2019 FDA TIDES (Peptides and Oligonucleotides) Harvest
by
 and
and
Danah Al Shaer
1,2,†, 
Othman Al Musaimi
1,2,†,
Fernando Albericio
2,3,* and
Beatriz G. de la Torre
1,* 1
KRISP, School of Laboratory of Medicine and Medical Science, College of Health Sciences, University of KwaZulu-Natal, Durban 4001, South Africa
2
School of Chemistry and Physics, University of KwaZulu-Natal, Durban 4001, South Africa
3
CIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine and Department of Organic Chemistry, University of Barcelona, 08028 Barcelona, Spain
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Pharmaceuticals 2020, 13(3), 40; https://doi.org/10.3390/ph13030040
Submission received: 11 February 2020
/
Revised: 2 March 2020
/
Accepted: 3 March 2020
/
Published: 5 March 2020
(This article belongs to the Special Issue The Story of Successful Drugs and Recent FDA-Approved Molecules)
Abstract
:2019 has been an excellent year in terms of peptides and oligonucleotides (TIDES) approved by the FDA. Despite the drop in the number of total drugs approved by the FDA in 2019 in comparison with 2018 (48 vs. 59), the total number of TIDES authorized increased (seven vs. three). Year after year, TIDES are increasingly present in therapy, as imaging agents, theragnostic and constituent moieties of other complex drugs, such as antibody drug conjugates. This means a consolidation of these kinds of drugs in the pharmaceutical arena, paving the way in the coming years for the approval of others for diverse medical indications. Here the TIDES approved in 2019 are analyzed in terms of chemical structure, medical target, mode of action, and adverse effects.
1. Introduction
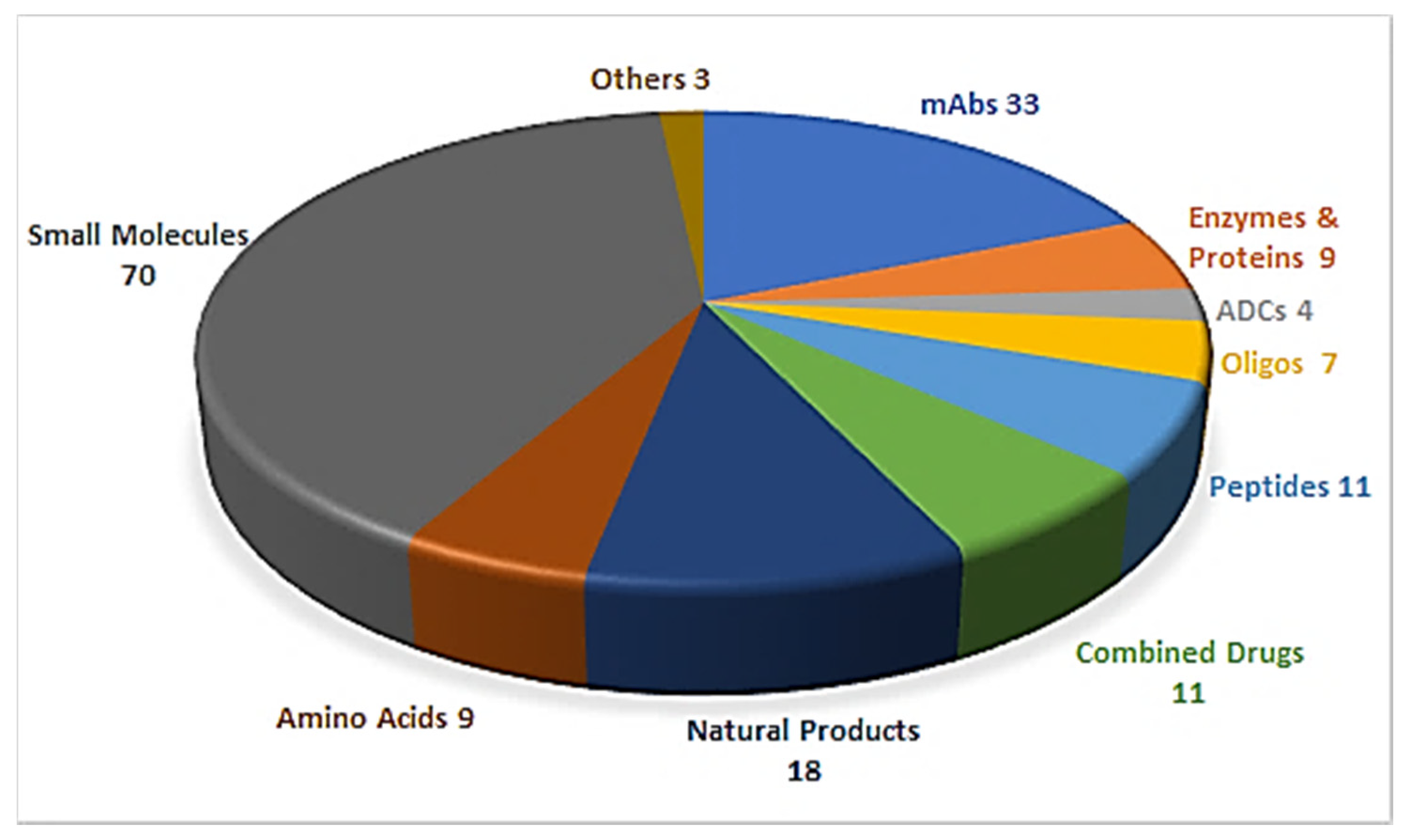
Drug discovery is a multifactorial activity involving the private and public sectors and, more importantly, society as a whole, which is represented by patients. From 2016 to 2019, the United States Food and Drug Administration (FDA) approved a total of 175 new drugs for commercialization (Figure 1) [1,2,3,4]. Forty-eight drugs were approved in 2019 [4], 10 of which were biologics and the remaining 38 new chemical entities (NCEs). The peptides and oligonucleotides (TIDES) class is manufactured chemically and thus belongs to NCEs; however, these molecules have a clear biological structure and could; therefore, be considered a transition between the two subclasses. In 2019, five TIDES (three peptides and two oligonucleotides) were authorized (Table 1). This figure accounts for approximately 10% of the total drugs approved and agrees with the total number approved in the period 2016–2019 (18 TIDES: 11 peptides and seven oligonucleotides, vs. 175). Furthermore, three antibody drug conjugates (ADCs) were approved. In two of these, namely enfortumab vedotin-ejfv and polatuzumab vedotin-piiq, the payload is the peptide monomethyl auristatin E (MMAE), derived from the marine mollusk dolastatin. Finally, peptides serve as linkers between the payload and the antibody in all three ADCs. Thus, TIDES are present in eight of the 48 drugs approved in 2019. These figures reflect the high relevance of such molecules for biomedical applications.
