
Machine Learning and Virtual Screening Methods to Discover Potential Cyclin-Dependent Kinase 2 (CDK2) Inhibitors



 and
and
Abstract
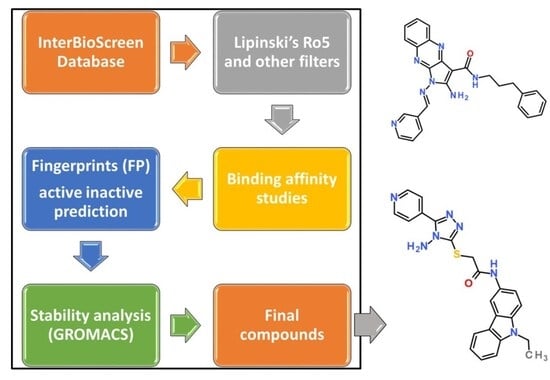
1. Introduction
2. Results
2.1. Virtual Screening and Lipinski’s Filtering
2.2. Model Generation and Prediction of New Compounds
2.3. Molecular Docking
2.4. Molecular Dynamics Simulation (MDS)
2.4.1. Stability Analysis by RMSD
2.4.2. Compactness Evaluation by Rg
2.4.3. Fluctuation Analysis by RMSF
2.5. Number of Hydrogen Bonds
2.6. Potential Energy
2.7. Total Interaction Energy
2.8. Binding Mode Analysis
2.9. Intermolecular Interactions
2.10. ADMET Analysis
3. Discussion
4. Materials and Methods
4.1. Selection of the Small Molecules
4.2. Selection of the Target
4.3. ML and Model Generation
4.3.1. Data Curation
4.3.2. Generation of the Fingerprints (FPs)
4.3.3. Model Generation
4.4. Evaluation Matrix
4.5. Confusion Matrix
4.6. Accuracy
4.7. Precision
4.8. Recall
4.9. F1 Score
4.10. AUC-ROC
4.11. Molecular Docking Studies
4.12. Molecular Dynamics Simulations (MDSs)
4.13. ADMET Assessment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sudhakar, A. History of Cancer, Ancient and Modern Treatment Methods. J. Cancer Sci. Ther. 2009, 1, 1. [Google Scholar] [CrossRef]
- Hardcastle, I.R.; Golding, B.T.; Griffin, R.J. Designing Inhibitors of Cyclin-Dependent Kinases. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 325–348. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Dong, P.; Gassler, N.; Taheri, M.; Baniahmad, A.; Dilmaghani, N.A. A Review on the Role of Cyclin Dependent Kinases in Cancers. Cancer Cell Int. 2022, 22, 325. [Google Scholar] [CrossRef]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in Cancer: Challenges and Opportunities for Therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A Complex Leads to Autophagic Degradation of CDK2 in Cancer Cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Gao, W.; Zhang, L.; Pan, Y.; Zhang, S.; Wang, Y. Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2. Int. J. Mol. Sci. 2015, 16, 9314–9340. [Google Scholar] [CrossRef] [PubMed]
- Talapati, S.R.; Nataraj, V.; Pothuganti, M.; Gore, S.; Ramachandra, M.; Antony, T.; More, S.S.; Krishnamurthy, N.R. Structure of Cyclin-Dependent Kinase 2 (CDK2) in Complex with the Specific and Potent Inhibitor CVT-313. Struct. Biol. Cryst. Commun. 2020, 76, 350–356. [Google Scholar] [CrossRef]
- Steinberg, S.F. Post-Translational Modifications at the ATP-Positioning G-Loop That Regulate Protein Kinase Activity. Pharmacol. Res. 2018, 135, 181–187. [Google Scholar] [CrossRef]
- Bártová, I.; Otyepka, M.; Kríz, Z.; Koca, J. Activation and Inhibition of Cyclin-Dependent Kinase-2 by Phosphorylation; a Molecular Dynamics Study Reveals the Functional Importance of the Glycine-Rich Loop. Protein Sci. 2004, 13, 1449–1457. [Google Scholar] [CrossRef]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the Human WEE1Hu CDK Tyrosine 15-Kinase during the Cell Cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Shah, M.; Patel, M.; Shah, M.; Patel, M.; Prajapati, M. Computational Transformation in Drug Discovery: A Comprehensive Study on Molecular Docking and Quantitative Structure Activity Relationship (QSAR). Intell. Pharm. 2024, 2, 589–595. [Google Scholar] [CrossRef]
