
Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties

 ,
,  , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
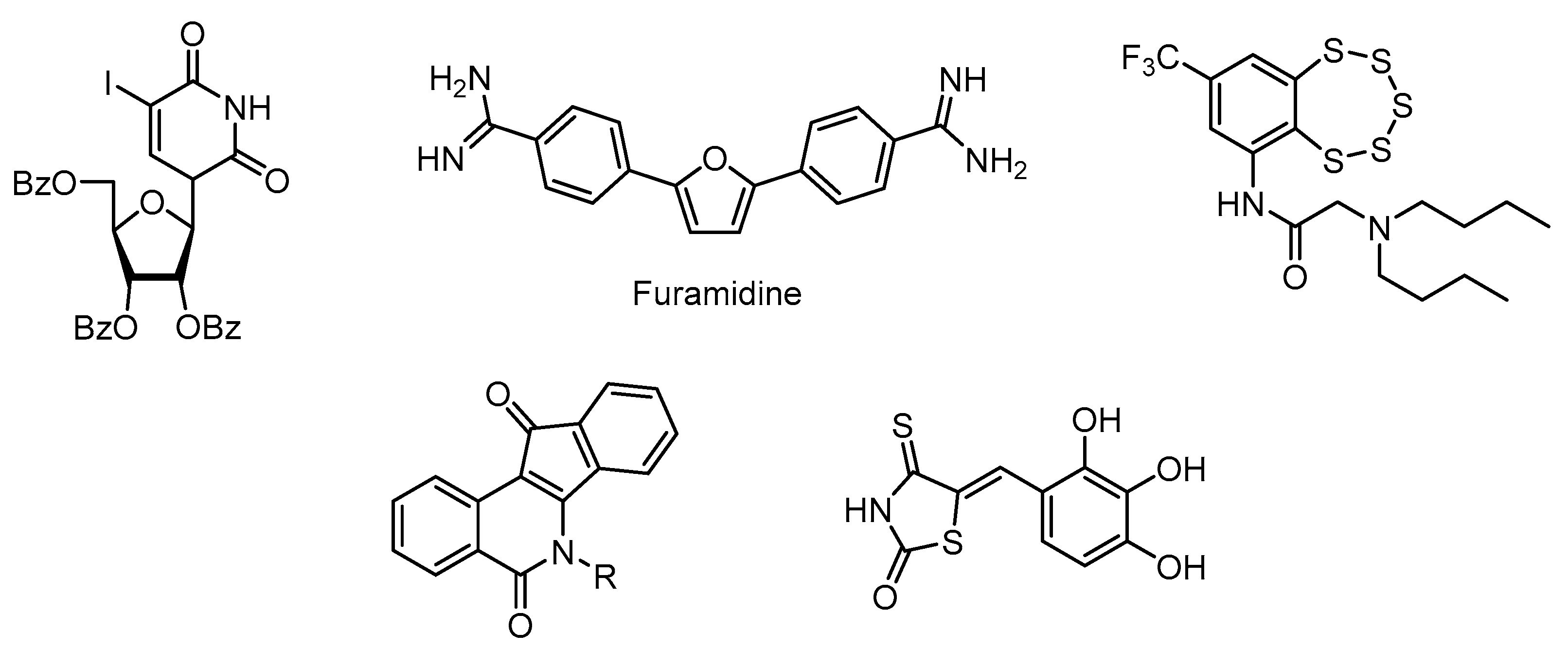
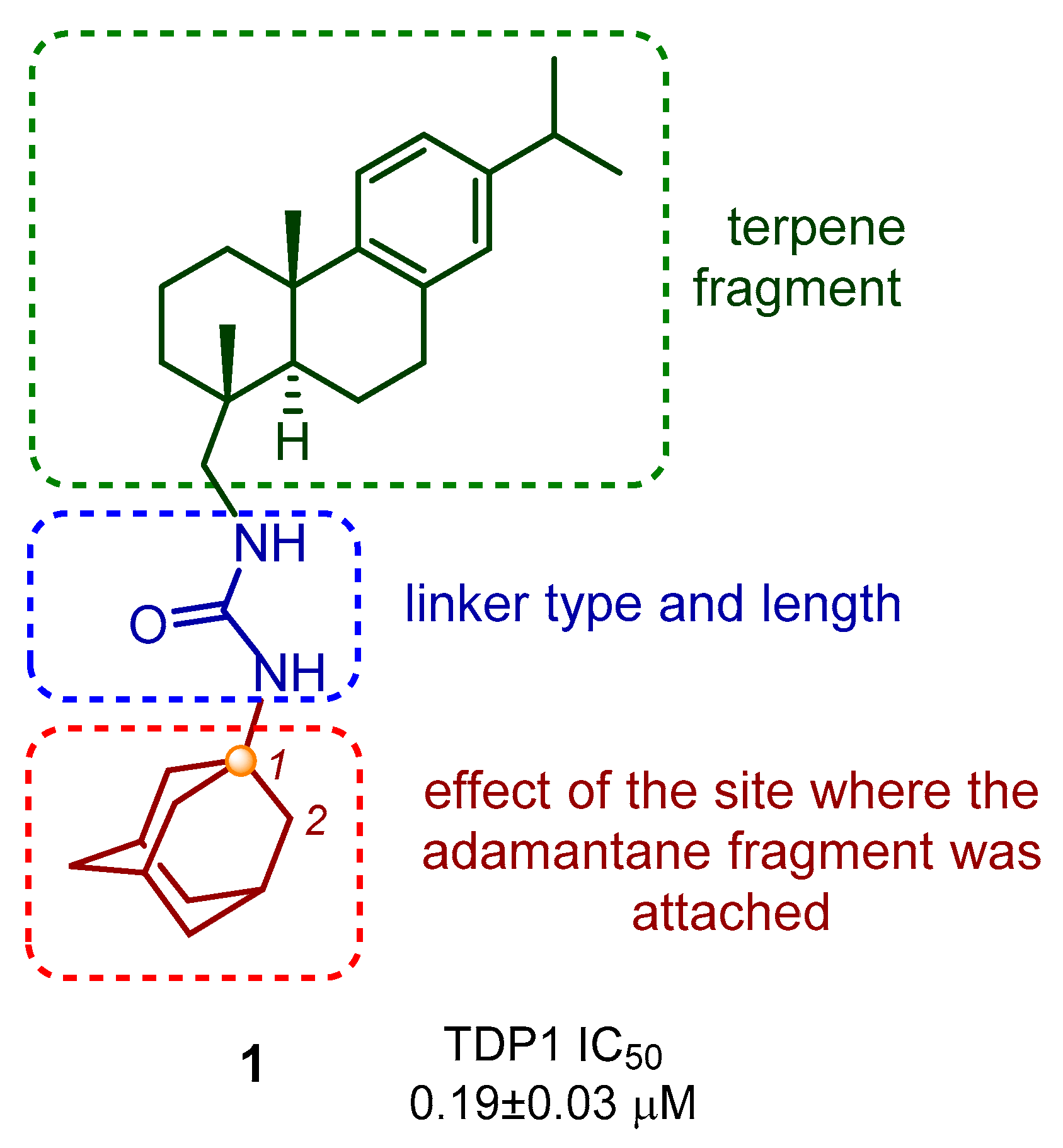
2. Results and Discussion
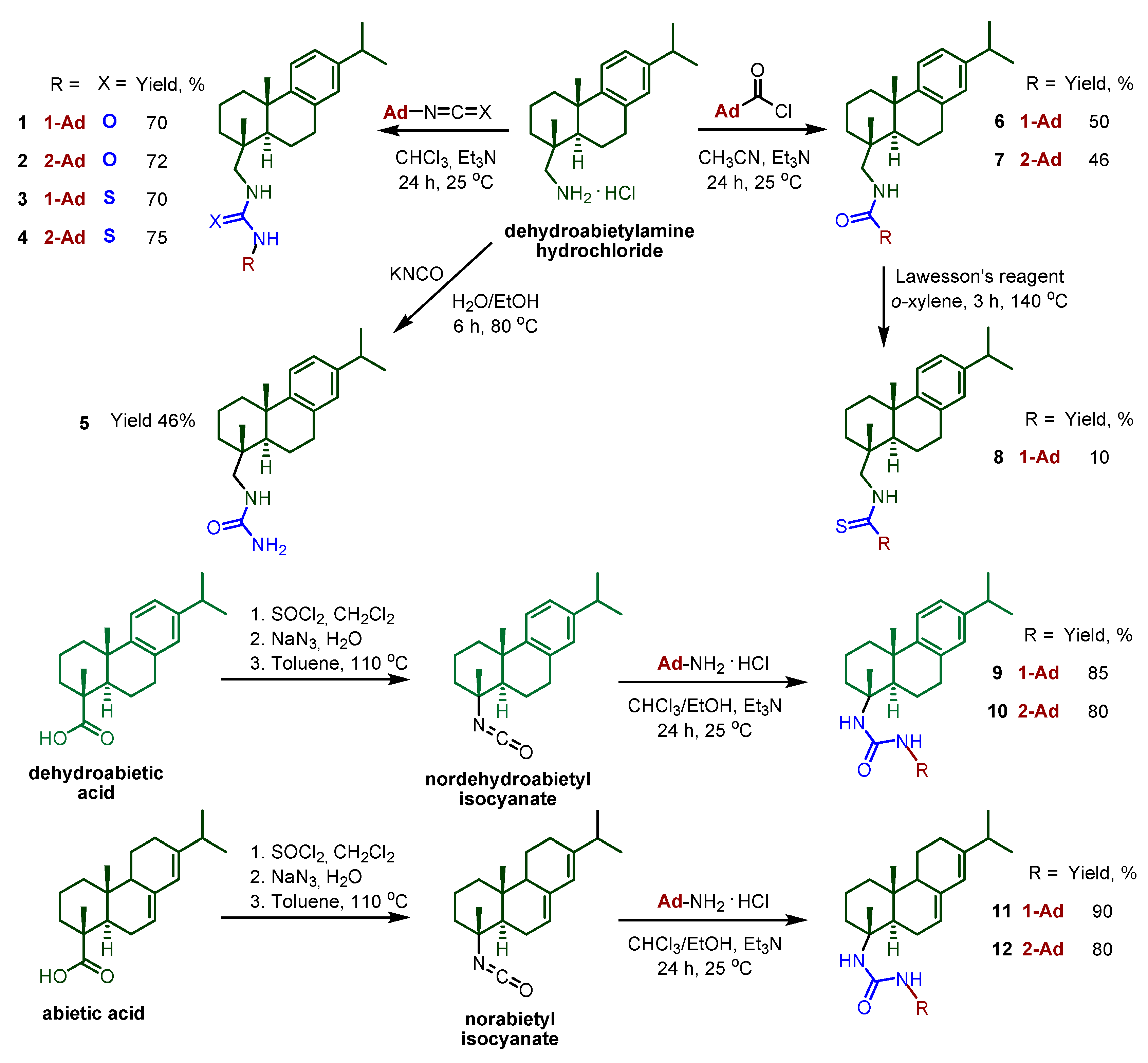
2.1. Chemistry
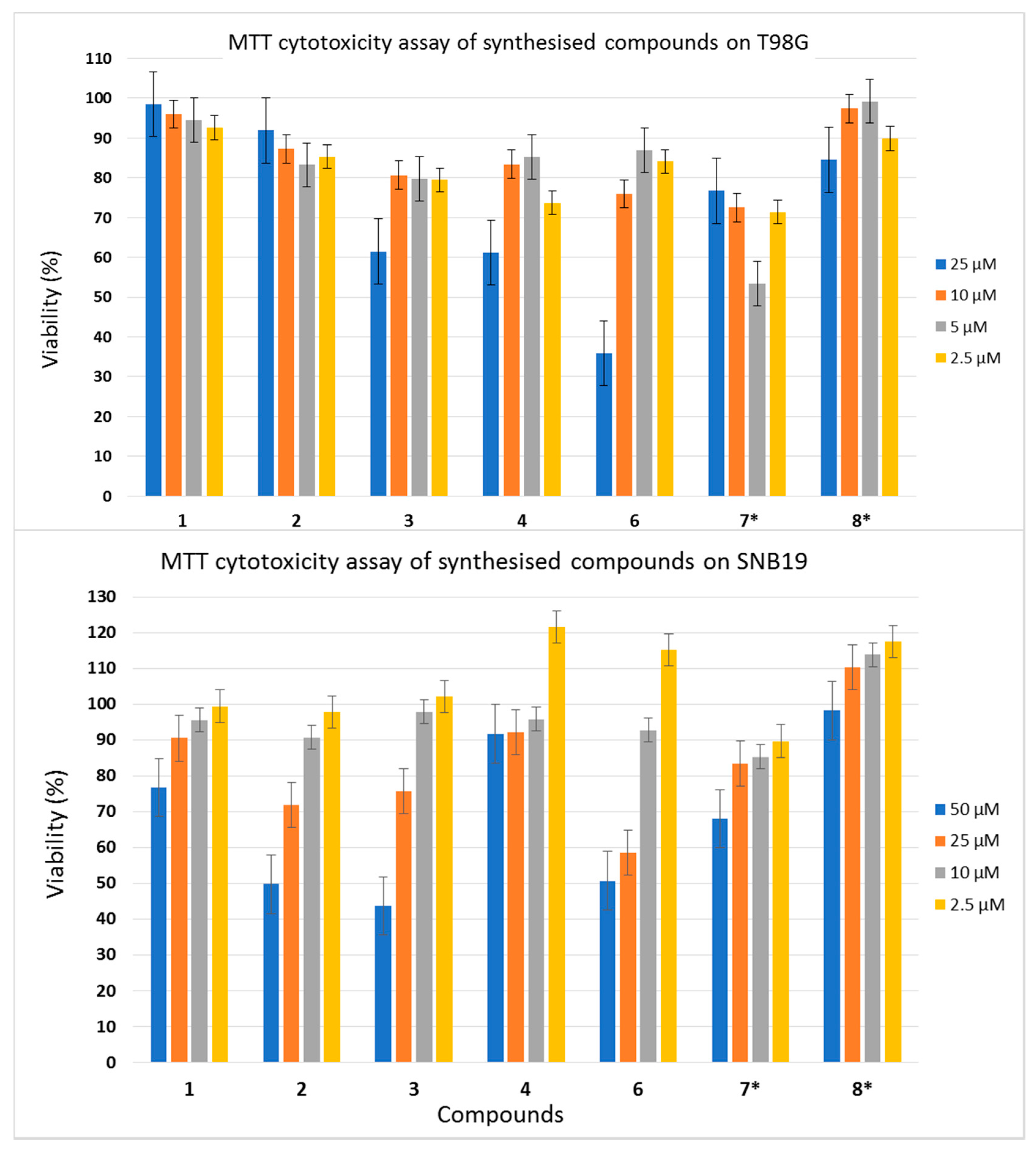
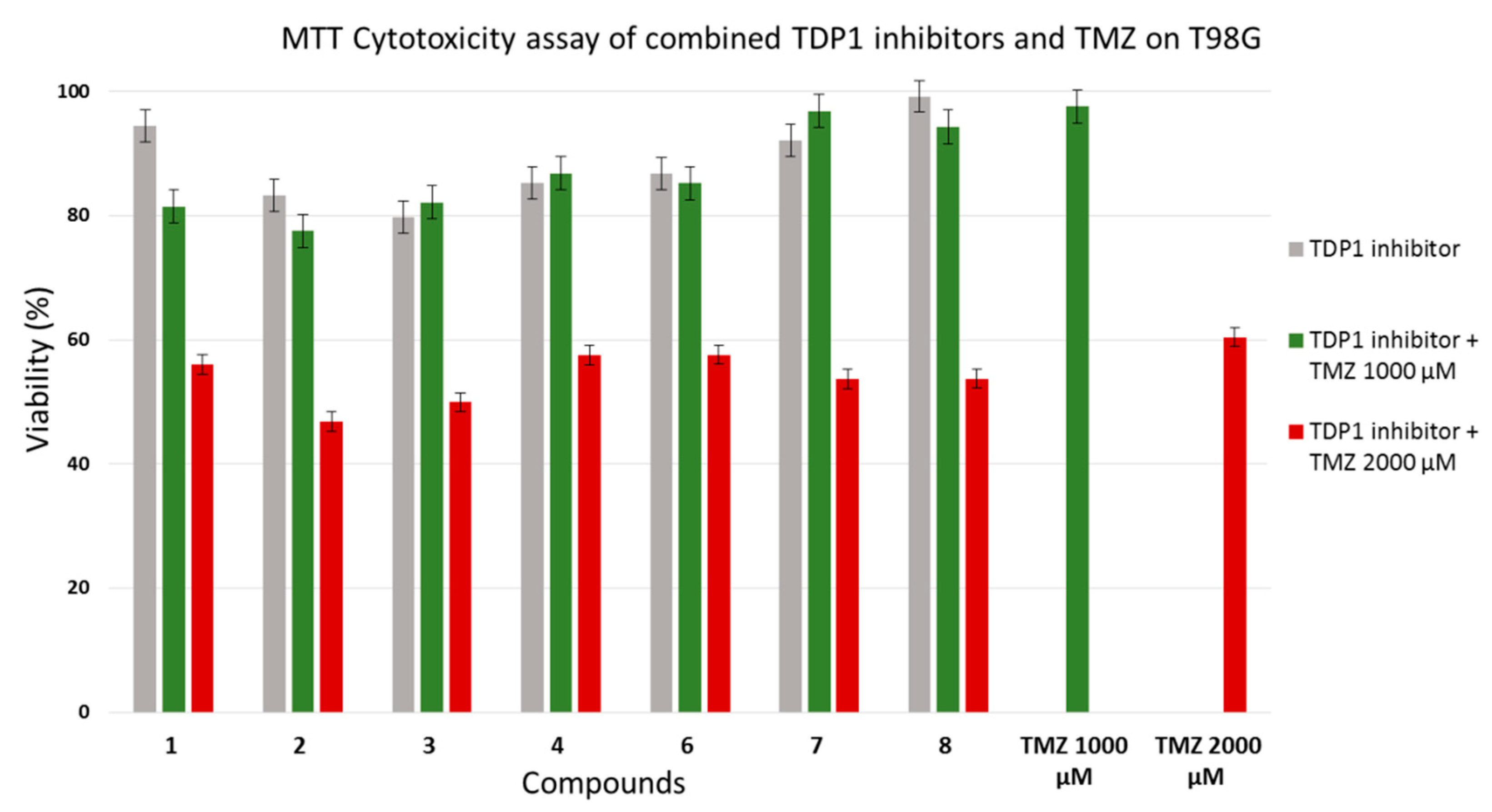
2.2. TDP1 Assay and Cytotoxicity Studies
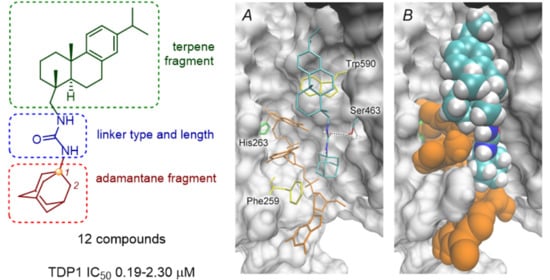
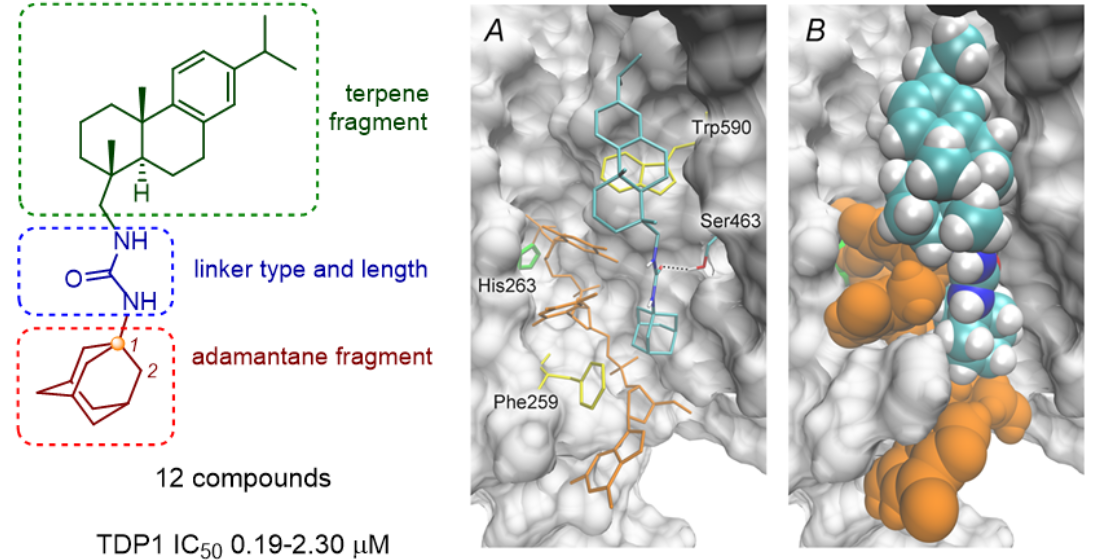
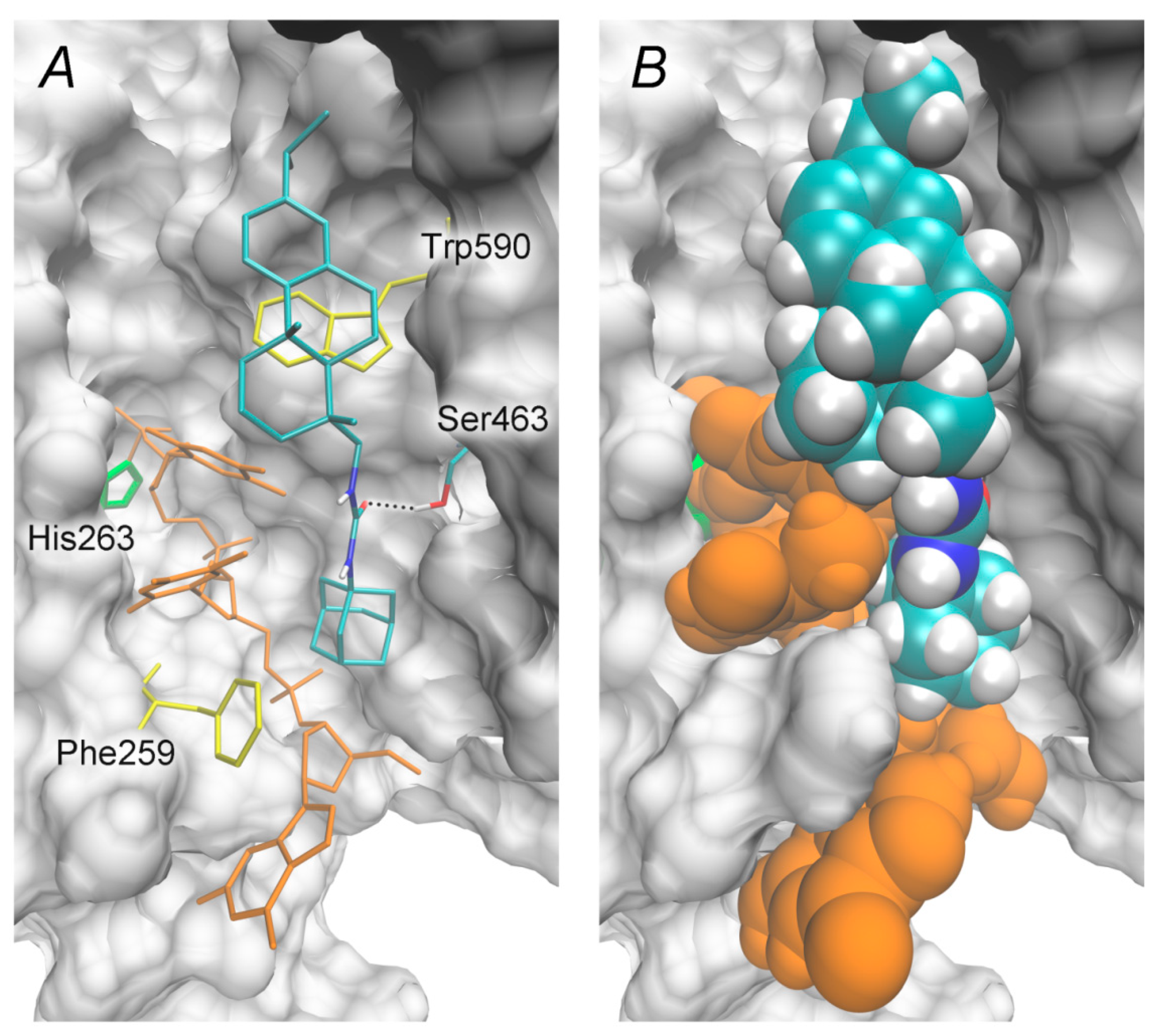
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Ureas and Thioureas 1–4
3.1.2. Synthesis of Urea 5
3.1.3. General Procedure for the Synthesis of Amides 6–7
3.1.4. Synthesis of Thioamide 8
3.1.5. General Procedure for the Synthesis of Norabietyl and Nordehydroabietyl Ureas
3.2. TDP1 Assay
3.3. Cytotoxicity Experiments
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.G.; Patel, B.D. Medicinal chemistry approaches of poly ADP-Ribose polymerase 1 (PARP1) inhibitors as anticancer—A recent update. Eur. J. Med. Chem. 2019, 165, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorganic Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA topoisomerases as molecular targets for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Boccard, S.G.; Marand, S.V.; Geraci, S.; Pycroft, L.; Berger, F.R.; Pelletier, L.A. Inhibition of DNA-repair genes Ercc1 and Mgmt enhances temozolomide efficacy in gliomas treatment: A pre-clinical study. Oncotarget 2015, 6, 29456–29468. [Google Scholar] [CrossRef]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.M.