Here, TIDES from the 2019 harvest are discussed from a molecular perspective, application as a drug, mode of action, and adverse effects.
2. Oligonucleotides
2.1. Golodirsen (Vyondys 53TM)
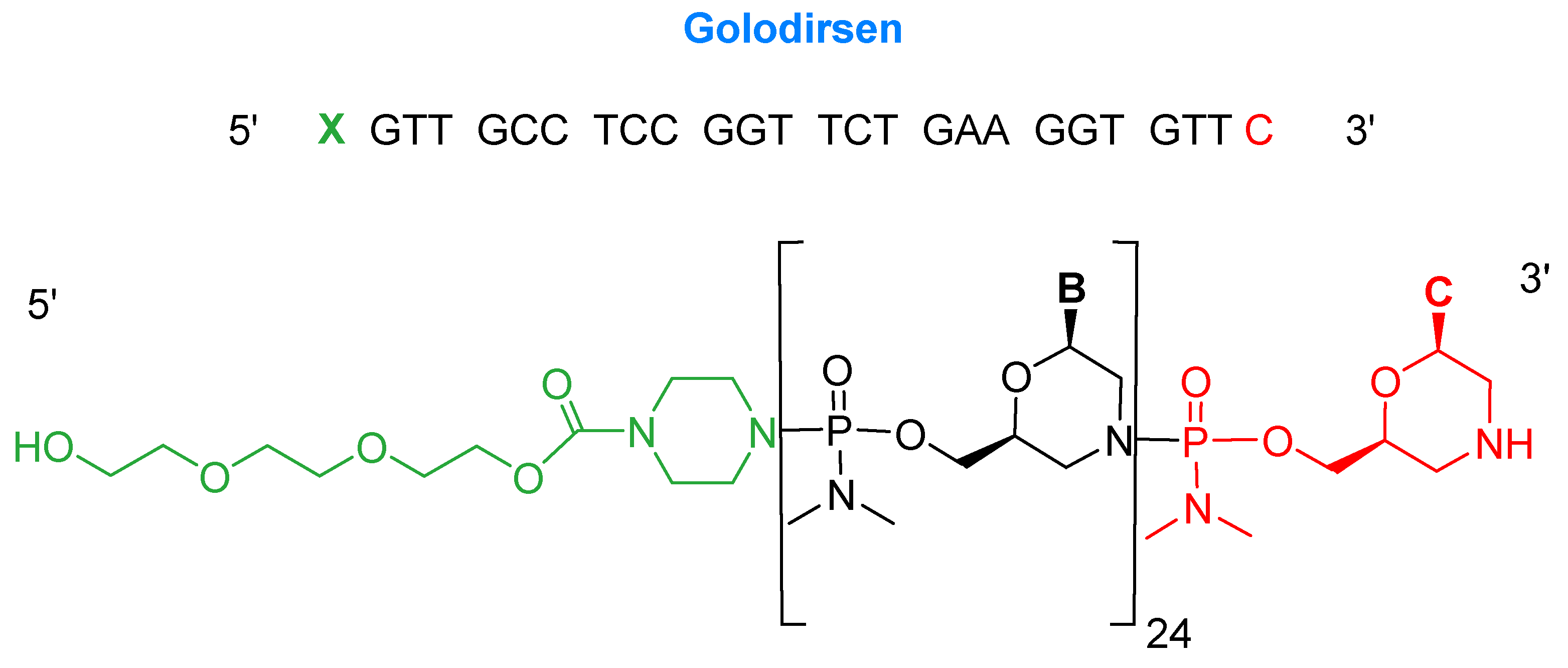
Golodirsen is an antisense oligonucleotide with a single strand of 25 monomers [6]. The subunits are linked through a synthetic neutral phosphorodiamidate morpholino oligomer (PMO) backbone. This neutral backbone confers greater stability to the strand than the natural negatively-charged phosphodiester linkage. The nitrogenous bases are incorporated on a morpholine six-membered heterocycle instead of the natural five-membered ribose (or deoxyribose) ring. The strand ends with a small hydrophilic triethylene glycol chain. Golodirsen has a molecular weight of 8647.4 Da (Figure 2).
