
Small-Molecule Factor Xa Inhibitors: Translational SAR, Assay-Aware Data Quality, and QSAR-Readiness for CADD-Oriented Discovery

 ,
,  , , and
, , and 
Abstract
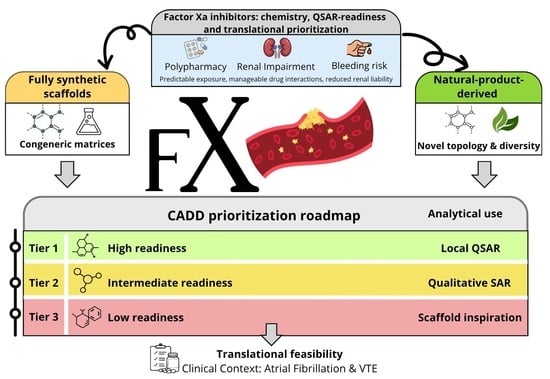
1. Introduction
2. Scope and Inclusion Strategy
2.1. Search Scope and Eligibility Criteria
2.2. Multi-Layer Data Extraction and Translational Depth
2.3. Operationalizing QSAR-Readiness
- ▪
- A sufficiently populated and chemically congeneric analog set;
- ▪
- Exact and predominantly uncensored potency endpoints;
- ▪
- Internally comparable assay conditions;
- ▪
- Clearly defined chemical structures and stereochemistry;
- ▪
- Limited ambiguity during data curation and descriptor generation before model development.
3. FXa Binding Logic Revisited
3.1. Why FXa Remains Chemically Designable
3.2. The S1 Pocket: Potency Anchor and Property Liability
3.3. The S4 Aromatic Box as a Selectivity and Affinity Lever
3.4. The Intervening Region: Geometry, Preorganization, and Hidden SAR
3.5. From Pocket Occupancy to Translational SAR
4. Fully Synthetic Chemotypes
4.1. From Scaffold Novelty to Modellable Analog Series
4.2. Anthranilamide Lineages as a Particularly Instructive Synthetic Platform
4.3. Piperazinylanthranilamides and the Value of Congeneric Expansion
4.4. Potent but Smaller Exploratory Series: Useful SAR, Limited Modeling Depth
4.5. What Synthetic Chemotypes Contribute to Translational SAR
5. Natural-Product-Derived and Semisynthetic Chemotypes
5.1. Isosteviol as the Most Instructive Semisynthetic FXa Platform
5.2. Beyond Isosteviol: Diversity and Modeling Constraints in Diterpenoids
5.3. Coumarin-Based Systems: Chemically Familiar, Mechanistically More Fragmented
5.4. What Natural-Product-Derived Chemotypes Contribute to Translational SAR
6. Integrated QSAR-Readiness Map, Modellability, and Critical Gaps
6.1. Readiness as a Review-Level Triage Tool
6.2. Why Some Series Are Modellable, and Others Are Not
6.2.1. Scaffold Similarity, Endpoint Consistency, and Activity Range
6.2.2. Endpoint Heterogeneity and Curation Burden
6.3. Critical Gaps and Future Directions
6.3.1. From Potency Catalogs to Evidence-Aware Design
6.3.2. Reporting Standards and Balanced Scaffold Interpretation
6.3.3. Benchmarking Against Marketed FXa Inhibitors
6.3.4. Natural-Product-Derived Series: From Inspiration to Programmatic Expansion
6.3.5. AI/ML-Enabled CADD: Using Readiness as a Dataset Triage
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.K.; Player, M.R. Developments in factor Xa inhibitors for the treatment of thromboembolic disorders. Med. Res. Rev. 2011, 31, 202–283. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Perzborn, E.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY 59-7939): An oral, direct factor Xa inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Fredenburgh, J.C.; Weitz, J.I. Oral direct factor Xa inhibitors. Circ. Res. 2012, 111, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.J.P.; Orwat, M.J.; Koch, S.; Rossi, K.A.; Alexander, R.S.; Smallwood, A.; Wong, P.C.; Rendina, A.R.; Luettgen, J.M.; Knabb, R.M.; et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo [3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J. Med. Chem. 2007, 50, 5339–5356. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Su, G.; Ren, Y.; Chen, Y. Design, synthesis and evaluation of isoxazolo [5,4-d]pyrimidin-4(5H)-one derivatives as antithrombotic agents. Bioorg. Med. Chem. Lett. 2015, 25, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Su, G.; Ren, Y.; Chen, Y. Synthesis of 3,4-diaminobenzoyl derivatives as factor Xa inhibitors. Eur. J. Med. Chem. 2015, 101, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Yang, L.; Li, H.; Li, Q.; Zhao, L.; Wang, X.; Zhang, Y.; Zhou, M.; Zhou, J.; Zhang, H. Identification of anthranilamide derivatives as potential factor Xa inhibitors: Drug design, synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 95, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Yang, L.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of anthranilamide derivatives as potential factor Xa (fXa) inhibitors. Bioorg. Med. Chem. 2018, 26, 5987–5999. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, D.; Li, M.; Wu, Q.; Lam, Y.P.Y.; Guo, Y.; Chen, C.; Bai, N.; Malhotra, S.; Li, W.; et al. Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur. J. Med. Chem. 2019, 183, 111722. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pan, B.W.; Li, W.C.; Wang, Q.; Wu, Q.; Pan, M.; Fu, H.-Z. Synthesis and biological evaluation of isosteviol derivatives as FXa inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126585. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hong, Z.; Liu, M.; Guo, S.; Yang, D.; Wang, Y.; Lan, T.; Gao, L.; Qi, H.; Gong, P.; et al. Design, synthesis, and biological activity of novel tetrahydropyrazolopyridone derivatives as FXa inhibitors with potent anticoagulant activity. Bioorg. Med. Chem. 2017, 25, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Yang, L.; Yang, Y.; Zhao, L.; Wei, Q.; Zhang, J.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of novel 2,3-dihydroquinazolin-4(1H)-one derivatives as potential fXa inhibitors. Eur. J. Med. Chem. 2017, 125, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Campbell, M.; Chaudry, L.; Chung, C.-W.; Convery, M.A.; Hamblin, J.N.; Johnstone, L.; et al. Factor Xa inhibitors: S1 binding interactions of a series of N-{(3S)-1-[(1S)-1-methyl-2-morpholin-4-yl-2-oxoethyl]-2-oxopyrrolidin-3-yl}sulfonamides. J. Med. Chem. 2007, 50, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, I.C.; Rienstra, M.; Bunting, K.V.; Casado-Arroyo, R.; Caso, V.; Crijns, H.J.G.M.; De Potter, T.J.R.; Dwight, J.; Guasti, L.; Hanke, T.; et al. 2024 ESC Guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2024, 45, 3314–3414. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Gawalko, M.; Betz, K.; Hendriks, J.M.; Lip, G.Y.H.; Vinter, N.; Guo, Y.; Johnsen, S. Atrial fibrillation: Epidemiology, screening and digital health. Lancet Reg. Health Eur. 2024, 37, 100786. [Google Scholar] [CrossRef] [PubMed]
- Engbers, M.J.; van Hylckama Vlieg, A.; Rosendaal, F.R. Venous thrombosis in the elderly: Incidence, risk factors and risk groups. J. Thromb. Haemost. 2010, 8, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Chopard, R.; Albertsen, I.E.; Piazza, G. Diagnosis and treatment of lower extremity venous thromboembolism: A review. JAMA 2020, 324, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Spruit, J.R.; de Vries, T.A.C.; Hemels, M.E.W.; Pisters, R.; de Groot, J.R.; Jansen, R.W.M.M. Direct oral anticoagulants in older and frail patients with atrial fibrillation: A decade of experience. Drugs Aging 2024, 41, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, C.; Hendriks, J.M.L.; Myrstad, M.; Van Gelder, I.C.; Rienstra, M. Managing elderly patients with atrial fibrillation and multimorbidity: Call for a systematic approach. Expert Rev. Cardiovasc. Ther. 2024, 22, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Badescu, M.C.; Popescu, D.; Gosav, E.M.; Costache, A.D.; Cosau, D.E.; Chetran, A.; Duca, Ș.-T.; Cucută, S.; Șerban, I.L.; Enache, I.I.C.; et al. The use of direct oral anticoagulants (DOACs) in the geriatric population—How to overcome the challenges of geriatric syndromes. J. Clin. Med. 2025, 14, 4396. [Google Scholar] [CrossRef] [PubMed]
- Sadlon, A.H.; Tsakiris, D.A. Direct oral anticoagulants in the elderly: Systematic review and meta-analysis of evidence, current and future directions. Swiss Med. Wkly. 2016, 146, w14356. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. Factor Xa inhibitors: Critical considerations for clinical development and testing. J. Thromb. Thrombolysis 2021, 52, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Maignan, S.; Guilloteau, J.-P.; Choi-Sledeski, Y.M.; Becker, M.R.; Ewing, W.R.; Pauls, H.W.; Spada, A.P.; Mikol, V. Molecular structures of human factor Xa complexed with ketopiperazine inhibitors: Preference for a neutral group in the S1 pocket. J. Med. Chem. 2003, 46, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Matter, H.; Will, D.W.; Nazare, M.; Schreuder, H.; Laux, V.; Wehner, V. Structural requirements for factor Xa inhibition by 3-oxybenzamides with neutral P1 substituents: Combining X-ray crystallography, 3D-QSAR and tailored scoring functions. J. Med. Chem. 2005, 48, 3290–3312. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, H.; Fu, H. Semisynthesis of ent-norstrobane diterpenoids as potential inhibitor for factor Xa. Bioorg. Med. Chem. Lett. 2018, 28, 3813–3815. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, M.; Gao, C.; Fu, H. Semisynthesis of epoxy-pimarane diterpenoids from kirenol and their FXa inhibition activities. Bioorg. Med. Chem. 2019, 27, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Skoptsova, A.A.; Geronikaki, A.; Novichikhina, N.P.; Sulimov, A.V.; Ilin, I.S.; Sulimov, V.B.; Bykov, G.A.; Podoplelova, N.A.; Pyankov, O.V.; Shikhaliev, K.S. Design, synthesis, and evaluation of new hybrid derivatives of 5,6-dihydro-4H-pyrrolo [3,2,1-ij]quinolin-2(1H)-one as potential dual inhibitors of blood coagulation factors Xa and XIa. Molecules 2024, 29, 373. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Dai, X.; Xu, B.; Tian, W.; Shi, J. Discovery and development of factor Xa inhibitors (2015–2022). Front. Pharmacol. 2023, 14, 1105880. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yuan, J.; Fu, X.; Huang, C. Novel anthranilamide-based FXa inhibitors: Drug design, synthesis and biological evaluation. Molecules 2016, 21, 491. [Google Scholar] [CrossRef] [PubMed]