- Aparoy, P.; Reddy, K.K.; Reddanna, P. Structure and Ligand Based Drug Design Strategies in the Development of Novel 5- LOX Inhibitors. Curr. Med. Chem. 2012, 19, 3763–3778. [Google Scholar] [CrossRef]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein-Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef]
- Bajad, N.G.; Rayala, S.; Gutti, G.; Sharma, A.; Singh, M.; Kumar, A.; Singh, S.K. Systematic Review on Role of Structure Based Drug Design (SBDD) in the Identification of Anti-Viral Leads against SARS-CoV-2. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100026. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef]
- Bacilieri, M.; Moro, S. Ligand-Based Drug Design Methodologies in Drug Discovery Process: An Overview. Curr. Drug Discov. Technol. 2006, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; MacKerell, A.D. Recent Advances in Ligand-Based Drug Design: Relevance and Utility of the Conformationally Sampled Pharmacophore Approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Ajjarapu, S.M.; Tiwari, A.; Ramteke, P.W.; Singh, D.B.; Kumar, S. Ligand-Based Drug Designing. In Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2022; pp. 233–252. [Google Scholar]
- Kumar, R.; Bavi, R.; Jo, M.G.; Arulalapperumal, V.; Baek, A.; Rampogu, S.; Kim, M.O.; Lee, K.W. New Compounds Identified through in Silico Approaches Reduce the α-Synuclein Expression by Inhibiting Prolyl Oligopeptidase in Vitro. Sci. Rep. 2017, 7, 10827. [Google Scholar] [CrossRef]
- Shi, X.-N.; Li, H.; Yao, H.; Liu, X.; Li, L.; Leung, K.-S.; Kung, H.; Lu, D.; Wong, M.-H.; Lin, M.C. In Silico Identification and In Vitro and In Vivo Validation of Anti-Psychotic Drug Fluspirilene as a Potential CDK2 Inhibitor and a Candidate Anti-Cancer Drug. PLoS ONE 2015, 10, e0132072. [Google Scholar] [CrossRef]
- Zia, K.; Ashraf, S.; Jabeen, A.; Saeed, M.; Nur-e-Alam, M.; Ahmed, S.; Al-Rehaily, A.J.; Ul-Haq, Z. Identification of Potential TNF-α Inhibitors: From in Silico to in Vitro Studies. Sci. Rep. 2020, 10, 20974. [Google Scholar] [CrossRef]
- Blanco-Gonzalez, A.; Cabezon, A.; Seco-Gonzalez, A.; Conde-Torres, D.; Antelo-Riveiro, P.; Pineiro, A.; Garcia-Fandino, R. The Role of AI in Drug Discovery: Challenges, Opportunities, and Strategies. Pharmaceuticals 2023, 16, 891. [Google Scholar] [CrossRef]
- Mak, K.-K.; Pichika, M.R. Artificial Intelligence in Drug Development: Present Status and Future Prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef]
- Farghali, H.; Canová, N.K.; Arora, M. The Potential Applications of Artificial Intelligence in Drug Discovery and Development. Physiol. Res. 2021, 70, S715. [Google Scholar] [CrossRef]
- Kang, L.; Gao, X.-H.; Liu, H.-R.; Men, X.; Wu, H.-N.; Cui, P.-W.; Oldfield, E.; Yan, J.-Y. Structure–Activity Relationship Investigation of Coumarin–Chalcone Hybrids with Diverse Side-Chains as Acetylcholinesterase and Butyrylcholinesterase Inhibitors. Mol. Divers. 2018, 22, 893–906. [Google Scholar] [CrossRef]
- Zeki, N.M.; Mustafa, Y.F. Digital Alchemy: Exploring the Pharmacokinetic and Toxicity Profiles of Selected Coumarin-Heterocycle Hybrids. Results Chem. 2024, 10, 101754. [Google Scholar] [CrossRef]
- Al Azzam, K.M.; Negim, E.-S.; Aboul-Enein, H.Y. ADME Studies of TUG-770 (a GPR-40 Agonist) for the Treatment of Type 2 Diabetes Using SwissADME Predictor: In Silico Study. J. Appl. Pharm. Sci. 2022, 12, 159–169. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2024, 52, W513–W520. [Google Scholar] [CrossRef]