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I–dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA Repair and Neurodegenerative Disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Drenichev, M.S.; Dyrkheeva, N.S.; Ivanov, G.A.; Oslovsky, V.E.; Ilina, E.S.; Chernyshova, I.A.; Lavrik, O.I.; Mikhailov, S.N. Inhibition of Tyrosyl-DNA Phosphodiesterase 1 by Lipophilic Pyrimidine Nucleosides. Molecules 2020, 25, 3694. [Google Scholar] [CrossRef]
- Antony, S.; Marchand, C.; Stephen, A.G.; Thibaut, L.; Agama, K.K.; Fisher, R.J.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [Google Scholar] [CrossRef]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Beck, D.E.; Reddy, P.V.N.; Lv, W.; Abdelmalak, M.; Tender, G.S.; Lopez, S.; Agama, K.; Marchand, C.; Pommier, Y.; Cushman, M. Investigation of the Structure–Activity Relationships of Aza-A-Ring Indenoisoquinoline Topoisomerase I Poisons. J. Med. Chem. 2015, 59, 3840–3853. [Google Scholar] [CrossRef]
- Sirivolu, V.R.; Vernekar, S.K.V.; Marchand, C.; Naumova, A.; Chergui, A.; Renaud, A.; Stephen, A.G.; Chen, F.; Sham, Y.Y.; Pommier, Y.; et al. 5-Arylidenethioxothiazolidinones as Inhibitors of Tyrosyl–DNA Phosphodiesterase I. J. Med. Chem. 2012, 55, 8671–8684. [Google Scholar] [CrossRef] [PubMed]
- Salakhutdinov, N.F.; Volcho, K.P.; Yarovaya, O.I. Monoterpenes as a renewable source of biologically active compounds. Pure Appl. Chem. 2017, 89, 1105–1117. [Google Scholar] [CrossRef]