- Gackowski, M.; Jedrzejewski, M.; Medicharla, S.S.; Kondabala, R.; Madriwala, B.; Madra-Gackowska, K.; Studzinska, R. Novel thiourea and oxime ether isosteviol-based anticoagulants: MD simulation and ADMET prediction. Pharmaceuticals 2024, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Khadse, A.N.; Savsani, H.H.; Chikhale, R.V.; Ghuge, R.B.; Prajapati, D.R.; Kureshi, G.; Murumkar, P.R.; Patel, K.V.; Rajput, S.J.; Yadav, M.R. Design, synthesis and biological evaluation of piperazinylanthranilamides as potential factor Xa inhibitors. J. Mol. Struct. 2022, 1270, 133974. [Google Scholar] [CrossRef]
- Milanovic, Z. Structural properties of newly 4,7-dihydroxycoumarin derivatives as potential inhibitors of XIIa, Xa, IIa factors of coagulation. J. Mol. Struct. 2024, 1298, 137049. [Google Scholar] [CrossRef]
- Rayani, R.H.; Soni, J.Y.; Parmar, D.R.; Kusurkar, R.V.; Eissa, I.H.; Metwaly, A.M.; Khalil, A.; Zunjar, V.; Battula, S.; Niazi, S. Identification of new pyrazolyl piperidine molecules as factor Xa inhibitors: Design, synthesis, in silico, and biological evaluation. Results Chem. 2022, 4, 100355. [Google Scholar] [CrossRef]
- Verhoef, D.; Visscher, K.M.; Vosmeer, C.R.; Cheung, K.L.; Reitsma, P.H.; Geerke, D.P.; Bos, M.H.A. Engineered factor Xa variants retain procoagulant activity independent of direct factor Xa inhibitors. Nat. Commun. 2017, 8, 528. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.; Marquart, A.L.; Joseph, S.; Waid, P.; Yee, Y.K.; Tebbe, A.L.; Ratz, A.M.; Herron, D.K.; Goodson, T.; Masters, J.J.; et al. Anthranilamide inhibitors of factor Xa. Bioorg. Med. Chem. Lett. 2007, 17, 4832–4836. [Google Scholar] [CrossRef] [PubMed]
- Gackowski, M.; Szewczyk-Golec, K.; Madra-Gackowska, K.; Pluskota, R.; Koba, M. Quantitative structure-activity relationship analysis of isosteviol-related compounds as activated coagulation factor X (FXa) inhibitors. Nutrients 2022, 14, 3521. [Google Scholar] [CrossRef] [PubMed]
- Gackowski, M.; Madriwala, B.; Studzinska, R.; Koba, M. Novel isosteviol-based FXa inhibitors: Molecular modeling, in silico design and docking simulation. Molecules 2023, 28, 4977. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, S.S.; Roy, K.K.; Saxena, A.K. Profiling the structural determinants for the selectivity of representative factor-Xa and thrombin inhibitors using combined ligand-based and structure-based approaches. J. Chem. Inf. Model. 2011, 51, 1966–1985. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, Z.; Ojima, I.; Samaras, D.; Wang, F. Artificial intelligence in drug discovery: Applications and techniques. Brief. Bioinform. 2022, 23, bbab430. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Domain | What Is Recorded | Why It Matters | Representative References |
|---|---|---|---|
| Series architecture | Core scaffold, series size, substitution pattern, analog progression, and degree of congenericity | Distinguishes well-defined medicinal-chemistry series from mixed or heterogeneous chemotype collections and supports the classification of local modellability | [5,7,8,12] |
| Primary FXa endpoint | Ki, IC50, pKi/pIC50, percentage inhibition at a fixed concentration, threshold values, or censored values | Determines whether quantitative comparison across compounds is meaningful and whether activity data can support local SAR or QSAR-oriented analysis | [5,7,9,10,28,29,30,31] |
| Assay context | Enzyme source, substrate system, assay format, concentration range, and internal consistency of experimental conditions | Defines whether activity values can be compared within a series and whether cross-series pooling would be methodologically defensible | [5,9,10,12,28,29,30,31] |
| Activity range/response variance | Distribution and span of exact potency values within a series, proportion of censored values, and whether the series contains sufficient variation for informative ranking or regression | Determines whether a chemically coherent and assay-consistent dataset can support informative regression rather than only qualitative SAR interpretation | [28,29,30,31] |