- Rampogu, S.; Lee, K.W. Pharmacophore Modelling-Based Drug Repurposing Approaches for SARS-CoV-2 Therapeutics. Front. Chem. 2021, 9, 38. [Google Scholar] [CrossRef]
- Rampogu, S.; Lee, G.; Park, J.S.; Lee, K.W.; Kim, M.O. Molecular Docking and Molecular Dynamics Simulations Discover Curcumin Analogue as a Plausible Dual Inhibitor for SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 1771. [Google Scholar] [CrossRef] [PubMed]
- Głowacki, E.D.; Irimia-Vladu, M.; Bauer, S.; Sariciftci, N.S. Hydrogen-Bonds in Molecular Solids–from Biological Systems to Organic Electronics. J. Mater. Chem. B 2013, 1, 3742–3753. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.S.; Saikia, B.J. Cancer and Cure: A Critical Analysis. Indian J. Cancer 2016, 53, 441–442. [Google Scholar] [CrossRef]
- Dietrich, C.; Trub, A.; Ahn, A.; Taylor, M.; Ambani, K.; Chan, K.T.; Lu, K.-H.; Mahendra, C.A.; Blyth, C.; Coulson, R. INX-315, a Selective CDK2 Inhibitor, Induces Cell Cycle Arrest and Senescence in Solid Tumors. Cancer Discov. 2024, 14, 446–467. [Google Scholar] [CrossRef]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef]
- Gerosa, R.; De Sanctis, R.; Jacobs, F.; Benvenuti, C.; Gaudio, M.; Saltalamacchia, G.; Torrisi, R.; Masci, G.; Miggiano, C.; Agustoni, F.; et al. Cyclin-Dependent Kinase 2 (CDK2) Inhibitors and Others Novel CDK Inhibitors (CDKi) in Breast Cancer: Clinical Trials, Current Impact, and Future Directions. Crit. Rev. Oncol. Hematol. 2024, 196, 104324. [Google Scholar] [CrossRef]
- Martin, M.P.; Endicott, J.A.; Noble, M.E.M. Structure-Based Discovery of Cyclin-Dependent Protein Kinase Inhibitors. Essays Biochem. 2017, 61, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M. The Role of Artificial Intelligence in Pharmaceutical Drug Discovery. Am. J. Pharm. Pharmacol. 2020, 1, 1–6. [Google Scholar] [CrossRef]
- Abbas, M.K.G.; Rassam, A.; Karamshahi, F.; Abunora, R.; Abouseada, M. The Role of AI in Drug Discovery. Chembiochem 2024, 25, e202300816. [Google Scholar] [CrossRef]
- Mak, K.-K.; Wong, Y.-H.; Pichika, M.R. Artificial Intelligence in Drug Discovery and Development. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; Springer: Berlin/Heidelberg, Germany, 2024; pp. 1461–1498. [Google Scholar]
- Visan, A.I.; Negut, I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life 2024, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Elgawish, M.S.; Almatary, A.M.; Zaitone, S.A.; Salem, M.S.H. Leveraging Artificial Intelligence and Machine Learning in Kinase Inhibitor Development: Advances, Challenges, and Future Prospects. RSC Med. Chem. 2025, 16, 4698–4720. [Google Scholar] [CrossRef]
- Abdelbaky, I.; Tayara, H.; Chong, K.T. Prediction of Kinase Inhibitors Binding Modes with Machine Learning and Reduced Descriptor Sets. Sci. Rep. 2021, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.; Deifallah, M.A.; Zaghloul, A.; Elgohary, R. Artificial Intelligence Strategies for Predicting Kinase Inhibitor Resistance: A Comprehensive Review of Methods, Challenges, and Future Perspectives. J. Intell. Med. 2025, 3, 26–46. [Google Scholar] [CrossRef]