Golodirsen was developed for the treatment of Duchenne’s muscular dystrophy (DMD), which is a progressive muscle deterioration that starts in early childhood and in most cases ends up crippling patients before adolescence. After several years, patients die mainly from heart failure [7,8]. This disorder is caused by a deletion mutation in the dystrophin gene, which transcribes for the production of dystrophin, a huge protein that covers muscular fibers, protecting them from damage upon contraction and enhancing muscle performance. The genetic disorder causes the production of a non-functioning dystrophin protein and consequently muscle wasting. This gene is linked to the X chromosome, thus making DMD disorder more noticeable in male infants [7,9].
The dystrophin gene consists of 79 exons. Some genetic mutations cause the deletion of exon 52, which blocks the translation process [7]. Golodersin conceals exon 53, allowing translation to take place and producing a protein with some missing parts but still functional [9,10]. A similar previous drug, eteplirsen (Exondys 51), was the first FDA-approved antisense (in 2016) therapy for the treatment of the same disorder by exon 51 skipping [10].
2.2. Givosiran (GivlaariTM)
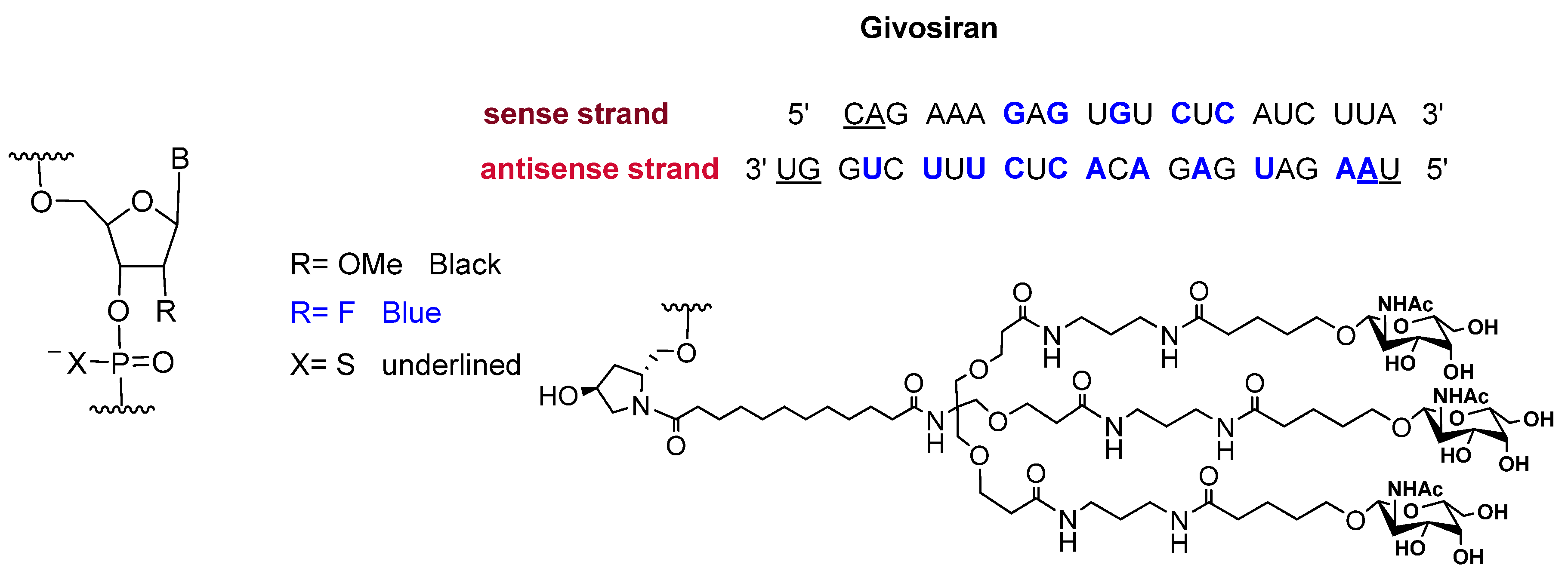
Givosiran is the second small interfering RNA (siRNA) drug to be approved by FDA [13] (the first one was patisiran (OnpattroTM), which was authorized in 2018 for the treatment of hereditary transthyretin-mediated amyloidosis and targets hepatic cells [14,15]). Additionally, givosiran is the first approved drug that demonstrates the enhanced stabilization chemistry (ESC)-GalNAc-siRNA conjugate technology. This technology involves several synthetic RNA stabilization chemistries. The 2′-OH of the ribose in some monomers are methylated (forming 2′-O-methyl-ribonucleoside), while in others they are substituted by the highly electronegative fluorine atom (2′-F-ribonucleoside) in order to boost the stability of the double strands against the nuclease [16]. In addition, the hepatocyte-targeting ligand that is attached to the 3′ terminal of the sense strand has three N-acetylgalactosamine moieties. The other three terminals (5′ of the sense strand, and 3′,5′ of the antisense strand) have thiophosphate linkages in the last two subunits for each side. This conjugation (ESC-GalNAc-siRNA) confers enhanced stability upon subcutaneous administration of the siRNA and offers a 10-fold increased potency of the drug over the standard template chemistry (STC) [13,17]. Givosiran is prepared as the sodium salt of a double-strand oligonucleotide (sense and antisense strands) (Figure 3) and it has a molecular weight of 17,245.56 Da.
