Lissowska, J.; Yang, H.P.; Khan, J.; et al. Detection of somatic mutations by high-resolution DNA melting (HRM) analysis in multiple cancers. PLoS ONE 2011, 6, e14522. [Google Scholar] [CrossRef]
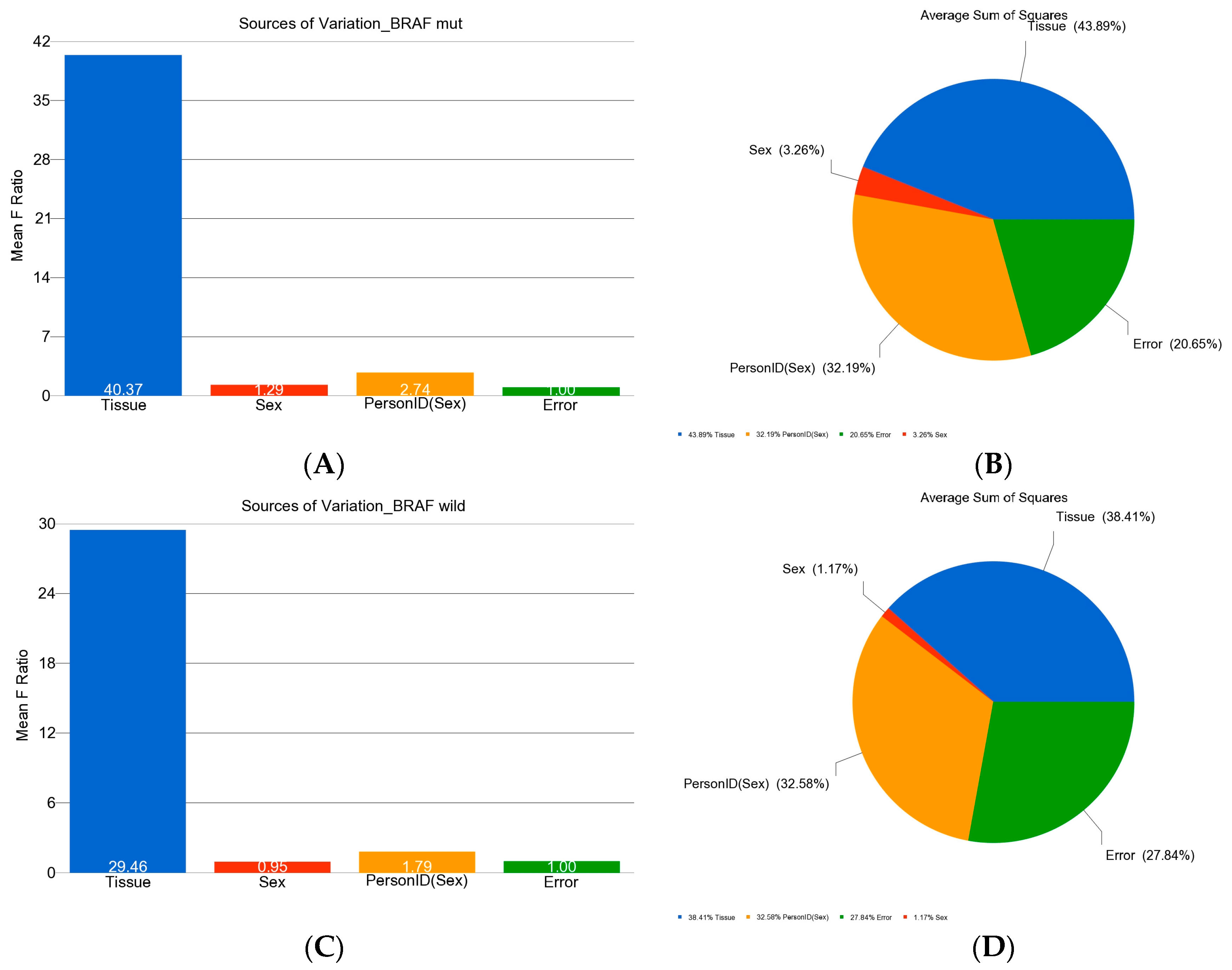
- Eisenhart, C. The assumptions underlying the analysis of variance. Biometrics 1947, 3, 1–21. [Google Scholar] [CrossRef]
- Brait, M.; Loyo, M.; Rosenbaum, E.; Ostrow, K.L.; Markova, A.; Papagerakis, S.; Zahurak, M.; Goodman, S.M.; Zeiger, M.; Sidransky, D.; et al. Correlation between BRAF mutation and promoter methylation of TIMP3, RARβ2 and RASSF1A in thyroid cancer. Epigenetics 2012, 7, 710–719. [Google Scholar] [CrossRef]
- Chang, H.; Shin, B.K.; Kim, A.; Kim, H.K.; Kim, B.H. DNA methylation analysis for the diagnosis of thyroid nodules—A pilot study with reference to BRAF(V) (600E) mutation and cytopathology results. Cytopathology 2016, 27, 122–130. [Google Scholar] [CrossRef]
- Khatami, F.; Larijani, B.; Heshmat, R.; Nasiri, S.; Haddadi-Aghdam, M.; Teimoori-Toolabi, L.; Tavangar, S.M. Hypermethylated RASSF1 and SLC5A8 promoters alongside BRAF(V600E) mutation as biomarkers for papillary thyroid carcinoma. J. Cell Physiol. 2020, 235, 6954–6968. [Google Scholar] [CrossRef]
- Santoro, A.; Pannone, G.; Carosi, M.A.; Francesconi, A.; Pescarmona, E.; Russo, G.M.; Feola, A.; Losito, S.; Franco, R.; Nappi, L.; et al. BRAF mutation and RASSF1A expression in thyroid carcinoma of southern Italy. J. Cell Biochem. 2013, 114, 1174–1182. [Google Scholar] [CrossRef]
- Sheikholeslami, S.; Azizi, F.; Ghasemi, A.; Alibakhshi, A.; Parsa, H.; Shivaee, S.; Zarif-Yeganeh, M.; Hedayati, M.; Teimoori-Toolabi, L. The Epigenetic Modification of SLC5A8 in Papillary Thyroid Carcinoma and its Effects on Clinic-Pathological Features. Iran. J. Public Health 2022, 51, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, Z.; Wu, Z.; Liu, C.; Yu, M.; Wen, M.; Zhang, L.; Wang, X. DDAH2 suppresses RLR-MAVS-mediated innate antiviral immunity by stimulating nitric oxide-activated, Drp1-induced mitochondrial fission. Sci. Signal. 2021, 14, eabc7931. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Kim, Y.; Jeong, Y.M.; Bae, J.S.; Jung, C.K. CCND1 Splice Variant as A Novel Diagnostic and Predictive Biomarker for Thyroid Cancer. Cancers 2018, 10, 437. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef]