- Shahab, M.; Zheng, G.; Khan, A.; Wei, D.; Novikov, A.S. Machine Learning-Based Virtual Screening and Molecular Simulation Approaches Identified Novel Potential Inhibitors for Cancer Therapy. Biomedicines 2023, 11, 2251. [Google Scholar] [CrossRef]
- Veit-Acosta, M.; de Azevedo Junior, W.F. Computational Prediction of Binding Affinity for CDK2-Ligand Complexes. A Protein Target for Cancer Drug Discovery. Curr. Med. Chem. 2022, 29, 2438–2455. [Google Scholar] [CrossRef]
- Li, M.-M.; Song, M.; Wu, S.-X.; Ren, X.-Y. Identification of CDK2 as a Key Apoptotic Gene for Predicting Cervical Cancer Prognosis Using Bioinformatics and Machine Learning. Am. J. Cancer Res. 2025, 15, 2750. [Google Scholar] [CrossRef]
- Matore, B.W.; Murmu, A.; Roy, P.P.; Singh, J. Computational Exploration and Discovery of Dual EGFR-CDK2 Kinase Inhibitors: AI-ML Powered Bioisosteric Design, 3D QSAR, Docking, DFT and ADMET Analysis of Novel Phthalimide Derivatives. SAR QSAR Environ. Res. 2026, 37, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Solanki, P.; Abdul Amin, S.; Manhas, A. Integrating Machine Learning with in Silico Studies and Quantum Chemistry: Exploring Novel Compounds through Multiscale Screening Targeting the CDK2 Enzyme. Comput. Biol. Med. 2025, 196, 110712. [Google Scholar] [CrossRef]
- Olaharski, A.J.; Gonzaludo, N.; Bitter, H.; Goldstein, D.; Kirchner, S.; Uppal, H.; Kolaja, K. Identification of a Kinase Profile That Predicts Chromosome Damage Induced by Small Molecule Kinase Inhibitors. PLoS Comput. Biol. 2009, 5, e1000446. [Google Scholar] [CrossRef]
- Ali, M.A.; Sarker, H.; Khan, T.; Sheikh, H.; Saif, A.; Farid, F.B.; Afrin, S.; Khatun, M.A.; Kumar, N. Multi-Omics Pan-Cancer Profiling of CDK2 and in Silico Identification of Plant-Derived Inhibitors Using Machine Learning Approaches. RSC Adv. 2025, 15, 36938–36968. [Google Scholar] [CrossRef]
- Sułek, A.; Klimczak, J.; Jończyk, J.; Kosciolek, T.; Danel, T.; Pucelik, B. Explainable Artificial Intelligence for Bioactivity Prediction: Unveiling the Challenges with Curated CDK2/4/6 Breast Cancer Dataset. In Proceedings of the International Conference on Computational Science; Springer: Berlin/Heidelberg, Germany, 2025; pp. 18–32. [Google Scholar]
- Pehlivan, S.N.; da Silva, A.D.; de Azevedo, W.F., Jr. Combining MVD and Ridge Method to Predict CDK2 Inhibition. In Docking Screens for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2025; pp. 35–49. [Google Scholar]
- Ikhsanurahman, R.Y.; Ikhsan, N.; Kurniawan, I. Classification of Cdk2 Inhibitor as Anti-Cancer Agent by Using Simulated Annealing-Support Vector Machine Methods. In Proceedings of the 2022 International Conference on Data Science and Its Applications (ICoDSA), Bandung, Indonesia, 6–7 July 2022; pp. 82–86. [Google Scholar]
- Bijral, R.K.; Singh, I.; Manhas, J.; Sharma, V.; Sharma, V. Exploring Computational Methods to Identify Inhibitors for CDK2 Kinase. Res. Sq. 2024, 58. [Google Scholar] [CrossRef]
- VA, A.K.; Mohan, K.; Riyaz, S. Structure Guided Inhibitor Designing of CDK2 and Discovery of Potential Leads against Cancer. J. Mol. Model. 2013, 19, 3581–3589. [Google Scholar] [CrossRef] [PubMed]