| Selectivity profile | Activity versus thrombin and other related serine proteases | Informs target differentiation, mechanism, and bleeding-relevant pharmacological interpretation | [2,4,5,12,23,24] |
| Translational pharmacology | PT/aPTT, ex vivo effects, in vivo antithrombotic activity, bleeding observations, and antiplatelet or multi-endpoint pharmacology when reported | Links enzyme inhibition to broader anticoagulant behavior, but should be interpreted as complementary pharmacological evidence rather than as a direct substitute for primary FXa potency. | [5,6,9,10,12,33] |
| Developability signals | PK/ADME properties, solubility, permeability, metabolic stability, plasma exposure, bioavailability, or computational developability predictions | Supports scaffold prioritization beyond potency and helps connect medicinal-chemistry optimization with translational feasibility | [3,9,22,32,34] |
| Curation burden | Structural ambiguity, stereochemistry, salt form, tautomerism, compound identifiers, naming inconsistencies, and availability of complete series-level data | Affects descriptor reliability, reproducibility, and suitability for downstream computational modeling | [28,29,30,31] |
| Extraction confidence | Whether full series-level data were available, partially available, or limited to accessible publication metadata | Prevents overclassification of datasets when only abstract-level, publication-level, or incomplete series-level information was available | [28,29,30,31,35,36] |
| Criterion | High Readiness | Intermediate Readiness | Low Readiness | Methodological/Illustrative References |
|---|---|---|---|---|
| Series size and congenericity | Approximately ≥20 closely related analogs within a single medicinal-chemistry core | Approximately 10–20 analogs or modest scaffold variation within one chemical family. | Fewer than 10 analogs or a heterogeneous set of weakly related scaffolds | [7,8,28,29,30,31] |
| Endpoint quality | Predominantly exact Ki, IC50, pKi, or pIC50 values | Mixture of exact values and censored or semi-quantitative data | Mostly single-point inhibition data, threshold-only readouts, or sparsely reported activity values | [5,7,28,29,30,31,37] |
| Assay consistency | Same or clearly comparable FXa assay format across the series | Minor assay differences that remain partially comparable or potentially harmonizable. | Multiple non-equivalent assay settings that limit or prevent direct comparison | [28,29,30,31] |
| Activity range/response variance | Sufficient potency range to support informative regression and meaningful structure–activity discrimination | Moderate activity range that may support cautious local modeling or qualitative SAR. | Narrow or poorly distributed activity range that limits regression value despite apparent dataset cleanliness | [28,29,30,31] |
| Structural clarity and curation burden | Well-defined chemical structures with clear stereochemistry, salt form, and minimal ambiguity after routine curation | Some structural or annotation ambiguity, but largely resolvable without major information loss. | Significant ambiguity, undefined mixtures, unclear stereochemistry, or uncertain structure assignment | [28,29,30,31] |
| Data-extraction confidence | Full series-level data available for extraction, including structures, endpoints, assay context, and compound-level activity annotation | Partial but usable publication-level data available; some fields require cautious interpretation or verification. | Only limited metadata, abstract-level information, or incomplete series-level detail available; readiness placement should be regarded as provisional. | [28,29,30,31,35,36] |
| Orthogonal biological context | Selectivity, PT/aPTT, PK/ADME, ex vivo, or in vivo data available and interpretable as complementary evidence | Limited orthogonal biological or pharmacological characterization. | No information beyond isolated FXa inhibition data | [5,6,9,10,12,33] |
| Best analytical use | Local QSAR-oriented modeling, regression analysis, and docking-informed analog design after validation | Qualitative SAR analysis, focused analog comparison, and cautious local modeling after curation. | Descriptive SAR, scaffold inspiration, or hypothesis generation only | [28,29,30,31] |
| Established QSAR/Data-Quality Principle | How It Is Translated in This Review | Practical Implications for FXa Inhibitor Literature |
|---|---|---|
