

Potential of Triazines as Antidiabetic Agents—A Review of Structures and Pharmacological Activity


Abstract
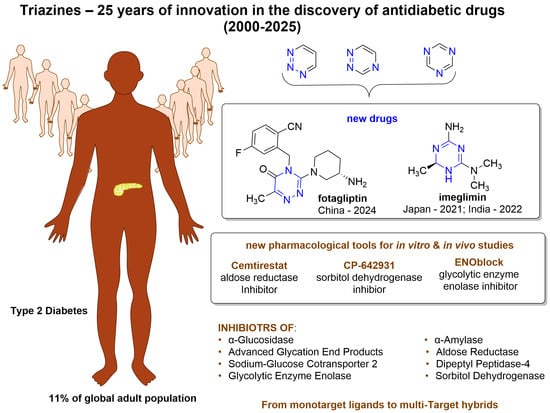
1. Introduction
2. 1,2,3-Triazines as Antidiabetic Agents
1,2,3-Triazines as α-Glucosidase Inhibitors
3. 1,2,4-Triazines as Antidiabetic Agents
3.1. 1,2,4-Triazines as Monotarget Ligands
3.1.1. 1,2,4-Triazines as α-Glucosidase Inhibitors
3.1.2. 1,2,4-Triazines as Inhibitors of Advanced Glycation End Products
3.1.3. 1,2,4-Triazines as Aldose Reductase Inhibitors
3.1.4. 1,2,4-Triazines as Sodium-Glucose Cotransporter 2 Inhibitors
3.1.5. 1,2,4-Triazines as Dipeptyl Peptidase-4 Inhibitors
3.1.6. 1,2,4-Triazine as Glucagon-like Peptide-1 Receptor Agonists
3.2. 1,2,4-Triazines as Dual or Multi-Target Ligands
3.2.1. 1,2,4-Triazines as Dual α-Amylase and α-Glucosidase Inhibitors
3.2.2. 1,2,4-Triazines as Multi-Target Ligands
4. Fused 1,2,4-Triazines as Antidiabetic Agents
4.1. 1,2,4-Triazolo-Fused 1,2,4-Triazines
4.1.1. 1,2,4-Triazolo-1,2,4-triazines as Dual α-Amylase and α-Glucosidase Inhibitors
4.1.2. 1,2,4-Triazolo-1,2,4-triazines as Dipeptyl Peptidase-4 Inhibitors
4.2. Pyrimido-1,2,4-Triazines
4.3. 1,2,4-Triazinoindoles
4.3.1. 1,2,4-Triazinoindoles as α-Glucosidase Inhibitors
4.3.2. 1,2,4-Triazinoindoles as α-Amylase Inhibitors
4.3.3. 1,2,4-Triazinoindoles as Aldose Reductase Inhibitors
5. 1,3,5-Triazines as Antidiabetic Agents
5.1. Imeglimin
5.2. Arylated Imeglimin Derivatives as Potential Antidiabetic Agents
5.3. 1,3,5-Triazines with Thiazolidinedione Moiety as Antidiabetic Agents
5.4. 1,3,5-Triazines as DPP-4 Inhibitors
5.5. 1,3,5-Triazines as Glycolytic Enzyme Enolase Inhibitors
5.6. 1,3,5-Triazine as Sorbitol Dehydrogenase Inhibitors
5.7. 1,3,5-Triazines as Potential Agents for Diabetic Nephropathy
5.8. Triazine-Based Insulin Mimetics Identified by Fluorescent Glucose Uptake Screening
5.9. 1,3,5-Triazines as Multi-Target Antidiabetic Agents
5.9.1. 1,3,5-Triazines with Antibacterial Activity
5.9.2. 1,3,5-Triazines with Inhibitory Activity for α-Glucosidase, Carbonic Anhydrase, and Acetylcholinesterase
5.9.3. 1,3,5-Triazines with Anti-Inflammatory and Antidiabetic Activities
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3T3-L1 | Mouse 3T3 Fibroblast-Derived Adipocyte Cell Line |
| 6-NBDG | 6-(N-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-Yl)Amino)-6-Deoxyglucose |
| ADME | Absorption, Distribution, Metabolism, and Excretion |
| AGE | Advanced Glycation End Product |
| AKR1B1 | Aldo-Keto Reductase Family 1 Member B1 |
| ALR | Aldose Reductase |
| ALR1 | Aldehyde Reductase 1 |
| ALR2 | Aldose Reductase 2 |
| AMPK | AMP-Activated Protein Kinase |
| APC | Article Processing Charge |
| AR | Aldose Reductase |
| ATP | Adenosine Triphosphate |
| ATPase | Adenosine Triphosphatase |
| CA | Carbonic Anhydrase |
| Caco-2 | Human Epithelial Colorectal Adenocarcinoma Cell Line |
| CHO | Chinese Hamster Ovary |
| CONSORT | Consolidated Standards of Reporting Trials |
| COX-2 | Cyclooxygenase-2 |
| CRP | C-Reactive Protein |
| DCFH-DA | 2,7-Dichlorodihydrofluorescein Diacetate |
| DMSO | Dimethyl Sulfoxide |
| DPP-4 | Dipeptidyl Peptidase-4 |
| DPP-8 | Dipeptidyl Peptidase-8 |
| DPP-9 | Dipeptidyl Peptidase-9 |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| FDA | Food and Drug Administration |
| FBS | Fasting Blood Sugar |
| FFA | Free Fatty Acid |
| GIP | Glucose-Dependent Insulinotropic Polypeptide |
| GLP-1 | Glucagon-Like Peptide-1 |
| GLUT1 | Glucose Transporter 1 |
| GLUT2 | Glucose Transporter 2 |
| GLUT4 | Glucose Transporter 4 |
| GSIS | Glucose-Stimulated Insulin Secretion |
| GSK-3β | Glycogen Synthase Kinase-3 Beta |
| hALR | Human Aldose Reductase |
| HbA1c | Glycated Haemoglobin |
| HDAC4 | Histone Deacetylase 4 |
| HDL | High-Density Lipoprotein |
| HG | High Glucose |
| IL-6 | Interleukin-6 |
| LDL | Low-Density Lipoprotein |
| MAPK | Mitogen-Activated Protein Kinase |
| MDA-MB-231 | Human Breast Cancer Cell Line |
| MD | Molecular Dynamics |
| MGO | Methylglyoxal |
| MM/GBSA | Molecular Mechanics/Generalised Born Surface Area |
| MTT | 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide |
| NAD+ | Nicotinamide Adenine Dinucleotide |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NEFA | Non-Esterified Fatty Acid |
| NF-κB | Nuclear Factor Kappa B |
| PDB | Protein Data Bank |
| PGE2 | Prostaglandin E2 |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| PPH | Postprandial Hyperglycaemia |
| RAGE | Receptor for Advanced Glycation End Products |
| ROS | Reactive Oxygen Species |