- de Ávila, M.B.; Xavier, M.M.; Pintro, V.O.; de Azevedo, W.F., Jr. Supervised Machine Learning Techniques to Predict Binding Affinity. A Study for Cyclin-Dependent Kinase 2. Biochem. Biophys. Res. Commun. 2017, 494, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, T.; Tachikawa, M. Collaborative Approach between Explainable Artificial Intelligence and Simplified Chemical Interactions to Explore Active Ligands for Cyclin-Dependent Kinase 2. ACS Omega 2022, 7, 10372–10381. [Google Scholar] [CrossRef]
- Belenahalli Shekarappa, S.; Kandagalla, S.; Lee, J. Development of Machine Learning Models Based on Molecular Fingerprints for Selection of Small Molecule Inhibitors against JAK2 Protein. J. Comput. Chem. 2023, 44, 1493–1504. [Google Scholar] [CrossRef]
- Liu, Y.; Bi, M.; Zhang, X.; Zhang, N.; Sun, G.; Zhou, Y.; Zhao, L.; Zhong, R. Machine Learning Models for the Classification of CK2 Natural Products Inhibitors with Molecular Fingerprint Descriptors. Processes 2021, 9, 2074. [Google Scholar] [CrossRef]
- Tondar, A.; Sánchez-Herrero, S.; Bepari, A.K.; Bahmani, A.; Calvet Liñán, L.; Hervás-Marín, D. Virtual Screening of Small Molecules Targeting BCL2 with Machine Learning, Molecular Docking, and MD Simulation. Biomolecules 2024, 14, 544. [Google Scholar] [CrossRef]
- Liang, J.-W.; Wang, M.-Y.; Wang, S.; Li, S.-L.; Li, W.-Q.; Meng, F.-H. Identification of Novel CDK2 Inhibitors by a Multistage Virtual Screening Method Based on SVM, Pharmacophore and Docking Model. J. Enzym. Inhib. Med. Chem. 2020, 35, 235–244. [Google Scholar] [CrossRef]
- Zhang, G.; Ren, Y. Molecular Modeling and Design Studies of Purine Derivatives as Novel CDK2 Inhibitors. Molecules 2018, 23, 2924. [Google Scholar] [CrossRef]
- Adasme-Carreño, F.; Muñoz-Gutierrez, C.; Caballero, J.; Alzate-Morales, J.H. Performance of the MM/GBSA Scoring Using a Binding Site Hydrogen Bond Network-Based Frame Selection: The Protein Kinase Case. Phys. Chem. Chem. Phys. 2014, 16, 14047–14058. [Google Scholar] [CrossRef]
- Omran, H.A.; Majed, A.A.; Hussein, K.; Abid, D.S.; Abdel-Maksoud, M.A.; Elwahsh, A.; Aufy, M.; Kotob, M.H. Anti-Cancer Activity, DFT and Molecular Docking Study of New BisThiazolidine Amide. Results Chem. 2024, 12, 101835. [Google Scholar] [CrossRef]
- Elgogary, S.R.; Khidre, R.E.; El-Telbani, E.M. Regioselective Synthesis and Evaluation of Novel Sulfonamide 1, 2, 3-Triazole Derivatives as Antitumor Agents. J. Iran. Chem. Soc. 2020, 17, 765–776. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S. Inhibition of Cyclin-dependent Kinases by Purine Analogues: Crystal Structure of Human Cdk2 Complexed with Roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Dinaciclib, a Novel CDK Inhibitor, Demonstrates Encouraging Single-Agent Activity in Patients with Relapsed Multiple Myeloma. Blood J. Am. Soc. Hematol. 2015, 125, 443–448. [Google Scholar] [CrossRef]
- Brooks, E.E.; Gray, N.S.; Joly, A.; Kerwar, S.S.; Lum, R.; Mackman, R.L.; Norman, T.C.; Rosete, J.; Rowe, M.; Schow, S.R. CVT-313, a Specific and Potent Inhibitor of CDK2 That Prevents Neointimal Proliferation. J. Biol. Chem. 1997, 272, 29207–29211. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cyclin-Dependent Kinases: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Copeland, R.A.; Walsh, C. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; Wiley Online Library: Hoboken, NJ, USA, 2005; Volume 46, ISBN 0471686964. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. CDK Inhibitors: Positive and Negative Regulators of G1-Phase Progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Son, M.; Park, C.; Yoon, S.; Parate, S.; Lee, K.W. Discovery of Lonafarnib-like Compounds: Pharmacophore Modeling and Molecular Dynamics Studies. ACS Omega 2020, 5, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Miebs, G.; Mielniczuk, A.; Kadziński, M.; Bachorz, R.A. Beyond the Arbitrariness of Drug-Likeness Rules: Rough Set Theory and Decision Rules in the Service of Drug Design. Appl. Sci. 2024, 14, 9966. [Google Scholar] [CrossRef]