| Chemical structure curation | Structures, stereochemistry, salt/tautomer state, and compound identifiers are checked for clarity and extraction confidence. | Ambiguous or only partially extractable series are retained for qualitative SAR but not upgraded to high readiness. |
| Endpoint and assay harmonization | Ki, IC50, pKi/pIC50, single-point inhibition, and censored values are treated as non-equivalent unless the assay context is internally comparable. | Series with mixed endpoints or heterogeneous assay formats are not pooled into a single broad regression dataset. |
| Applicability-domain-aware modeling | Congenericity is interpreted as shared binding logic plus comparable chemistry, not only as a common scaffold label. | Local subseries are preferred over global FXa datasets when scaffold drift or different S1/S4 presentation is likely. |
| Robustness and external validation | Readiness is limited to suitability for future local modeling; it is not presented as proof of predictive performance. | A high-readiness placement means that modeling may be justified after curation and validation, not that a validated model already exists. |
| Data uncertainty and curation burden | Publication-level or metadata-level extraction is explicitly marked as provisional. | This reduces the risk of overinterpreting small, incompletely documented, or exploratory studies. |
| Chemotype/Series | Reference | What Makes It Useful | Translational Depth | Working Readiness |
|---|---|---|---|---|
| Isoxazolo[5,4-d]pyrimidin-4(5H)-ones | [5] | Focused synthetic series; compound 6g showed IC50 = 0.013 μm, 2 × PT = 2.12 μm, and high selectivity over thrombin and trypsin | FXa inhibition, PT prolongation in human plasma, and selectivity over thrombin/trypsin | Intermediate |
| 3,4-Diaminobenzoyl derivatives | [6] | Congeneric series with a clear FXa optimization strategy; compound 7b was reported as a potent and selective direct FXa inhibitor | FXa inhibition, selectivity, and in vivo antithrombotic activity for lead compound 7b | Intermediate |
| Anthranilamide derivatives | [7] | Fragment-based identification followed by analog expansion; compound 9b showed an FXa IC50 of 23 nM | FXa inhibition, thrombin selectivity, and PT prolongation | Intermediate-to-high |
| Anthranilamide derivatives, expanded series | [8] | Expanded optimization series; compound 16g combined potent/selective FXa inhibition with broader translational evaluation | In vitro FXa inhibition, in vivo antithrombotic activity, bleeding-risk evaluation, PK data, and cellular safety testing | High |
| Piperazinylanthranilamides | [35] | Anthranilamide-centered extension reported as a potential FXa inhibitor series; useful for qualitative SAR, but quantitative reuse should remain conservative because only accessible publication-level information could be extracted in this review | FXa inhibition, thrombin selectivity, and rat anticoagulant activity are reported in accessible publication metadata. | Low-to-intermediate |
| Pyrazolyl piperidines | [37] | Compact pyrazolyl piperidine series; compound 4a showed IC50 = 13.4 nM against FXa | FXa inhibition with PT/aPTT clotting readouts for selected compounds | Low |
| Anthranilamide-based FXa inhibitors | [33] | Selective anthranilamide-based series with exact inhibitory values and thrombin selectivity | FXa inhibition, thrombin selectivity, and PT prolongation in plasma | Intermediate |
| 2,3-Dihydroquinazolin-4(1H)-ones | [12] | Cyclized anthranilamide-related scaffold; compound 8e showed IC50 = 21 nM against FXa and thrombin IC50 = 67 μm | FXa inhibition, thrombin selectivity, PT/aPTT, AV-shunt activity, and bleeding-risk evaluation | Intermediate-to-high |
| Tetrahydropyrazolopyridones | [11] | Compact synthetic series; compound 15c showed IC50(FXa) = 0.14 μm and strong enzymatic FXa inhibition | FXa inhibition, anticoagulant activity, and selected in vivo venous thrombosis evaluation | Low-to-intermediate |
| Dual FXa/XIa pyrroloquinolinone hybrids | [27] | Dual-target exploratory design probing FXa/XIa activity relationships with thrombin counter-screening | FXa/XIa screening with thrombin counter-screening | Low |
| Chemotype/Series | Reference | What Makes It Useful | Translational Depth | Working Readiness |
|---|---|---|---|---|
| Isosteviol-based antithrombotic lead series | [9] | Semisynthetic diterpenoid scaffold; compound 6k selectively inhibited FXa with Ki = 0.015 μm | FXa inhibition, selectivity against a serine-protease panel, docking, ex vivo PT/aPTT prolongation, antiplatelet activity, PK data, and ex vivo/in vivo antithrombotic activity | Intermediate-to-high |