- Luzina, O.; Filimonov, A.; Zakharenko, A.; Chepanova, A.; Zakharova, O.; Ilina, E.; Dyrkheeva, N.; Likhatskaya, G.; Salakhutdinov, N.; Lavrik, O. Usnic Acid Conjugates with Monoterpenoids as Potent Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. J. Nat. Prod. 2020, 83, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising New Inhibitors of Tyrosyl-DNA Phosphodiesterase I (Tdp 1) Combining 4-Arylcoumarin and Monoterpenoid Moieties as Components of Complex Antitumor Therapy. Int. J. Mol. Sci. 2019, 21, 126. [Google Scholar] [CrossRef] [PubMed]
- Salomatina, O.V.; Popadyuk, I.I.; Zakharenko, A.L.; Zakharova, O.D.; Chepanova, A.A.; Dyrkheeva, N.; Komarova, N.I.; Reynisson, J.; Anarbaev, R.O.; Salakhutdinov, N.F.; et al. Deoxycholic acid as a molecular scaffold for tyrosyl-DNA phosphodiesterase 1 inhibition: A synthesis, structure–activity relationship and molecular modeling study. Steroids 2021, 165, 108771. [Google Scholar] [CrossRef] [PubMed]
- Mozhaitsev, E.S.; Zakharenko, A.L.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Vasil’eva, I.A.; Chepanova, A.A.; Black, E.; Patel, J.; Chand, R.; et al. Novel Inhibitors of DNA Repair Enzyme TDP1 Combining Monoterpenoid and Adamantane Fragments. Anti-Cancer Agents Med. Chem. 2019, 19, 463–472. [Google Scholar] [CrossRef]
- Kovaleva, K.; Oleshko, O.; Mamontova, E.; Yarovaya, O.; Zakharova, O.; Zakharenko, A.; Kononova, A.; Dyrkheeva, N.; Cheresiz, S.; Pokrovsky, A.; et al. Dehydroabietylamine Ureas and Thioureas as Tyrosyl-DNA Phosphodiesterase 1 Inhibitors That Enhance the Antitumor Effect of Temozolomide on Glioblastoma Cells. J. Nat. Prod. 2019, 82, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, K.; Mamontova, E.; Yarovaya, O.; Zakharova, O.; Zakharenko, A.; Lavrik, O.; Salakhutdinov, N. Dehydroabietylamine-based thiazolidin-4-ones and 2-thioxoimidazolidin-4-ones as novel tyrosyl-DNA phosphodiesterase 1 inhibitors. Mol. Divers. 2020, 1–9. [Google Scholar] [CrossRef]
- Chepanova, A.A.; Mozhaitsev, E.S.; Munkuev, A.A.; Suslov, E.V.; Korchagina, D.V.; Zakharova, O.D.; Zakharenko, A.L.; Patel, J.; Ayine-Tora, D.M.; Reynisson, J.; et al. The Development of Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Combination of Monoterpene and Adamantine Moieties via Amide or Thioamide Bridges. Appl. Sci. 2019, 9, 2767. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Ponomarev, K.U.; Suslov, E.V.; Korchagina, D.V.; Volcho, K.P.; Vasil’Eva, I.A.; Salakhutdinov, N.F.; Lavrik, O.I. Inhibitory properties of nitrogen-containing adamantane derivatives with monoterpenoid fragments against tyrosyl-DNA phosphodiesterase 1. Russ. J. Bioorg. Chem. 2015, 41, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, K.Y.; Suslov, E.V.; Zakharenko, A.L.; Zakharova, O.D.; Rogachev, A.D.; Korchagina, D.V.; Zafar, A.; Reynisson, J.; Nefedov, A.A.; Volcho, K.P.; et al. Aminoadamantanes containing monoterpene-derived fragments as potent tyrosyl-DNA phosphodiesterase 1 inhibitors. Bioorg. Chem. 2018, 76, 392–399. [Google Scholar] [CrossRef]
- Kugler, S.; Ossowicz, P.; Malarczyk-Matusiak, K.; Wierzbicka, E. Advances in Rosin-Based Chemicals: The Latest Recipes, Applications and Future Trends. Molecules 2019, 24, 1651. [Google Scholar] [CrossRef] [PubMed]