| SAA | Serum Amyloid A |
| SAR | Structure–Activity Relationship |
| SDH | Sorbitol Dehydrogenase |
| SGLT1 | Sodium–Glucose Cotransporter 1 |
| SGLT2 | Sodium–Glucose Cotransporter 2 |
| SIRT1 | Sirtuin 1 |
| T2D | Type 2 Diabetes |
| THP-1 | Human Monocytic Cell Line THP-1 |
| TIMES | Trials of Imeglimin for Efficacy and Safety |
| TNF-α | Tumour Necrosis Factor Alpha |
| TZD | Thiazolidinedione |
| VCAM-1 | Vascular Cell Adhesion Molecule 1 |
| WST-1 | Water-Soluble Tetrazolium Salt-1 Assay |
| ZDF | Zucker Diabetic Fatty |
References
- International Diabetes Federation. IDF Diabetes Atlas, 11th ed.; International Diabetes Federation: Brussels, Belgium, 2025; Available online: https://diabetesatlas.org (accessed on 15 May 2026).
- Antar, S.A.; Ashour, N.A.; Sharaky, M.; Khattab, M.; Ashour, N.A.; Zaid, R.T.; Roh, E.J.; Elkamhawy, A.; Al-Karmalawy, A.A. Diabetes mellitus: Classification, mediators, and complications; A gate to identify potential targets for the development of new effective treatments. Biomed. Pharmacother. 2023, 168, 115734. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xie, Q.; Pan, X.; Zhang, R.; Zhang, X.; Peng, G.; Zhang, Y.; Shen, S.; Tong, N. Type 2 diabetes mellitus in adults: Pathogenesis, prevention and therapy. Signal Transduct. Target. Ther. 2024, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Młynarska, E.; Czarnik, W.; Dzieża, N.; Jędraszak, W.; Majchrowicz, G.; Prusinowski, F.; Stabrawa, M.; Rysz, J.; Franczyk, B. Type 2 Diabetes Mellitus: New Pathogenetic Mechanisms, Treatment and the Most Important Complications. Int. J. Mol. Sci. 2025, 26, 1094. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ahmed, W.L.; Zhuo, L.; Yuan, H.; Wang, S.; Zhou, F. Association of Sleep Patterns with Type 2 Diabetes Mellitus: A Cross-Sectional Study Based on Latent Class Analysis. Int. J. Environ. Res. Public Health 2023, 20, 393. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Ahmed, T.H.; Mohamed, N.; Elhaj, M.S.; Mohammed, Z.; Paulsingh, C.N.; Mohamed, M.B.; Khan, S. Does Insufficient Sleep Increase the Risk of Developing Insulin Resistance: A Systematic Review. Cureus 2022, 14, e23501. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Fan, Y.; Zhao, B.; He, X.; Yang, J.; Chen, C.; Ma, X. Association Between Late Bedtime and Diabetes Mellitus: A Large Community-Based Study. J. Clin. Sleep Med. 2019, 15, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Yagyu, H.; Shimano, H. A Comprehensive Analysis of Diabetic Complications and Advances in Management Strategies. J. Atheroscler. Thromb. 2025, 32, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Cabré, F.; Centelles, J.J.; Cascante, M. From Current Therapeutics to Multitarget Ligands: A Review of Diabetes Pharmacological Treatments. Pharmaceutics 2025, 17, 1125. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Nowak, W.; Grzeszczak, W. Tirzepatide—A dual GIP/GLP-1 receptor agonist—A new antidiabetic drug with potential metabolic activity in the treatment of type 2 diabetes. Endokrynol. Pol. 2022, 73, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Kuo, B.; Li, J.H. Pharmacologic Therapies for Type 2 Diabetes. NEJM Evid. 2025, 4, EVIDra2400265. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ahmad, M.F.A.; Khan, S.; Alouffi, S.; Khan, M.; Prakash, C.; Khan, M.W.A.; Ansari, I.A. Exploring aldose reductase inhibitors as promising therapeutic targets for diabetes-linked disabilities. Int. J. Biol. Macromol. 2024, 280, 135761. [Google Scholar] [CrossRef] [PubMed]
- Lerma, E.; White, W.B.; Bakris, G. Effectiveness of nonsteroidal mineralocorticoid receptor antagonists in patients with diabetic kidney disease. Postgrad. Med. 2023, 135, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, Y.; Ke, C.; Jin, Y.; Lao, W.; Wu, Y.; Liu, Y.; Kong, X.; Qiao, J.; Zhai, A.; et al. HDAC4: An emerging target in diabetes mellitus and diabetic complications. Eur. J. Med. Res. 2025, 30, 429. [Google Scholar] [CrossRef]
- Mihanfar, A.; Akbarzadeh, M.; Darband, S.G.; Sadighparvar, S.; Majidinia, M. SIRT1: A promising therapeutic target in type 2 diabetes mellitus. Arch. Physiol. Biochem. 2024, 130, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Artasensi, A.; Pedretti, A.; Vistoli, G.; Fumagalli, L. Type 2 Diabetes Mellitus: A Review of Multi-Target Drugs. Molecules 2020, 25, 1987. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, A.D.; Singh, J.; Singh, H.; Roy, R.K.; Chaudhary, A. 1,2,3-Triazine scaffold as a potent biologically active moiety: A mini review. Mini Rev. Med. Chem. 2014, 14, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sirohi, T.S.; Singh, H.; Yadav, R.; Roy, R.K.; Chaudhary, A.; Pandeya, S.N. 1,2,4-triazine analogs as novel class of therapeutic agents. Mini Rev. Med. Chem. 2014, 14, 168–207. [Google Scholar] [CrossRef] [PubMed]