| Isosteviol derivatives evaluated as FXa inhibitors | [10] | Focused semisynthetic analog series; compounds 22, 35, and 38 showed stronger FXa inhibition than isosteviol | FXa inhibition, SPR evaluation for selected compounds, and PT/aPTT activity for compounds 22 and 35 | Intermediate |
| Isosteviol dataset, QSAR reinterpretation | [40] | Demonstrates that natural-product-derived series can support QSAR-oriented analysis when structurally congeneric | Retrospective QSAR analysis of experimentally reported isosteviol-related FXa inhibition data | Intermediate |
| ent-Norstrobane diterpenoids | [25] | Diterpenoid scaffold rearrangement; compound 7 showed IC50 = 81 ± 11 nM against FXa | Enzymatic FXa inhibition with limited additional translational data | Low-to-intermediate |
| Epoxy-pimarane diterpenoids from kirenol | [26] | Expands diterpenoid design space beyond isosteviol; reported FXa IC50 values ranged from 0.22 to 27.9 μm | Enzymatic FXa screening with moderate activity; edoxaban used as positive control | Low |
| 4,7-Dihydroxycoumarin derivatives | [36] | Natural-product-related scaffold evaluated across multiple coagulation factors; quantitative interpretation should be approached with caution, given the limited set of available analogs. | Multi-target coagulation-factor testing with limited analog-set depth based on available information | Low |
| Series/Chemotype | Domain | Readiness Tier | Main Reason for Placement | Best Analytical Use |
|---|---|---|---|---|
| Anthranilamide derivatives [7] | Fully synthetic | Intermediate-to-high | Interpretable common core, fragment-based design logic, exact FXa potency, thrombin selectivity, and PT readout for the lead compound | Local SAR, scaffold-focused follow-up design, and constrained QSAR-oriented analysis |
| Anthranilamide derivatives, expanded series [8] | Fully synthetic | High | Larger congeneric optimization series with exact inhibitory data, clear FXa-focused scaffold logic, in vivo antithrombotic activity, bleeding-risk evaluation, and PK data | Local QSAR, descriptor-based modeling, and docking-informed analog prioritization |
| Anthranilamide-based FXa inhibitors [33] | Fully synthetic | Intermediate | Selective anthranilamide-based series with exact inhibitory values, thrombin selectivity, and in vitro anticoagulant activity for selected compounds | Qualitative SAR, local scaffold-focused comparison, and support for anthranilamide design logic |
| 3,4-Diaminobenzoyl derivatives [6] | Fully synthetic | Intermediate | Congeneric series with a clear FXa optimization strategy; compound 7b was reported as a potent, selective direct FXa inhibitor with in vivo antithrombotic activity | Qualitative SAR, focused analog comparison, and translational SAR interpretation. |
| Piperazinylanthranilamides [35] | Fully synthetic | Low-to-intermediate | Anthranilamide-centered extension with reported FXa inhibition and thrombin selectivity; placement is provisional because detailed series-level extraction was limited to accessible publication-level information | Qualitative SAR and cautious hypothesis-generating comparison; not definitive local QSAR classification |
| Isoxazolo[5,4-d]pyrimidin-4(5H)-ones [5] | Fully synthetic | Intermediate | Focused synthetic series; compound 6g showed IC50 = 0.013 μm, 2 × PT = 2.12 μm, and high selectivity over thrombin and trypsin | Series-level SAR, docking-informed interpretation, and constrained local modeling |
| Isosteviol derivatives [9,10,40] | Natural-product-derived/semisynthetic | Intermediate | Strong semisynthetic isosteviol lineage with exact FXa potency, translational readouts, and retrospective QSAR reinterpretation, but higher curation burden than simpler synthetic series | Semisynthetic SAR, scaffold-specific CADD after curation, and focused QSAR reinterpretation. |
| Pyrazolyl piperidines [37] | Fully synthetic | Low | Compound 4a showed an IC50 of 13.4 nM and PT/aPTT readouts, but the analog count remains too small for robust regression modeling. | Qualitative SAR and lead-inspiration analysis |
| 4,7-Dihydroxycoumarin derivatives [36] | Natural-product-related | Low | Available information indicates a very limited set of derivatives with multi-target coagulation readouts, insufficient for local QSAR. | Mechanistic hypothesis generation and scaffold inspiration |
| Tetrahydropyrazolopyridones [11] | Fully synthetic | Low-to-intermediate/intermediate | Congeneric synthetic series with reported FXa inhibition and selected in vivo venous thrombosis evaluation; modellability depends on endpoint coverage and extractable activity range | Qualitative SAR, constrained local modeling, and translational SAR interpretation. |