- Rampogu, S.; Jung, T.S.; Ha, M.W.; Lee, K.W. Repurposing and Computational Design of PARP Inhibitors as SARS-CoV-2 Inhibitors. Sci. Rep. 2023, 13, 10583. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Lee, K.W. Old Drugs for New Purpose—Fast Pace Therapeutic Identification for SARS-CoV-2 Infections by Pharmacophore Guided Drug Repositioning Approach. Bull. Korean Chem. Soc. 2021, 42, 212–226. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Fan, Y.-W.; Liu, W.-H.; Chen, Y.-T.; Hsu, Y.-C.; Pathak, N.; Huang, Y.-W.; Yang, J.-M. Exploring Kinase Family Inhibitors and Their Moiety Preferences Using Deep SHapley Additive ExPlanations. BMC Bioinform. 2022, 23, 242. [Google Scholar] [CrossRef]
- Hermansyah, O.; Rahmawati, S.; Dwi Putri Masrijal, C.; Intan Perma Sari, R. Identification of DPP-4 Inhibitor Active Compounds Using Machine Learning Classification. Can. Int. J. Chem. Biochem. Sci. 2023, 24, 674–681. [Google Scholar]
- Yang, R.; Zha, X.; Gao, X.; Wang, K.; Cheng, B.; Yan, B. Multi-Stage Virtual Screening of Natural Products against P38α Mitogen-Activated Protein Kinase: Predictive Modeling by Machine Learning, Docking Study and Molecular Dynamics Simulation. Heliyon 2022, 8, e10495. [Google Scholar] [CrossRef]
- Egieyeh, S.; Syce, J.; Malan, S.F.; Christoffels, A. Predictive Classifier Models Built from Natural Products with Antimalarial Bioactivity Using Machine Learning Approach. PLoS ONE 2018, 13, e0204644. [Google Scholar] [CrossRef]
- Pereira, R.; Silva, A.M.S.; Ribeiro, D.; Silva, V.L.M.; Fernandes, E. Bis-Chalcones: A Review of Synthetic Methodologies and Anti-Inflammatory Effects. Eur. J. Med. Chem. 2023, 252, 115280. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Kuwahara, H.; Gao, X. Analysis of the Effects of Related Fingerprints on Molecular Similarity Using an Eigenvalue Entropy Approach. J. Cheminform. 2021, 13, 27. [Google Scholar] [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.-L. One Molecular Fingerprint to Rule Them All: Drugs, Biomolecules, and the Metabolome. J. Cheminform. 2020, 12, 43. [Google Scholar] [CrossRef]
- Boldini, D.; Ballabio, D.; Consonni, V.; Todeschini, R.; Grisoni, F.; Sieber, S.A. Effectiveness of Molecular Fingerprints for Exploring the Chemical Space of Natural Products. J. Cheminform. 2024, 16, 35. [Google Scholar] [CrossRef]
- Yuan, X.; Li, L.; Shi, Z.; Liang, H.; Li, S.; Qiao, Z. Molecular-Fingerprint Machine-Learning-Assisted Design and Prediction for High-Performance MOFs for Capture of NMHCs from Air. Adv. Powder Mater. 2022, 1, 100026. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; Volume 4, pp. 217–241. ISBN 1574-1400. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef]
- Pandya, V.J. Comparing Handwritten Character Recognition by AdaBoostClassifier and KNeighborsClassifier. In Proceedings of the 2016 8th International Conference on Computational Intelligence and Communication Networks (CICN), Tehri, India, 23–25 December 2016; pp. 271–274. [Google Scholar]