- Ganai, A.M.; Pathan, T.K.; Hampannavar, G.A.; Pawar, C.; Obakachi, V.A.; Kushwaha, B.; Kushwaha, N.D.; Karpoormath, R. Recent Advances on the s-Triazine Scaffold with Emphasis on Synthesis, Structure-Activity and Pharmacological Aspects: A Concise Review. ChemistrySelect 2021, 6, 1616. [Google Scholar] [CrossRef]
- Khalid, Z.; Alnuwaiser, M.A.; Ahmad, H.A.; Shafqat, S.S.; Munawar, M.A.; Kamran, K.; Abbas, M.M.; Kalam, M.A.; Ewida, M.A. Experimental and Computational Analysis of Newly Synthesized Benzotriazinone Sulfonamides as Alpha-Glucosidase Inhibitors. Molecules 2022, 27, 6783. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, J.; Xie, Z.; Li, L.; Chen, M.; Chen, S.; Peng, Y. Synthesis, molecular docking and α-glucosidase inhibition of 2-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)-N-arylacetamides. Bioorganic Med. Chem. Lett. 2017, 27, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Valipour, M.; Zakeri Khatir, Z.; Kiadaliry, K.; Mojtabavi, S.; Faramarzi, M.A.; Shokati Sayyad, M.; Seyedabadi, M.; Ghasemian, M.; Hashemi, S.M.; Irannejad, H. Design, synthesis, α-glucosidase inhibition and hypoglycemic activity of 3-aceto(benzo)hydrazide-1,2,4-triazines as potential anti-diabetic agents. Eur. J. Med. Chem. Rep. 2024, 12, 100207. [Google Scholar] [CrossRef]
- Jahan, H.; Tufail, P.; Shamim, S.; Khan, K.M.; Gennari, M.; Pizzi, M.; Choudhary, M.I. Antiglycation and anti-inflammatory potential of 1,2,4-triazine derivatives targeting AGE–RAGE-mediated signaling in THP-1 monocytes. Int. Immunopharmacol. 2024, 142, 113145. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.; Machowiak, A.; Rychter, A.M.; Ratajczak, A.E.; Szymczak-Tomczak, A.; Dobrowolska, A.; Krela-Kaźmierczak, I. Accumulation of Advanced Glycation End-Products in the Body and Dietary Habits. Nutrients 2022, 14, 3982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Tu, C.; Chen, X.; He, R. Advanced Glycation End Products in Disease Development and Potential Interventions. Antioxidants 2025, 14, 492. [Google Scholar] [CrossRef] [PubMed]
- Shamim, S.; Khan, K.M.; Ullah, N.; Chigurupati, S.; Wadood, A.; Rehman, A.U.; Ali, M.; Salar, U.; Alhowail, A.; Taha, M.; et al. Synthesis and screening of (E)-3-(2-benzylidenehydrazinyl)-5,6-diphenyl-1,2,4-triazine analogs as novel dual inhibitors of α-amylase and α-glucosidase. Bioorganic Chem. 2020, 101, 103979. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Choudhary, S.; Singh, P.K.; Silakari, O. Addressing selectivity issues of aldose reductase 2 inhibitors for the management of diabetic complications. Future Med. Chem. 2020, 12, 1327–1358. [Google Scholar] [CrossRef] [PubMed]
- Roney, M.; Issahaku, A.R.; Uddin, M.N.; Wilhelm, A.; Aluwi, M.F.F.M. Exploration of leads from bis-indole based triazine derivatives targeting human aldose reductase in diabetic type 2: In-silico approaches. 3 Biotech 2025, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Rehman, W.; Rahim, F.; Hussain, R.; Obaidullah, A.J.; Faris Alotaibi, H.; Alanazi, M.; Usman Khan, M.; Khan, Y. Bis-indole based triazine derivatives: Synthesis, characterization, in vitro β-glucuronidase, anti-cancer and anti-bacterial evaluation along with in silico molecular docking and ADME analysis. Arab. J. Chem. 2023, 16, 104970. [Google Scholar] [CrossRef]
- Hanke, J.; Romejko, K.; Niemczyk, S. Sodium-Glucose Cotransporter-2 Inhibitors in Diabetes and Beyond: Mechanisms, Pleiotropic Benefits, and Clinical Use—Reviewing Protective Effects Exceeding Glycemic Control. Molecules 2025, 30, 4125. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Zhao, L.J. Synthetic approaches and clinical application of small-molecule inhibitors of sodium-dependent glucose transporters 2 for the treatment of type 2 diabetes mellitus. Eur. J. Med. Chem. 2024, 269, 116343. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Kim, M.J.; Lee, J. 1,2,4-Triazinylmethylphenyl glucoside as novel C-aryl glucoside SGLT2 inhibitors. Bull. Korean Chem. Soc. 2011, 32, 2938–2940. [Google Scholar] [CrossRef][Green Version]
- Lee, J.; Lee, S.H.; Seo, H.J.; Kim, M.J.; Kang, S.Y.; Kim, J.; Lee, S.H.; Jung, M.E.; Son, E.J.; Song, K.S.; et al. C-Aryl Glucoside SGLT2 Inhibitors and Pharmaceutical Compositions Comprising Same. US Patent 8,541,380 B2, 24 September 2013. [Google Scholar]