| Dual FXa/XIa pyrroloquinolinone hybrids [27] | Fully synthetic/dual-target exploratory | Low | Dual-target exploratory design with FXa/XIa screening and thrombin counter-screening; not suitable for a clean single-target FXa QSAR model | Multi-target hypothesis generation and qualitative SAR |
| Readiness Determinant | What It Looks Like in Practice | Effect on QSAR-Readiness | Best Analytical Response | Supporting References |
|---|---|---|---|---|
| High congenericity with exact endpoints | One medicinal-chemistry core, clear analog progression, predominantly exact Ki/IC50 values, and internally consistent assay conditions | Supports reliable local QSAR modeling, interpretable SAR trends, and docking-informed prioritization | Retain as a single local series; validate carefully and preserve assay consistency. | [7,8,28,29,30,31] |
| Moderate scaffold drift within one family | Shared design intent but noticeable changes in linker geometry, ring system, or substituent vectors across subsets | Reduces the coherence of a global model while still allowing for qualitative SAR interpretation | Split into subseries or apply cautiously defined local QSAR models. | [11,12,28,29,30,31] |
| Endpoint heterogeneity | Mixture of Ki, IC50, pKi/pIC50, single-point inhibition, and censored values within one dataset | Introduces artificial variance and reduces the interpretability of regression-based QSAR models | Restrict analysis to one endpoint class or treat the remaining data qualitatively. | [7,28,29,30,31,37] |
| Activity-cliff-like behavior and geometry shifts | Minor structural changes produce disproportionate differences in potency, selectivity, or translational readouts. | Disrupts the continuity required for smooth QSAR relationships, even within congeneric series | Use matched-pair analysis, local modeling, and discontinuity-focused SAR interpretation. | [2,4,13,23,24,28,29,30,31] |
| High curation burden | Ambiguous stereochemistry, unresolved salt forms or tautomers, inconsistent compound identifiers, and discrepancies between reported structures | Reduces the descriptor reliability and reproducibility of computational models | Perform rigorous curation or exclude from quantitative modeling workflows. | [28,29,30,31] |
| Limited data-extraction confidence | Full series-level structures, assay details, or compound-level activity annotations are unavailable or accessible only through limited publication metadata. | Makes readiness placement provisional and increases the risk of overclassifying a dataset as modellable | Classify conservatively; use for qualitative SAR unless full series-level extraction can be verified. | [28,29,30,31,35,36] |
| Rich translational pharmacology with limited series coherence | PT/aPTT, in vivo, or PK data reported for small, semisynthetic, or structurally complex analog sets | Enhances biological interpretation but does not directly improve QSAR suitability | Use for translational SAR and medicinal-chemistry decision-making, not as a pooled QSAR dataset. | [9,10,12,28,29,30,31] |
| Agent/Class | Clinical Status and Main Use Context | Key Pharmacological/Developability Considerations | Design-Relevant Benchmark for New FXa Inhibitor Series |
|---|---|---|---|
| Rivaroxaban | Approved/marketed oral direct FXa inhibitor; used across major thromboembolic indications, including stroke prevention in atrial fibrillation and VTE treatment or prevention [2,3,14,17,18,19,20,21,22,32]. | Oral direct FXa inhibition with clinically established anticoagulant use; interpretation of new series should consider renal function, interaction liability, exposure predictability, and bleeding-risk context. | Sets a benchmark for oral FXa inhibition, predictable anticoagulation, and clinically manageable dosing, while still requiring attention to renal function, drug interactions, and bleeding risk in older or multimorbid patients. |
| Apixaban | Approved/marketed oral direct FXa inhibitor with broad use in atrial fibrillation and VTE-related indications [3,4,14,17,18,19,20,21,22,32]. | Oral FXa inhibition with a clinically established balance of potency, exposure, and safety; population-specific factors such as renal function, concomitant therapy, and bleeding vulnerability remain important comparators. | Provides a clinically established reference for balancing potency, selectivity, oral exposure, and safety; early series should not be judged by enzyme potency alone. |