- Butov, G.M.; Burmistrov, V.; Saad, K.R. Synthesis of Adamantyl-Containing Compounds: Structure Elements of Rotaxanes and Supramolecular Polymers. Mod. Org. Chem. Res. 2017, 4, 1910–1914. [Google Scholar] [CrossRef]
- Claremon, D.A.; Zhuang, L.; Ye, Y.; Singh, S.B.; Tice, C.M. Carbamate and urea inhibitors of 11beta-hydroxysteroid dehydrogenase. Patent Number WO2009131669A2, 29 October 2009. [Google Scholar]
- Burmistrov, V.V.; Butov, G.M.; Pitushkin, D.A. Synthesis of adamantyl-containing isothiocyanates. Russ. J. Org. Chem. 2015, 51, 1795–1796. [Google Scholar] [CrossRef]
- Pitushkin, D.A.; Burmistrov, V.V.; Butov, G.M. Synthesis of Homologs of 1-Isothiocyanatoadamantane. Russ. J. Org. Chem. 2018, 54, 1475–1479. [Google Scholar] [CrossRef]
- Zeiss, H.H.; Martin, W.B. Synthesis and Stereochemistry of the 3-Keto-Δ4-steroidal System from Diterpenic Acids1a,b. J. Am. Chem. Soc. 1953, 75, 5935–5940. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445. [Google Scholar] [CrossRef] [PubMed]
- Francesconi, V.; Cichero, E.; Schenone, S.; Naesens, L.; Tonelli, M. Synthesis and Biological Evaluation of Novel (thio)semicarbazone-Based Benzimidazoles as Antiviral Agents against Human Respiratory Viruses. Molecules 2020, 25, 1487. [Google Scholar] [CrossRef]
- Pallicer, J.M.; Rosés, M.; Ràfols, C.; Bosch, E.; Pascual, R.; Port, A. Evaluation of log Po/w values of drugs from some molecular structure calculation softwares. ADMET DMPK 2014, 2, 107–114. [Google Scholar] [CrossRef]