- Lee, J.J.; Au, A.Y.; Foukakis, T.; Barbaro, M.; Kiss, N.; Clifton-Bligh, R.; Staaf, J.; Borg, A.; Delbridge, L.; Robinson, B.G.; et al. Array-CGH identifies cyclin D1 and UBCH10 amplicons in anaplastic thyroid carcinoma. Endocr. Relat. Cancer 2008, 15, 801–815. [Google Scholar] [CrossRef]
- Takahashi, H.; Yoshimatsu, G.; Faustman, D.L. The Roles of TNFR2 Signaling in Cancer Cells and the Tumor Microenvironment and the Potency of TNFR2 Targeted Therapy. Cells 2022, 11, 1952. [Google Scholar] [CrossRef]
- Vanamee, É.S.; Faustman, D.L. TNFR2: A Novel Target for Cancer Immunotherapy. Trends Mol. Med. 2017, 23, 1037–1046. [Google Scholar] [CrossRef]
- Li, M.; Zhang, X.; Bai, X.; Liang, T. Targeting TNFR2: A Novel Breakthrough in the Treatment of Cancer. Front. Oncol. 2022, 12, 862154. [Google Scholar] [CrossRef]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal. 2017, 10, eaaf8608. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shen, X.; Zhu, G.; Xing, M. REC8 is a novel tumor suppressor gene epigenetically robustly targeted by the PI3K pathway in thyroid cancer. Oncotarget 2015, 6, 39211–39224. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Arigami, T.; Okumura, H.; Uchikado, Y.; Kita, Y.; Kurahara, H.; Maemura, K.; Kijima, Y.; Ishihara, Y.; Sasaki, K.; et al. Human leukocyte antigen (HLA)-E and HLA-F expression in gastric cancer. Anticancer Res. 2015, 35, 2279–2285. [Google Scholar]
- Huang, G.; Chen, J.; Zhou, J.; Xiao, S.; Zeng, W.; Xia, J.; Zeng, X. Epigenetic modification and BRAF gene mutation in thyroid carcinoma. Cancer Cell Int. 2021, 21, 687. [Google Scholar] [CrossRef]
- Hou, P.; Liu, D.; Xing, M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 687–697. [Google Scholar] [CrossRef]