- Shao, D.W.; Zhao, L.J.; Sun, J.F. Synthesis and clinical application of representative small-molecule dipeptidyl Peptidase-4 (DPP-4) inhibitors for the treatment of type 2 diabetes mellitus (T2DM). Eur. J. Med. Chem. 2024, 272, 116464. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.; Gujarathi, D. DPP—4 inhibitors in Type 2 Diabetes mellitus—A Panoramic Review. Int. J. Clin. Med. Case Rep. 2023, 2, 1–7. [Google Scholar]
- Deacon, C.F. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, H.; Li, C.; Zheng, W.; Wang, M.; Li, Y.; Sun, H.; Wu, M. Safety and pharmacokinetic interaction between fotagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin in healthy subjects. Expert Opin. Drug Metab. Toxicol. 2021, 17, 725–731. [Google Scholar] [CrossRef]
- Wu, M.; Li, Q.Q.; Zhang, H.; Zhu, X.X.; Li, X.J.; Li, Y.; Sun, H.G.; Ding, Y.H. Safety, Pharmacokinetics, and Pharmacodynamics of a Dipeptidyl Peptidase-4 Inhibitor: A Randomized, Double-Blinded, Placebo-Controlled Daily Administration of Fotagliptin Benzoate for 14 Days for Type 2 Diabetes Mellitus. Clin. Pharmacol. Drug Dev. 2021, 10, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Sun, K.; Xu, W.; Wang, C.; Yan, D.; Li, S.; Cong, L.; Pi, Y.; Song, W.; Sun, Q.; et al. Fotagliptin monotherapy with alogliptin as an active comparator in patients with uncontrolled type 2 diabetes mellitus: A randomized, multicenter, double-blind, placebo-controlled, phase 3 trial. BMC Med. 2023, 21, 388. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Xu, W.; Guo, X.; Xu, M.; Zhang, W.; Yan, C.; Pang, S.; Shu, X.; Peng, W.; Zhang, Y.; et al. Fotagliptin add-on therapy to metformin in patients with uncontrolled type 2 diabetes: A randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Diabetes Obes. Metab. 2025, 27, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yuan, L.; Gong, Z.; Li, M.; Yuan, Y.; Geng, J. New drugs approved by the NMPA in 2024: Synthesis and clinical applications. Eur. J. Med. Chem. 2025, 291, 117643. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yun, Y.; Guo, S.; Wang, X.; Xiong, M.; Zhao, T.; Xu, T.; Shen, J.; Xie, X.; Wang, K. Discovery of Novel 5,6-Dihydro-1,2,4-triazine Derivatives as Efficacious Glucagon-Like Peptide-1 Receptor Agonists. J. Med. Chem. 2023, 66, 7988–8010. [Google Scholar] [CrossRef] [PubMed]
- Buckeridge, C.; Cobain, S.; Bays, H.E.; Matsuoka, O.; Fukushima, Y.; Halstead, P.; Tsamandouras, N.; Sherry, N.; Gorman, D.N.; Saxena, A.R. Efficacy and safety of danuglipron (PF-06882961) in adults with obesity: A randomized, placebo-controlled, dose-ranging phase 2b study. Diabetes Obes. Metab. 2025, 27, 4915–4926. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-provides-update-oral-glp-1-receptor-agonist (accessed on 18 May 2026).
- Jha, R.; Goyal, K.; Mehan, S.; Singh, G. Dual α-amylase and α-glucosidase inhibitors: Recent progress from natural and synthetic resources. Bioorganic Chem. 2025, 163, 108762. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Hussain, R.; Khan, Y.; Iqbal, T.; Shoaib, K.; Jehangir, U.; Khowdiary, M.M.; Felemban, S. Developing Correlation Between In Silico Molecular Modeling and In Vitro α-Amylase, Anti-Cancer and SARS-COV-2 Studies of Bis-Indole Derived Triazine-Based Thiazolidinone Hybrid Derivatives. ChemistrySelect 2024, 9, e202400686. [Google Scholar] [CrossRef]
- Seyfi, S.; Salarinejad, S.; Moghimi, S.; Toolabi, M.; Sadeghian, N.; Tüzün, B.; Firoozpour, L.; Ketabforoosh, S.H.M.E.; Taslimi, P.; Foroumadi, A. Synthesis, biological activities, and molecular docking studies of triazolo[4,3-b]triazine derivatives as a novel class of α-glucosidase and α-amylase inhibitors. Arch. Pharm. 2024, 357, e2300628. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.D.; Bhadada, S.V.; Ghate, M.D. Design, synthesis and anti-diabetic activity of triazolotriazine derivatives as dipeptidyl peptidase-4 inhibitors. Bioorganic Chem. 2017, 72, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Guertin, K.R.; Setti, L.; Qi, L.; Dunsdon, R.M.; Dymock, B.W.; Jones, P.S.; Overton, H.; Taylor, M.; Williams, G.; Sergi, J.A.; et al. Identification of a novel class of orally active pyrimido[5,4-3][1,2,4]triazine-5,7-diamine-based hypoglycemic agents with protein tyrosine phosphatase inhibitory activity. Bioorganic Med. Chem. Lett. 2003, 13, 2895–2898. [Google Scholar] [CrossRef] [PubMed]