- Gowda, R.; Madhunapantula, S.V.; Kuzu, O.F.; Sharma, A.; Robertson, G.P. Targeting Multiple Key Signaling Pathways in Melanoma Using Leelamine. Mol. Cancer Ther. 2014, 13, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, K.S.; Kononova, A.A.; Korobeynikov, A.V.; Cheresiz, S.V.; Zarubaev, V.V.; Shtro, A.A.; Orshanskaya, Y.R.; Yarovaya, O.I.; Pokrovsky, A.G.; Salakhutdinov, N.F. Cytotoxic and Antiviral Properties of Novel Dehydroabietylamine Salts. Med. Chem. 2016, 6, 642–646. [Google Scholar] [CrossRef]
- Chen, H.; Qiao, C.; Miao, T.T.; Li, A.L.; Wang, W.Y.; Gu, W. Synthesis and biological evaluation of novel N-(piperazin-1-yl)alkyl-1H-dibenzo[a,c]carbazole derivatives of dehydroabietic acid as potential MEK inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1544–1561. [Google Scholar] [CrossRef]
- Davies, U.R.; Interthal, H.; Champoux, J.J.; Hol, W.G.J. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure 2002, 10, 237–248. [Google Scholar] [CrossRef]
- Gajewski, S.; Comeaux, E.Q.; Jafari, N.; Bharatham, N.; Bashford, D.; White, S.W.; Van Waardenburg, R.C. Analysis of the Active-Site Mechanism of Tyrosyl-DNA Phosphodiesterase I: A Member of the Phospholipase D Superfamily. J. Mol. Biol. 2012, 415, 741–758. [Google Scholar] [CrossRef]
- DeYonker, N.J.; Webster, C.E. A Theoretical Study of Phosphoryl Transfers of Tyrosyl-DNA Phosphodiesterase I (Tdp1) and the Possibility of a “Dead-End” Phosphohistidine Intermediate. Biochemistry 2015, 54, 4236–4247. [Google Scholar] [CrossRef] [PubMed]