- Jasmine, F.; Rahaman, R.; Roy, S.; Raza, M.; Paul, R.; Rakibuz-Zaman, M.; Paul-Brutus, R.; Dodsworth, C.; Kamal, M.; Ahsan, H.; et al. Interpretation of genome-wide infinium methylation data from ligated DNA in formalin-fixed, paraffin-embedded paired tumor and normal tissue. BMC Res. Notes 2012, 5, 117. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Category | BRAF Mutation | BRAF Wild | p-Value |
|---|---|---|---|---|
| Sex | Female | 13 | 15 | 1 |
| Male | 5 | 7 | ||
| Age | Mean | 38.56 | 35.68 | 0.395 |
| SD | 15.58 | 14.42 | ||
| Pathology | PTC | 16 | 13 | 0.072 |
| FVPTC | 2 | 5 | ||
| FTC | 0 | 4 | ||
| Cervical lymph node | Absent | 12 | 15 | 1 |
| Present | 6 | 7 | ||
| Surgical procedure | Lobectomy | 2 | 6 | 0.258 |
| Total thyroidectomy | 16 | 16 | ||
| Tumor size (cm) | Mean | 2.11 | 2.82 | 0.54 |
| SD | 1.16 | 1.92 | ||
| Duration of illness (months) | Mean | 7.33 | 7.23 | 0.854 |
| SD | 2.51 | 2.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasmine, F.; Aschebrook-Kilfoy, B.; Rahman, M.M.; Zaagman, G.; Grogan, R.H.; Kamal, M.; Ahsan, H.; Kibriya, M.G. Association of DNA Promoter Methylation and BRAF Mutation in Thyroid Cancer. Curr. Oncol. 2023, 30, 2978-2996. https://doi.org/10.3390/curroncol30030227
Jasmine F, Aschebrook-Kilfoy B, Rahman MM, Zaagman G, Grogan RH, Kamal M, Ahsan H, Kibriya MG. Association of DNA Promoter Methylation and BRAF Mutation in Thyroid Cancer. Current Oncology. 2023; 30(3):2978-2996. https://doi.org/10.3390/curroncol30030227
Chicago/Turabian StyleJasmine, Farzana, Briseis Aschebrook-Kilfoy, Mohammad M. Rahman, Garrett Zaagman, Raymon H. Grogan, Mohammed Kamal, Habibul Ahsan, and Muhammad G. Kibriya. 2023. "Association of DNA Promoter Methylation and BRAF Mutation in Thyroid Cancer" Current Oncology 30, no. 3: 2978-2996. https://doi.org/10.3390/curroncol30030227
APA StyleJasmine, F., Aschebrook-Kilfoy, B., Rahman, M. M., Zaagman, G., Grogan, R. H., Kamal, M., Ahsan, H., & Kibriya, M. G. (2023). Association of DNA Promoter Methylation and BRAF Mutation in Thyroid Cancer. Current Oncology, 30(3), 2978-2996. https://doi.org/10.3390/curroncol30030227