- Coronell-Tovar, A.; Pardo, J.P.; Rodríguez-Romero, A.; Sosa-Peinado, A.; Vásquez-Bochm, L.; Cano-Sánchez, P.; Álvarez-Añorve, L.I.; González-Andrade, M. Protein tyrosine phosphatase 1B (PTP1B) function, structure, and inhibition strategies to develop antidiabetic drugs. FEBS Lett. 2024, 598, 1811–1838. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, K.; Ullah, H.; Wadood, A.; Taha, M.; Rehman, A.U.; Uddin, I.; Ashraf, M.; Shaukat, A.; Rehman, W.; et al. Triazinoindole analogs as potent inhibitors of α-glucosidase: Synthesis, biological evaluation and molecular docking studies. Bioorganic Chem. 2015, 58, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Alshamrani, F.J.; Rahim, F.; Hayat, S.; Ullah, H.; Zaman, K.; Imran, S.; Khan, K.M.; Naz, F. Synthesis of Novel Triazinoindole-Based Thiourea Hybrid: A Study on α-Glucosidase Inhibitors and Their Molecular Docking. Molecules 2019, 24, 3819. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Tariq, S.; Taha, M.; Ullah, H.; Zaman, K.; Uddin, I.; Wadood, A.; Khan, A.A.; Rehman, A.U.; Uddin, N.; et al. New triazinoindole bearing thiazole/oxazole analogues: Synthesis, α-amylase inhibitory potential and molecular docking study. Bioorganic Chem. 2019, 92, 103284. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Kumar, P.; Hooda, M.; Singh, R.; Kumar, P. Efficient synthesis of promising antidiabetic triazinoindole analogues via a solvent-free method: Investigating the reaction of 1,3-diketones and 2,5-dihydro-3H-[1,2,4]triazino[5,6-b]indole-3-thione. Org. Biomol. Chem. 2025, 23, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://calculator.academy/%CE%BCg-ml-to-%C2%B5m-calculator/ (accessed on 10 May 2026).
- Stefek, M.; Soltesova Prnova, M.; Majekova, M.; Rechlin, C.; Heine, A.; Klebe, G. Identification of novel aldose reductase inhibitors based on carboxymethylated mercaptotriazinoindole scaffold. J. Med. Chem. 2015, 58, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Prnová, M.Š.; Račková, L.; Kováčiková, L.; Balleková, J.; Viskupičová, J.; Micháliková, S.; Taşkoparan, B.; Elmazoğlu, Z.; Rižner, T.L.; Karasu, C.; et al. General toxicity assessment of the novel aldose reductase inhibitor cemtirestat. Interdiscip. Toxicol. 2019, 12, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Prnova, M.S.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Colín-González, A.L.; Piedra-García, F.; Rangel-López, E.; Kovacikova, L.; Ceylan, A.; Karasu, C.; Santamaria, A.; et al. Antioxidant Mechanisms in the Neuroprotective Action of Cemtirestat: Studies in Chemical Models, Liposomes and Rat Brain Cortical Slices. Neuroscience 2020, 443, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Reihanifar, T.; Şahin, M.; Stefek, M.; Ceylan, A.F.; Karasu, Ç. Antioxidants in Diabetes-Induced Complications (ADIC) Study Group. Cemtirestat, an aldose reductase inhibitor and antioxidant compound, induces ocular defense against oxidative and inflammatory stress in rat models for glycotoxicity. Cell Biochem. Funct. 2023, 41, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Elmazoglu, Z.; Prnova, M.S.; Santamaria, A.; Stefek, M.; Karasu, C. Combatting Nitrosative Stress and Inflammation with Novel Substituted Triazinoindole Inhibitors of Aldose Reductase in PC12 Cells Exposed to 6-Hydroxydopamine Plus High Glucose. Neurotox. Res. 2021, 39, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-T.; Guo, Q.-Q.; Chen, Z.-L.; Wang, L.-L.; Du, Y.; Chen, R.; Mao, Y.-H.; Yang, S.-G.; Huang, J.; Wang, J.-T.; et al. Design, synthesis and bioevaluation of novel substituted triazines as potential dual PI3K/mTOR inhibitors. Eur. J. Med. Chem. 2020, 204, 112637. [Google Scholar] [CrossRef]
- Lamb, Y.N. Imeglimin Hydrochloride: First Approval. Drugs 2021, 81, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.C.; Gupta, S. Imeglimin: The new kid on the block. Curr. Diabetes Rep. 2024, 24, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Grzeszczak, W. Imeglimin: A new antidiabetic drug with potential for the treatment of patients with type 2 diabetes. Endokrynol. Pol. 2022, 73, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Shireen, S.M.; Bhavya, E.; Parthiban, R. Imeglimin systematic review: A novel therapeutic approach for type 2 diabetes—Unveiling benefits on β-cell function, insulin sensitivity, and potential long-term glycaemic control (HbA1c). Eur. J. Clin. Pharmacol. 2025, 81, 1713–1731. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Weiskirchen, R. Imeglimin: Discovery, Pharmacology, and Trials. J. Endocrinol. Metab. 2026, 16, 1–14. [Google Scholar] [CrossRef]