Givosiran was developed for the treatment of acute hepatic porphyria in adults, which is a genetic disorder that results in the accumulation of the neurotoxic intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG) during the hemes production cycle (the hemoglobin oxygen binding site) in hepatic cells. This disorder causes severe abdominal pain, nausea, vomiting, and constipation and it can be triggered by several factors, such as certain drugs, low sugar intake due to fasting, smoking, and stress [18,19].
The N-acetylgalactosamine ligand bound to the sense strand facilitates the uptake into liver cells [19]. After entering hepatocytes, it binds to and silences aminolevulinate synthase 1 (ALAS1) mRNA, thereby halting ALA production and consequently preventing the accumulation of the toxic intermediates in body tissues [18].
Givosiran is administered subcutaneously and is well-tolerated. However, regular check-ups for liver and kidney function are highly recommended [19]. It was developed by Alnylam Pharmaceuticals Inc. (Cambridge, Massachusetts, United States) (the same company that developed patisiran) and approved by the FDA on 20 November 2019 [20].
3. Peptide-Based Drugs
3.1. [68Ga]Ga-DOTATOC ([[68Ga]Ga-DOTA, Tyr3]-Octreotide)
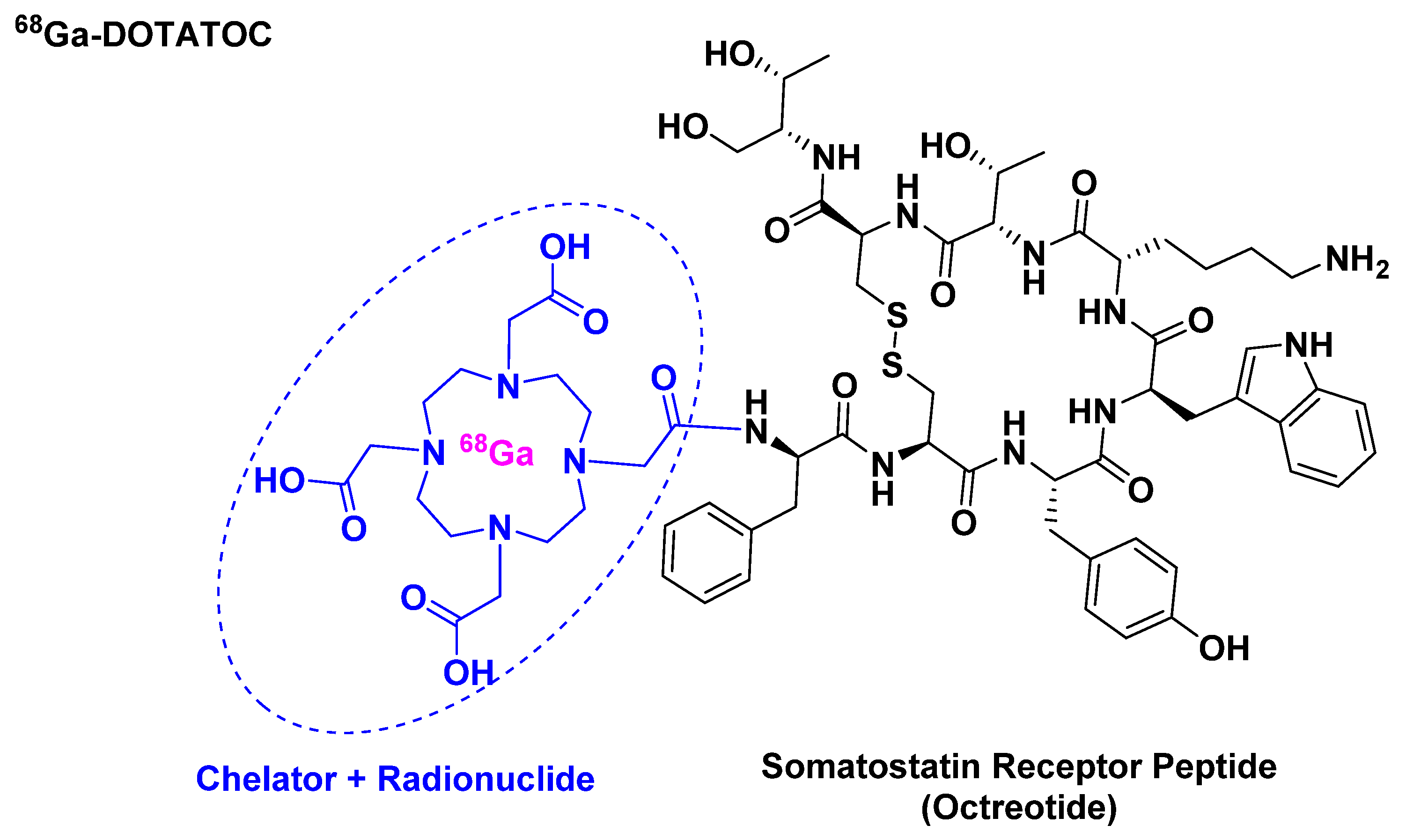
68Ga-DOTATOC belongs to the peptide receptor radionuclide therapy (PRRT) class and it is used for scintigraphic imaging [21]. It is composed of 68Ga, which has a half-life of 68 min and a high positron abundance [22,23], as radionuclide, which is chelated by DOTA that in turn is attached to [Tyr3]-octreotide (Figure 4). It is used mainly in positron imaging therapy (PET) for the detection of somatostatin receptor-positive neuroendocrine tumors (NETs).