| Edoxaban | Approved/marketed oral direct FXa inhibitor in many regions; clinically used for atrial fibrillation and VTE treatment contexts [3,14,17,18,19,20,21,22,32]. | Oral FXa inhibition in AF/VTE-related settings: renal function dependence, systemic exposure, and interaction risk are key developability considerations when comparing with experimental compounds. | Highlights the importance of exposure predictability, renal-function considerations, and clinically interpretable efficacy–safety trade-offs. |
| Betrixaban | Approved in the United States for extended VTE prophylaxis in acutely ill medical patients, with regional availability and regulatory status differing from the more widely used oral FXa inhibitors [22,32]. | Indication-specific clinical use illustrates that the exposure profile, renal elimination considerations, and regulatory context can strongly shape the practical value of an FXa inhibitor. | Illustrates that indication selection, renal elimination profile, half-life, and regulatory context can be as important as biochemical target engagement. |
| Representative non-marketed clinical candidates | Earlier clinical FXa inhibitor candidates such as darexaban, otamixaban, letaxaban, or eribaxaban progressed beyond discovery but did not become routinely marketed anticoagulants [22,32]. | These candidates illustrate that biochemical FXa inhibition and clinical progression do not guarantee translation when the route, exposure, bleeding liability, efficacy-safety balance, or population-specific utility is insufficiently favorable. | Their trajectories emphasize that FXa potency and even clinical progression are insufficient without a favorable efficacy, bleeding, exposure, route-of-administration, and population-specific risk–benefit profile. |
| AI/ML Task | Dataset Requirement | How Readiness Triage Helps |
|---|---|---|
| Regression modeling of potency | Exact, harmonized Ki/IC50 or transformed pKi/pIC50 values with sufficient response variance. | Separates regression-ready local subsets from qualitative or censored activity data. |
| Classification or prioritization | Clear active/inactive thresholds and assay context that justify label assignment. | Prevents arbitrary binning of compounds measured under incompatible assay formats. |
| Docking rescoring/consensus CADD | Chemically coherent series with plausible shared binding mode and interpretable S1/S4 engagement. | Reduces the risk that docking scores are trained or interpreted across non-equivalent binding solutions. |
| Active learning and analog selection | Constrained chemical domain with reliable labels and feasible prospective analogs. | Identifies series in which new compounds would genuinely reduce model uncertainty rather than introduce noise. |
| Generative or scaffold-hopping workflows | Explicit boundaries between modellable local SAR and exploratory scaffold inspiration. | Keeps generative suggestions tied to the applicability domain and flags when experimental validation is essential. |
| ADMET and translational filtering | Potency data linked to selectivity, PT/aPTT, PK/ADME, or bleeding-relevant signals where available. | Encourages multilayer decision-making without merging mechanistically distinct endpoints into one undifferentiated model. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Gordon, P.; Janiak, M.; Mądra-Gackowska, K.; Wydeheft, L.; Hołyńska-Iwan, I.; Gackowski, M. Small-Molecule Factor Xa Inhibitors: Translational SAR, Assay-Aware Data Quality, and QSAR-Readiness for CADD-Oriented Discovery. Pharmaceuticals 2026, 19, 1017. https://doi.org/10.3390/ph19071017
Gordon P, Janiak M, Mądra-Gackowska K, Wydeheft L, Hołyńska-Iwan I, Gackowski M. Small-Molecule Factor Xa Inhibitors: Translational SAR, Assay-Aware Data Quality, and QSAR-Readiness for CADD-Oriented Discovery. Pharmaceuticals. 2026; 19(7):1017. https://doi.org/10.3390/ph19071017
Chicago/Turabian StyleGordon, Paweł, Michał Janiak, Katarzyna Mądra-Gackowska, Lidia Wydeheft, Iga Hołyńska-Iwan, and Marcin Gackowski. 2026. "Small-Molecule Factor Xa Inhibitors: Translational SAR, Assay-Aware Data Quality, and QSAR-Readiness for CADD-Oriented Discovery" Pharmaceuticals 19, no. 7: 1017. https://doi.org/10.3390/ph19071017
APA StyleGordon, P., Janiak, M., Mądra-Gackowska, K., Wydeheft, L., Hołyńska-Iwan, I., & Gackowski, M. (2026). Small-Molecule Factor Xa Inhibitors: Translational SAR, Assay-Aware Data Quality, and QSAR-Readiness for CADD-Oriented Discovery. Pharmaceuticals, 19(7), 1017. https://doi.org/10.3390/ph19071017