- Tewari, J.; Qidwai, K.A.; Tewari, A.; Rana, A.; Singh, V.; Tewari, V.; Mateen, R.; Khatoon, S.; Ahmad, F.; Haque, S. Efficacy and safety of imeglimin, a novel oral agent in the management of type 2 diabetes mellitus: A systematic review and meta-analysis. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2025, 398, 14501–14513. [Google Scholar] [CrossRef] [PubMed]
- Khodakhah, A.; Mohammadi, H.; Abdoli, S.; Zarei, I.; Palimi, M.; Ekhtiari, Z.; Talebi, M.; Biglar, M.; Khorramizadeh, M.R.; Amanlou, M. Synthesis and molecular docking studies of new aryl imeglimin derivatives as a potent antidiabetic agent in a diabetic zebrafish model. Sci. Rep. 2024, 14, 9410. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Murmu, A.; Matore, B.W.; Roy, P.P.; Singh, J. Thiazolidinedione an auspicious scaffold as PPAR-γ agonist: Its possible mechanism to Manoeuvre against insulin resistant diabetes mellitus. Eur. J. Med. Chem. Rep. 2024, 11, 100160. [Google Scholar] [CrossRef]
- Ahmadi, A.; Khalili, M.; Samavat, S.; Shahbazi, E.; Nahri-Niknafs, B. Synthesis and evaluation of the hypoglycemic and hypolipidemic activity of novel arylidene thiazolidinedione analogs on a type 2 diabetes model. Pharm. Chem. J. 2016, 50, 165–170. [Google Scholar] [CrossRef]
- Andrews, K.M.; Beebe, D.A.; Benbow, J.W.; Boyer, D.A.; Doran, S.D.; Hui, Y.; Liu, S.; McPherson, R.K.; Neagu, C.; Parker, J.C.; et al. 1-((3S,4S)-4-amino-1-(4-substituted-1,3,5-triazin-2-yl) pyrrolidin-3-yl)-5,5-difluoropiperidin-2-one inhibitors of DPP-4 for the treatment of type 2 diabetes. Bioorganic Med. Chem. Lett. 2011, 21, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.D.; Liu, P.; Yang, Y.; Gao, H. Sulfonamide-1,3,5-triazine–thiazoles: Discovery of a novel class of antidiabetic agents via inhibition of DPP-4. RSC Adv. 2016, 6, 83438–83447. [Google Scholar] [CrossRef]
- Cho, H.; Um, J.; Lee, J.-H.; Kim, W.-H.; Kang, W.S.; Kim, S.H.; Ha, H.-H.; Kim, Y.-C.; Ahn, Y.-K.; Jung, D.-W.; et al. ENOblock, a unique small molecule inhibitor of the non-glycolytic functions of enolase, alleviates the symptoms of type 2 diabetes. Sci. Rep. 2017, 7, 44186. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Kim, W.H.; Park, S.H.; Lee, J.; Kim, J.; Su, D.; Ha, H.H.; Chang, Y.T.; Williams, D.R. A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes. ACS Chem. Biol. 2013, 8, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- El-Kabbani, O.; Darmanin, C.; Chung, R.P. Sorbitol dehydrogenase: Structure, function and ligand design. Curr. Med. Chem. 2004, 11, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Mylari, B.L.; Oates, P.J.; Zembrowski, W.J.; Beebe, D.A.; Conn, E.L.; Coutcher, J.B.; O’Gorman, M.T.; Linhares, M.C.; Withbroe, G.J. A Sorbitol Dehydrogenase Inhibitor of Exceptional in Vivo Potency with a Long Duration of Action: 1-(R)-{4-[4-(4,6-Dimethyl[1,3,5]triazin-2-yl)-2R,6S-dimethylpiperazin-1-yl]pyrimidin-2-yl}ethanol. J. Med. Chem. 2002, 45, 4398–4401. [Google Scholar] [CrossRef] [PubMed]
- Chu-Moyer, M.Y.; Ballinger, W.E.; Beebe, D.A.; Coutcher, J.B.; Day, W.W.; Li, J.; Oates, P.J.; Weekly, R.M. SAR and species/stereo-selective metabolism of the sorbitol dehydrogenase inhibitor, CP-470,711. Bioorganic Med. Chem. Lett. 2002, 12, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Landau, Z.; Novotny, M.J.; Preston, G.M.; Wright, K.; Freeman, T.; Dai, H.; Thompson, J.; Oates, P.J.; Calle, R.A. Pharmacokinetics, Pharmacodynamics, Tolerability, and Safety of a Novel Sorbitol Dehydrogenase Inhibitor in Healthy Participants. J. Clin. Pharmacol. 2010, 50, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Mylari, B.L.; Withbroe, G.J.; Beebe, D.A.; Brackett, N.S.; Conn, E.L.; Coutcher, J.B.; Oates, P.J.; Zembrowski, W.J. Design and Synthesis of a Novel Family of Triazine-Based Inhibitors of Sorbitol Dehydrogenase with Oral Activity: 1-{4-[3R,5S-Dimethyl-4-(4-methyl-[1,3,5]triazin-2-yl)-piperazin-1-yl]-[1,3,5]triazin-2-yl}-(R)ethanol. Bioorganic Med. Chem. 2003, 11, 4179–4188. [Google Scholar] [CrossRef] [PubMed]
- El-Harakeh, M.D.; Njeim, R.; Youssef, A.; Youssef, N.; Eid, A.A.; Bouhadir, K.H. Novel triazine-based pyrimidines suppress glomerular mesangial cells proliferation and matrix protein accumulation through a ROS-dependent mechanism in the diabetic milieu. Bioorganic Med. Chem. Lett. 2019, 29, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.-W.; Ha, H.-H.; Zheng, X.; Chang, Y.-T.; Williams, D.R. Novel use of fluorescent glucose analogues to identify a new class of triazine-based insulin mimetics possessing useful secondary effects. Mol. Biosyst. 2011, 7, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.K.; Dubey, P.; Singh, S.; Bhat, H.R.; Kumawat, M.K.; Singh, U.P. Discovery of Novel 1,3,5-Triazine-Thiazolidine-2,4-Diones as Dipeptidyl Peptidase-4 Inhibitors with Antibacterial Activity Targeting the S1 Pocket for the Treatment of Type 2 Diabetes. RSC Adv. 2015, 5, 14095–14102. [Google Scholar]