68Ga-DOTATOC is related to 68Ga-DOTATATE, which was approved in 2016. Both share almost the same affinity towards the somatostatin receptor (sstr). The in vitro affinity of 68Ga-DOTATATE towards sstr subtype 2 (sstr2) is about 10-fold that of 68Ga-DOTATOC [24]. Nevertheless, 68Ga-DOTATOC is able to detect more NET lesions than the DOTATATE analogue with highly reproducible imaging efficiency [22]. Furthermore, the higher in vitro affinity of the DOTATATE analogue has not proven to be relevant clinically. These studies showed that 68Ga-DOTATOC has a higher affinity towards sstr2 receptor than the 68Ga-DOTATATE as demonstrated by higher tumor uptake values. This could explain the superiority of 68Ga-DOTATOC for more lesion detection [24].
A study by L. K. Khor et al. has shown that 68Ga-DOTATOC is a good biomarker for newly-diagnosed undifferentiated nasopharyngeal carcinomas (NPCs), and to a lesser extent for recurrent NPC and metastatic nodes [25].
Prior to 68Ga-DOTATOC, 111In-octreotide was the only FDA-approved imaging agent for somatostatin receptor scintigraphy of NETs [22]. 111In-octreotide utilizes the single-photon emission computed tomography (SPECT) technique.
Using [111In]In-octreotide for imaging of NETs results in a higher dose to the patient (12 mSv) as compared to the use of [68Ga]Ga-DOTATOC (4.26 mSv) [22]. This is due to the longer half-life of 111In (T1/2 = 2.8 d) as compared to 68Ga (T1/2 = 68 min).
68Ga-DOTATOC is administered intravenously and has some adverse effects, including nausea, pruritis, and flushing [26]. It was developed by the University of Iowa Health Care (UIHC) and approved by the FDA on 21 August 2019 [27]. From the same family, in 2018 the FDA approved [177Lu]Lu-DOTA-TATE ([[177Lu]Lu-DOTA0, Tyr3]-octreotate) for the treatment of gastroenteropancreatic neuroendocrine tumors (GEP-NETs) [28].
3.2. Afamelanotide (Scenesse®)
Afamelanotide is a synthetic tridecapeptide structural analogue of α-melanocyte stimulating hormone (α-MSH) with a molecular weight of 1646.87 Da (Figure 5A) [29]. It differs from the natural analogue (Figure 5B) in its fourth and seventh amino acid residues, in which Met and Phe are replaced by norleucine (Nle) and D-Phe, respectively [30]. Such modifications play a key role in enhancing the properties of afamelanotide versus the physiological α-MSH analogue. In this regard, afamelanotide shows enhanced resistance against enzymatic degradation, increases biological activity, and prolongs the plasma half-life [30] by stimulating binding affinity with melanocortin 1 receptor (MC1R) [31]. Furthermore, unlike other small therapeutic peptides, afamelanotide has a minimal risk of inducing anti-drug antibodies even after six years of continuous treatment [31,32]. In contrast, anti-drug antibody induction has been reported for its α-MSH natural analogue, in which a noticeable increase of IgM autoantibodies against α-MSH was observed [33].
Afamelanotide is used for the treatment of erythropoietic protoporphyria (EPP) [29]. It provides photoprotection upon exposure to direct sunlight by increasing the density of eumelanin in the skin—this is called the skin tanning process [34]. It works like α-MSH, binding to the G-protein-coupled MC1R in dermal cells and stimulating the production of melanin, along with consecutive biological processes [35]. Despite stimulating melanin synthesis in the same way, afamelanotide is considered a preventive therapy. In which, unlike α-MSH natural analogue, afamelanotide induces eumelanin synthesis in advance and independently of having UV-damaged skin cells [32].
Afamelanotide is administered subcutaneously [36] through a poly(lactic-co-glycolic acid) (PLGA) biodegradable polymer [34]. It has a half-life of 30 to 50 min, after which it is hydrolyzed into shorter peptides and amino acid residues [30,32]. Most of the active materials are excreted within two days, and by 10 days its plasma levels are below the limit of quantification [30,32].
Afamelanotide has some adverse effects, including, but not limited to, nausea, vomiting, flushing, headache, cough, fatigue, and dizziness [36].
Its development was started in the 1980s by Tomi Sawyer and Victor Hruby at the University of Arizona [29], then Clinuvel Inc. performed the required clinical studies and brought the product to the market. It has been sold in Switzerland and Italy since 2013 [35], but was only approved by FDA on 8 October 2019 [37].