- Maulud, D.; Abdulazeez, A.M. A Review on Linear Regression Comprehensive in Machine Learning. J. Appl. Sci. Technol. Trends 2020, 1, 140–147. [Google Scholar] [CrossRef]
- Song, Y.-Y.; Lu, Y. Decision Tree Methods: Applications for Classification and Prediction. Shanghai Arch. Psychiatry 2015, 27, 130–135. [Google Scholar] [CrossRef]
- Suthaharan, S.; Suthaharan, S. Decision Tree Learning. In Machine Learning Models and Algorithms for Big Data Classification: Thinking with Examples for Effective Learning; Springer: Boston, MA, USA, 2016; pp. 237–269. [Google Scholar]
- Biau, G.; Scornet, E. A Random Forest Guided Tour. TEST 2016, 25, 197–227. [Google Scholar] [CrossRef]
- Ramraj, S.; Uzir, N.; Sunil, R.; Banerjee, S. Experimenting XGBoost Algorithm for Prediction and Classification of Different Datasets. Int. J. Control Theory Appl. 2016, 9, 651–662. [Google Scholar]
- Ahmetoglu, H.; Das, R. A Comprehensive Review on Detection of Cyber-Attacks: Data Sets, Methods, Challenges, and Future Research Directions. Internet Things 2022, 20, 100615. [Google Scholar] [CrossRef]
- Liew, X.Y.; Hameed, N.; Clos, J. An Investigation of XGBoost-Based Algorithm for Breast Cancer Classification. Mach. Learn. Appl. 2021, 6, 100154. [Google Scholar] [CrossRef]
- Mahesh, T.R.; Geman, O.; Margala, M.; Guduri, M. The Stratified K-Folds Cross-Validation and Class-Balancing Methods with High-Performance Ensemble Classifiers for Breast Cancer Classification. Healthc. Anal. 2023, 4, 100247. [Google Scholar]
- Fontanari, T.; Fróes, T.C.; Recamonde-Mendoza, M. Cross-Validation Strategies for Balanced and Imbalanced Datasets. In Proceedings of the Brazilian Conference on Intelligent Systems; Springer: Berlin/Heidelberg, Germany, 2022; pp. 626–640. [Google Scholar]
- Patil, P.A.; Kumbhar, B.V. Structure Based Drug Design and Machine Learning Approaches for Identifying Natural Inhibitors against the Human AβIII Tubulin Isotype. Sci. Rep. 2025, 15, 32716. [Google Scholar] [CrossRef]
- Sosnina, E.A.; Sosnin, S.; Nikitina, A.A.; Nazarov, I.; Osolodkin, D.I.; Fedorov, M.V. Recommender Systems in Antiviral Drug Discovery. ACS Omega 2020, 5, 15039–15051. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chong, D.; Batarseh, F.A. Foundations of Data Imbalance and Solutions for a Data Democracy. In Data Democracy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 83–106. [Google Scholar]
- Hossin, M.; Sulaiman, M.N. A Review on Evaluation Metrics for Data Classification Evaluations. Int. J. Data Min. Knowl. Manag. Process 2015, 5, 1. [Google Scholar]
- Narkhede, S. Understanding Auc-Roc Curve. Towards Data Sci. 2018, 26, 220–227. [Google Scholar]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2014; pp. 243–250. [Google Scholar]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Docking-Based Virtual Screening Using PyRx Tool: Autophagy Target Vps34 as a Case Study. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 463–477. [Google Scholar]