- Lolak, N.; Akocak, S.; Türkeş, C.; Taslimi, P.; Işık, M.; Beydemir, Ş.; Gülçin, İ.; Durgun, M. Synthesis, characterization, inhibition effects, and molecular docking studies as acetylcholinesterase, α-glycosidase, and carbonic anhydrase inhibitors of novel benzenesulfonamides incorporating 1,3,5-triazine structural motifs. Bioorganic Chem. 2020, 100, 103897. [Google Scholar] [CrossRef] [PubMed]
- Mallia, A.; Brocca, L.; Papaianni, G.G.; Banfi, C. Carbonic anhydrases inhibition in the management of cardiovascular and cardiometabolic disorders. Biomed. Pharmacother. 2025, 190, 118396. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Liao, S.; Zhong, W.; Xiao, X.; Zhu, J.; Li, W.; Wu, X.; Feng, Y. Synthesis, Characterization, and Biological Evaluations of 1,3,5-Triazine Derivatives of Metformin Cyclization with Berberine and Magnolol in the Presence of Sodium Methylate. Molecules 2017, 22, 1752. [Google Scholar] [CrossRef] [PubMed]
- Kurach, Ł. Methodological approaches for inducing diabetes-like metabolic conditions in zebrafish larvae. Curr. Issues Pharm. Med. Sci. 2025, 38, 150–154. [Google Scholar] [CrossRef]
- Vhora, N.; Naskar, U.; Hiray, A.; Kate, A.S.; Jain, A. Recent Advances in In-Vitro Assays for Type 2 Diabetes Mellitus: An Overview. Rev. Diabet. Stud. 2020, 16, 13–23. [Google Scholar] [CrossRef] [PubMed]




































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Reference | Isomer | Number of Described Compounds | Type of Studies | Biological Activity Range | Other Studies | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fused 1,2,3- Triazine | 1,2,4-Triazine | Fused 1,2,4-Triazine | 1,3,5-Triazine | In Vitro | In Vivo (or Ex Vivo) | |||||
| 1 | Khalid et al. [20] | Yes | - | - | - | 13 | Yes | α-Glucosidase inhibitors IC50: 29.8–102.2 μM | Molecular docking (UniProt:P53051) | |
| 2 | Wang et al. [21] | - | Yes | - | - | 17 | Yes | - | α-Glucosidase inhibitors IC50: 12.5–72.7 μM | Molecular docking (PBD:3AJ7) |
| 3 | Valipour et al. [22] | - | Yes | - | - | 10 | Yes | Yes | α-Glucosidase inhibitors IC50: 12.0–672.6 μM | Molecular docking (PDB:7P2Z) Enzyme kinetics Cytotoxicity (lines: HCT-116, MDA-MB-231, A549) in MTT assay |
| 4 | Jahan et al. [23] | - | Yes | - | - | 26 | Yes | - | Glycation inhibitors IC50: 91.0–259.0 μM | MTT assay, WST-1 assay, Western blot, immunocytochemistry |
| 5 | Roney et al. [28] | - | Yes | - | - | 12 | - | - | Aldose reductase inhibitors (not confirmed—only in silico studies) | Molecular docking (PBD:1US0), MD simulations (200 ns) MM/GBSA, MD, PCA |
| 6 | Kang et al. [32] | - | Yes | - | - | 2 | Yes | - | SGLT2 inhibitors IC50 = 24.9 nM & IC50 = 491 nM | --- |
| 7 | Chen et al. [43] | - | Yes | - | - | 41 | Yes | Yes | GLP-1R agonists EC50: 6 pM to >10,000 nM | hERG channel inhibition Molecular docking (PDB ID:6X1A) Pharmacokinetic studies in rats for compound 21: t1/2 = 1.1 h; Cmax = 130 ng/mL AUClast = 70 h·ng/mL In vivo (in human GLP-1R Knock-In mice): —oral glucose tolerance test —food intake test |
| 8 | Shamim et al. [26] | - | Yes | - | - | 24 | Yes | - | α-Amylase & α-glucosidase inhibitors IC50 < 50 μM IC50: 13.0–46.9 µM (α-amylase) IC50: 13.1–46.4 µM (α-glucosidase) | Molecular docking (PDB:3AJ7, α-glucosidase) (PDB:3BAJ, α-amylase) Kinetic studies |
| 9 | Khan et al. [47] | - | Yes | - | - | 12 | Yes | - | Multi-target ligands (α-amylase, anticancer & antiviral) IC50: 3.10–25.80 μM (α-amylase) IC50: 0.20–19.10 μM (anticancer), IC50: 3.20–16.20 μM (SARS-CoV-2) | Molecular docking (PBD:6LU7, SARS-CoV-2) |
| 10 | Seyfi et al. [48] | - | - | Yes | - | 15 | Yes | - | Dual α-amylase & α-glucosidase inhibitors IC50: 24.64–115.57 nM (α-amylase) IC50: 34.52–213.44 nM (α-glucosidase) | Molecular docking (PDB—no precise information) Kinetic studies |
| 11 | Patel et al. [49] | - | - | Yes | - | 17 | Yes | Yes | DPP-4 inhibitors (only 2 active) IC50 = 28.1 μM & IC50 = 166.4 μM | Molecular docking (PBD:3KWF) Oral glucose tolerance tests A chronic model of high-fat diet fed with streptozotocin in rats |
| 12 | Guertin et al. [50] | - | - | Yes | - | 15 | Yes | Yes | PTP1B inhibitors IC50 (300 nM dithiothreitol): 2.9 to >100 μM | Pharmacokinetic studies in C57BL/6J mice for compound 33: t1/2 = 1.1 h; CL = 104 mL/Kg/min Vss = 3.1 L/kg; Cmax = 4.0 μM F = 97% An ob/ob mouse model |