3.3. Bremelanotide (VYLEESITM)
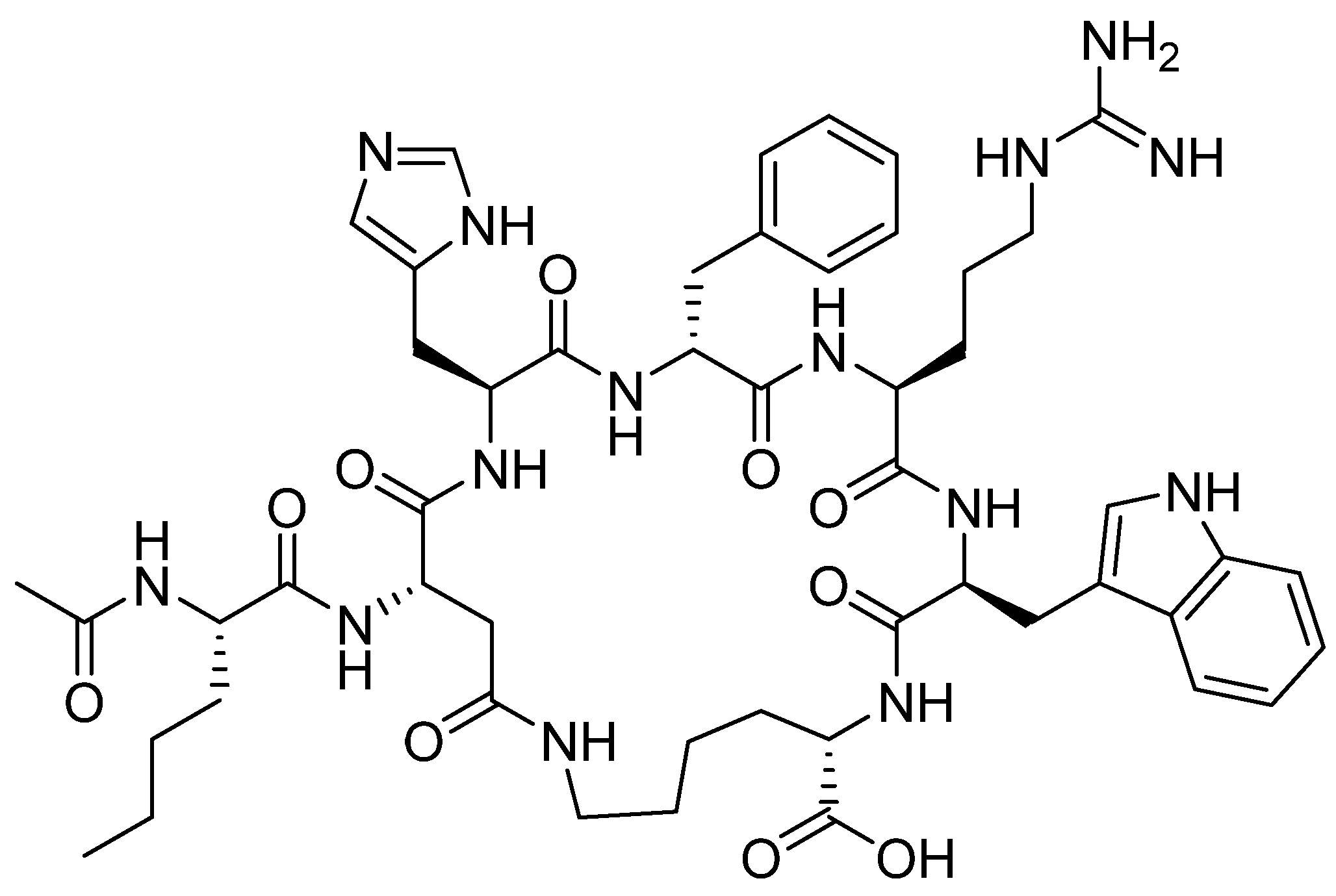
Bremelanotide is a homodetic side-chain to tail cyclic heptapeptide with the sequence of Ac-Nle-cyclo[Asp-His-DPhe-Arg-Trp-Lys]-OH [38]. The cycle through an amide bond is between the ß carboxylic acid of Asp ε amino of the Lys, which is the C-terminal residue. The exocyclic Nle is acetylated. It has a molecular weight of 1025.182 Da. The structure is shown in Figure 6.
Bremelanotide is an analogue of the natural α-MSH [39]. It works as an agonist of melanocortin receptors and is used to treat hypoactive sexual desire (HSDD) in women of fertile age. This disorder results in a low sex drive that is not caused by other factors such as medications or any medical or psychiatric condition [40,41].
The drug is administered subcutaneously and possible adverse effects include nausea, flushing, injection site reactions, headache, and vomiting [42,43].
Bremelanotide was first studied by Arizona Cancer Research Centre as a self-tanning inducer. However, increased sexual desire in patients was observed as a side effect [44]. Later on, it was developed by Palatin technology as a treatment for HSDD [44] and then out-licensed to AMAG PHARMS and approved by the FDA on 21 June 2019 [45].
Although bremelanotide and afamelanotide belong to the same α-MSH hormone-analogous family, their structures show some differences. Bremelanotide resembles the middle section of afamelanotide with the absence of the first three residues Ser-Tyr-Ser at the N-terminus and the last two residues Pro-Val at the C-terminus, in addition to the Gly, the tenth residue. Additionally, bremelanotide comprises a side-to-side amide bond that forms the cycle.