- Comeaux, E.Q.; Van Waardenburg, R.C.A.M. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Fujita, K.I.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 12234–12248. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; Lavrik, O.I. AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett. 2011, 585, 683–686. [Google Scholar] [CrossRef]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G. Crystal Structure of a Transition State Mimic for Tdp1 Assembled from Vanadate, DNA, and a Topoisomerase I-Derived Peptide. Chem. Biol. 2003, 10, 139–147. [Google Scholar] [CrossRef]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef] [PubMed]
- Gushchina, I.V.; Nilov, D.K.; Zakharenko, A.L.; Lavrik, O.I.; Švedas, V.K. Structure Modeling of Human TyrosylDNA Phosphodiesterase 1 and Screening for Its Inhibitors. Acta Nat. 2017, 9, 59–66. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead Finder: An Approach To Improve Accuracy of Protein-Ligand Docking, Binding Energy Estimation, and Virtual Screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Novikov, F.N.; Stroylov, V.S.; Zeifman, A.A.; Stroganov, O.V.; Kulkov, V.; Chilov, G.G. Lead Finder docking and virtual screening evaluation with Astex and DUD test sets. J. Comput. Mol. Des. 2012, 26, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound |  |  |  | IC50 (TDP1), μM | LogP |
|---|---|---|---|---|---|
| 1 | DGAAm |  | 1-Ad | 0.19 ± 0.03 | 7.50 |
| 2 | DGAAm |  | 2-Ad | 0.80 ± 0.20 | 7.31 |
| 3 | DGAAm |  | 1-Ad | 1.50 ± 0.50 | 7.60 |
| 4 | DGAAm |  | 2-Ad | 0.67 ± 0.19 | 7.38 |
| 5 | DGAAm |  | - | >15 | 5.45 |
| 6 | DGAAm |  | 1-Ad | 0.94 ± 0.18 | 7.48 |
| 7 | DGAAm |  | 2-Ad | 1.80 ± 0.40 | 7.34 |