| 13 | Rahim et al. [52] | - | - | Yes | - | 11 | Yes | - | α-Glucosidase inhibitors IC50: 2.3–312.8 μM | Molecular docking (PBD:2AJ7) |
| 14 | Taha et al. [53] | - | - | Yes | - | 25 | Yes | - | α-Glucosidase inhibitors IC50: 1.3–5.8 µM | Molecular docking (PBD:3W37) |
| 15 | Rahim et al. [54] | - | - | Yes | - | 21 | Yes | - | α-Amylase inhibitors IC50: 1.2–21.50 µM | Molecular docking (PBD:1OSE) |
| 16 | Aggarwal et al. [55] | - | - | Yes | - | 10 | Yes | - | α-Amylase inhibitors IC50: 16.14–27.69 μg/mL (IC50: 41.7–75.6 µM) | Molecular docking (PBD:7TAA) |
| 17 | Stefek et al. [57] | - | - | Yes | - | 15 | Yes | Yes (ex vivo) | Aldose reductase inhibitors ALR2 IC50: 0.097–53.5 µM ALR1 IC50: 1.2–80.2 µM | Crystal structure AKR1B10 inhibition Enzyme kinetics Sorbitol accumulation in isolated rat eye lenses cultivated with glucose (50 mM) |
| 18 | Hlaváč et al. [62] | - | - | Yes | - | 4 | Yes | Yes (ex vivo) | Aldose reductase inhibitors ALR2 IC50: 51–787 nM (1% DMSO) ALR2 IC50: 42–434 nM (H2O) | AKR1B1 inhibition AKR1B10 inhibition Sorbitol accumulation in isolated rat eye lenses cultivated with glucose (50 mM) MD simulations |
| 19 | Khodakhah et al. [69] | - | - | - | Yes | 10 | - | Yes | Antihyperglycaemic agents Fasting blood sugar of zebrafish diabetic model: 72.3–108.3 mg/dL | Molecular docking SIRT1 (PBD:5BTR) and GSK-3β (PBD:1Q4L) Zebrafish diabetic model |
| 20 | Ahmadi et al. [71] | - | - | - | Yes | 2 | - | Yes | Antihyperglycaemic agents Antihyperlipidaemic agents | Alloxan-induced diabetic rat model |
| 21 | Andrews et al. [72] | - | - | - | Yes | 16 | Yes | DPP-4 inhibitors IC50: 1.6–1400 nM | Many in vitro assays | |
| 22 | Gao et al. [73] | - | - | - | Yes | - | Yes | Yes | DPP-4 inhibitors IC50: 2.3–544.4 nM | DPP-8 inhibition, DPP-9 inhibition Molecular docking (PBD:2FJP) Oral glucose tolerance test (male IRC mice) STZ-induced diabetes in rats |
| 23 | Cho et al. [74] | - | - | - | Yes | 1 (ENOblock) | Yes | Yes | Glycolytic enzyme enolase modulator IC50 = 576 nM | Gene expression analysis and histology Diabetic db/db mice |
| 24 | Mylari et al. [76] | - | - | - | Yes | 10 | Yes | Yes | Sorbitol dehydrogenase inhibitors IC50: 5–39 nM (rat) IC50: 4–43 nM (human) | Pharmacokinetic studies (compound 56 serum half-lives: 7 h—rats; 10 h—dogs) Two streptozotocin diabetic rat models (acute and chronic) |
| 25 | Mylari et al. [80] | - | - | - | Yes | 10 | Yes | Yes | Sorbitol dehydrogenase inhibitors IC50: 36–400 nM (rat) IC50: 42–390 nM (human) | Two streptozotocin diabetic rat models (acute and chronic) |
| 26 | El-Harakeh et al. [81] | - | - | - | Yes | 8 | Yes | - | Antidiabetic nephropathy agent | MTT assay and Western blot |
| 27 | Jung et al. [82] | - | - | - | Yes | 6 | Yes | - | Insulin mimetic agents | Cytotoxicity and anti-inflammatory studies |
| 28 | Srivastava et al. [83] | - | - | - | Yes | 11 | Yes | - | DPP-4 inhibitors IC50: 6.4–49.2 μM & antibacterial activity | Molecular docking (PBD:2FJP) |
| 29 | Lolak et al. [84] | - | - | - | Yes | 11 | Yes | - | α-Glycosidase inhibitors (IC50 = 44.7–84.3 μM) Acetylcholinesterase inhibitors (IC50: 397.3–856.3 μM) Carbonic anhydrase inhibitors IC50: 45.2–207.2 μM (CA I) IC50: 37.8–194.5 μM (CA II) | Molecular docking (PBD:5NN8, α-glycosidase) (PBD:4M0F, acetylcholinesterase) (PBD:4WUQ, CA I) (PBD:4FU5, CA II) |
| 30 | Cao et al. [86] | - | - | - | Yes | 3 | Yes | - | Antidiabetic (INS-1 cells) & anti-inflammatory (RAW264.1 cells) | ---- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Łażewska, D.; Strelchuk, D.; Handzlik, J. Potential of Triazines as Antidiabetic Agents—A Review of Structures and Pharmacological Activity. Pharmaceuticals 2026, 19, 1018. https://doi.org/10.3390/ph19071018
Łażewska D, Strelchuk D, Handzlik J. Potential of Triazines as Antidiabetic Agents—A Review of Structures and Pharmacological Activity. Pharmaceuticals. 2026; 19(7):1018. https://doi.org/10.3390/ph19071018
Chicago/Turabian StyleŁażewska, Dorota, Diana Strelchuk, and Jadwiga Handzlik. 2026. "Potential of Triazines as Antidiabetic Agents—A Review of Structures and Pharmacological Activity" Pharmaceuticals 19, no. 7: 1018. https://doi.org/10.3390/ph19071018
APA StyleŁażewska, D., Strelchuk, D., & Handzlik, J. (2026). Potential of Triazines as Antidiabetic Agents—A Review of Structures and Pharmacological Activity. Pharmaceuticals, 19(7), 1018. https://doi.org/10.3390/ph19071018