4. Peptides as Payloads in ADCs
4.1. Enfortumab Vedotin-Ejfv (PADCEVTM)
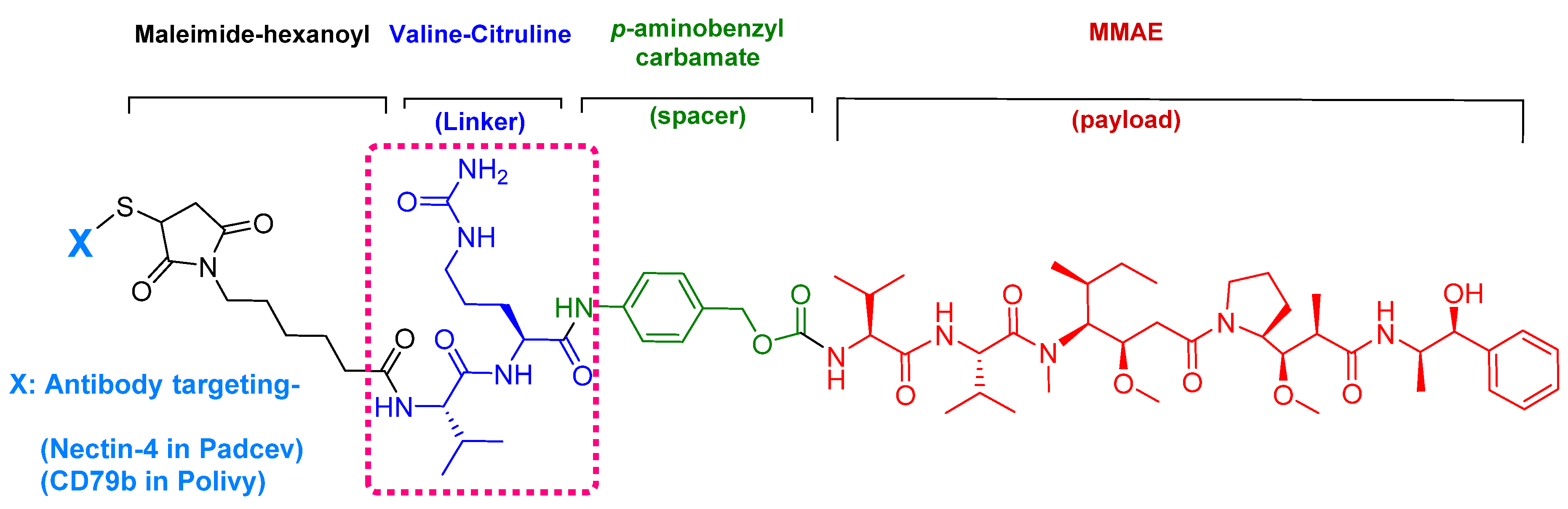
Enfortumab vedotin-ejfv is an ADC therapy [46], which is an emerging therapeutic strategy for transporting cytotoxic chemotherapeutic agents to certain tumors [47]. This drug has three main components: payload/cytotoxic drug, mAb, and linker [47]. The antibody comprises human monoclonal antibody (enfortumab) and targets nectin-4 (also known as poliovirus receptor-related protein 4 (PVLR4)), which is highly expressed in NETs [46]. Enfortumab is derived either from a Chinese hamster cell ovary line [ASG-22CE] or can be prepared via murine hybridoma technology (AGS-22M6E (or ASG-22ME)) [48]. Enfortumab is conjugated to MMAE (vedotin) via a cathepsin-cleavable linker (Figure 7) [46].
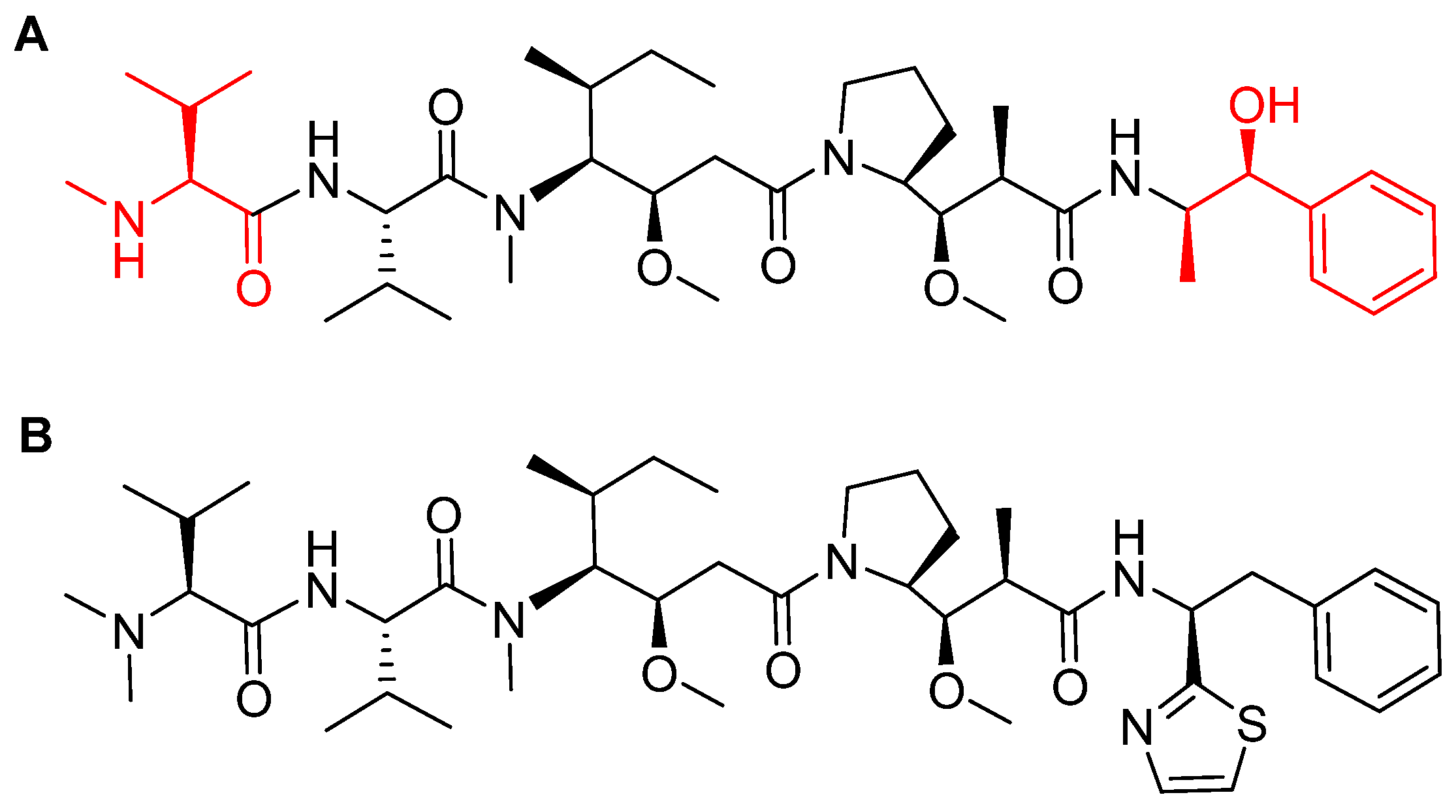
MMAE, the cytotoxic component [49], is a synthetic pentapeptide (717.99 Da) that works as a potent microtubule-disrupting agent [48]. It is a structurally-modified analogue of the natural dolastatin 10 [50], a potent antineoplastic pentapeptide (785.1 Da) isolated from the marine mollusk dolabella auricularia by Pettit et al. in 1987 [51]. MMAE comprises the following four amino acid residues: dolavaline (Dov), Val, dolaisoleuine (Dil), dolaproine (Dap), and the C-terminal amine dolaphenine (Doe) [52]. Figure 8 shows the structural differences between the synthetic MMAE analogue (A) and the natural pentapeptide dolastatin 10 (B).
Given that MMAE is a peptide, it is metabolized into smaller non-toxic amino acid fragments and then recycled or excreted by the body [53].
Nectin-4 is a 66 KDa protein that is expressed in several cancer tissues (breast, lung, bladder, among others) and highly expressed in urothelial cancer [54]. Among other nectins, it is considered a potential target due to its distinguished sequence in its family (1, 2, or 3) with low degree of similarity with other family members [55]. Furthermore, the limited expression of nectin-4 in normal tissues minimizes the possibility of these tissues being targeted during the course of the treatment [46].
Enfortumab vedotin-ejfv is a pan-fibroblast growth factor receptor (FGFR) suppressor [56]. It is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who previously received immune checkpoint therapy [programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor] [56]. After platinum-based chemotherapy, enfortumab vedotin-ejfv is prescribed as a second line treatment for patients with susceptible FGFR2 or FGFR3 [48,56].