| 8 | DGAAm |  | 1-Ad | 2.30 ± 0.80 | 8.28 |
| 9 | nor-DGAA |  | 1-Ad | 0.57 ± 0.16 | 7.19 |
| 10 | nor-DGAA |  | 2-Ad | 0.59 ± 0.15 | 7.27 |
| 11 | nor-AA |  | 1-Ad | 1.70 ± 0.60 | 6.72 |
| 12 | nor-AA |  | 2-Ad | 1.40 ± 0.30 | 6.30 |
| Furamidine | - | - | - | 1.20 ± 0.30 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovaleva, K.; Yarovaya, O.; Ponomarev, K.; Cheresiz, S.; Azimirad, A.; Chernyshova, I.; Zakharenko, A.; Konev, V.; Khlebnikova, T.; Mozhaytsev, E.; et al. Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties. Pharmaceuticals 2021, 14, 422. https://doi.org/10.3390/ph14050422
Kovaleva K, Yarovaya O, Ponomarev K, Cheresiz S, Azimirad A, Chernyshova I, Zakharenko A, Konev V, Khlebnikova T, Mozhaytsev E, et al. Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties. Pharmaceuticals. 2021; 14(5):422. https://doi.org/10.3390/ph14050422
Chicago/Turabian StyleKovaleva, Kseniya, Olga Yarovaya, Konstantin Ponomarev, Sergey Cheresiz, Amirhossein Azimirad, Irina Chernyshova, Alexandra Zakharenko, Vasily Konev, Tatiana Khlebnikova, Evgenii Mozhaytsev, and et al. 2021. "Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties" Pharmaceuticals 14, no. 5: 422. https://doi.org/10.3390/ph14050422
APA StyleKovaleva, K., Yarovaya, O., Ponomarev, K., Cheresiz, S., Azimirad, A., Chernyshova, I., Zakharenko, A., Konev, V., Khlebnikova, T., Mozhaytsev, E., Suslov, E., Nilov, D., Švedas, V., Pokrovsky, A., Lavrik, O., & Salakhutdinov, N. (2021). Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties. Pharmaceuticals, 14(5), 422. https://doi.org/10.3390/ph14050422