- Shamsian, S.; Sokouti, B.; Dastmalchi, S. Benchmarking Different Docking Protocols for Predicting the Binding Poses of Ligands Complexed with Cyclooxygenase Enzymes and Screening Chemical Libraries. Bioimpacts 2023, 14, 29955. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein–Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rampogu, S.; Balasubramaniyam, T.; Lee, J.-H. Curcumin Chalcone Derivatives Database (CCDD): A Python Framework for Natural Compound Derivatives Database. PeerJ 2023, 11, e15885. [Google Scholar] [CrossRef]
- Rampogu, S.; Shaik, M.R.; Khan, M.; Khan, M.; Oh, T.H.; Shaik, B. CBPDdb: A Curated Database of Compounds Derived from Coumarin–Benzothiazole–Pyrazole. Database 2023, 2023, baad062. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-ADMET: A Compendium of Fingerprint-Based ADMET Prediction Models. J. Cheminform. 2021, 13, 75. [Google Scholar] [CrossRef]
- Rampogu, S. Structure-Based Pharmacophore Modeling, Molecular Docking and ADMET Analysis to Discover Potential HMPV Inhibitors. In Silico Res. Biomed. 2025, 1, 100031. [Google Scholar] [CrossRef]
- Price, G.; Patel, D.A. Drug Bioavailability. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557852/ (accessed on 20 January 2026).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Hit1 | Hit2 | Ref. |
|---|---|---|---|
| Molecular weight | 449.51 g/mol | 443.52 g/mol | [30] |
| No. of hydrogen bond donors | 2 | 2 | |
| No. of hydrogen bond acceptors | 5 | 5 | |
| Rotatable bonds | 8 | 7 | |
| Consensus LogP (1–3 recommended) | 3.6 | 2.85 | |
| Solubility (ESOL) | moderately soluble | moderately soluble | |
| BBB permeant | no | No | |
| CYP1A2 inhibitor | yes | Yes | |
| GI absorption | high | High | |
| Consensus LogPo/w | 3.68 | 2.85 | |
| Hepatotoxicity | inactive | Inactive | [31,32] |
| Bioavailability Score | 0.55 | 0.55 | [30] |
| 2D structures |  |  | - |
| Compound | Hydrogen Bond | Carbon Hydrogen Bond | Alkyl, π- Alkyl Interactions | van der Waals Interactions |
|---|---|---|---|---|
| ref | Glu12:HN-lig:O3 Gln131:OE1-lig:H2 | Ile10, Gly11, Glu12, Gly13, Lys33, The160 | Val18, Tyr159 | His161, Glu162 |
| hit1 | Gln131:HE21-lig:O1 | Gln131 | Ile10, Val18, Ala31, Ala144, Leu134 | Gly11, Glu12, Glu18, Lys33, Val64, Phe80, Leu83, His84, Gln85, Phe82, Asn132, Asp145 |
| hit2 | Asp86:OD2-lig:H2 Gln131:HE21-lig:O1 | Gln85 | Ile10, Val18, Ala31, Phe80 | Gly11, Glu12, Gly13, Val64, Glu81, Phe82, Leu83, His84, Ala144, Asp145 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Rampogu, S.; Balasubramaniyam, T.; Kubiak, J.Z.; Lee, K.W. Machine Learning and Virtual Screening Methods to Discover Potential Cyclin-Dependent Kinase 2 (CDK2) Inhibitors. Pharmaceuticals 2026, 19, 1019. https://doi.org/10.3390/ph19071019
Rampogu S, Balasubramaniyam T, Kubiak JZ, Lee KW. Machine Learning and Virtual Screening Methods to Discover Potential Cyclin-Dependent Kinase 2 (CDK2) Inhibitors. Pharmaceuticals. 2026; 19(7):1019. https://doi.org/10.3390/ph19071019
Chicago/Turabian StyleRampogu, Shailima, Thananjeyan Balasubramaniyam, Jacek Z. Kubiak, and Keun Woo Lee. 2026. "Machine Learning and Virtual Screening Methods to Discover Potential Cyclin-Dependent Kinase 2 (CDK2) Inhibitors" Pharmaceuticals 19, no. 7: 1019. https://doi.org/10.3390/ph19071019
APA StyleRampogu, S., Balasubramaniyam, T., Kubiak, J. Z., & Lee, K. W. (2026). Machine Learning and Virtual Screening Methods to Discover Potential Cyclin-Dependent Kinase 2 (CDK2) Inhibitors. Pharmaceuticals, 19(7), 1019. https://doi.org/10.3390/ph19071019