The drug binds to nectin-4-expressing cells. The resulting complex is internalized into the cell and then the valine-citruline (Val-Cit) dipeptide linker is recognized and cleaved by cathepsin-B in the tumor cell [57]. Consequently, the cytotoxic MMAE is selectively released, thereby leading to apoptosis [46].
It is administered intravenously. Common adverse effects include fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, dysgeusia, diarrhea, dry eye, pruritus, and dry skin [58].
It was developed by Astellas Pharma and granted accelerated approval by the FDA on 18 December 2019 [59].
4.2. Polatuzumab Vedotin-Piiq (PolivyTM)
Polatuzumab vedotin-piiq is an ADC therapy [60]. It comprises the same linker and payload as in the previous drug (enfortumab vedotin-ejfv), but a different antibody (Figure 9). It is prescribed as a combination with bendamustine and rituximab (BR combination) [5] and is used for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma [61].
It selectively binds to CD79b that is overexpressed in mature B-cells [61]. Following the same mechanism in enfortumab vedotin-ejfv which ends by cell apoptosis.
Of note, the tolerability and safety profile of this drug was accepted for non-Hodgkin’s lymphoma (NHL) patients but not for those with chronic lymphocytic leukemia (CLL) [61].
It is administered intravenously. Common adverse effects include neutropenia, thrombocytopenia, anemia, peripheral neuropathy, fatigue, diarrhea, pyrexia, decreased appetite, and pneumonia [62].
It was developed by Roche and granted accelerated approval by the FDA on 10 June 2019 [63].
5. Peptides as Linkers in ADCs
5.1. Val-Cit
The choice of a suitable linker is a highly sensitive step in ADC manufacturing. First, the conjugate should be stable enough during its circulation in blood serum to avoid damaging body tissues. Second, the programable release of cargo should be easily triggered once the conjugate reaches its target. Thus, a suitable linker should successfully combine serum stability and in-target lability without adversely affecting the stability of the antibody itself upon conjugation [64,65].
Among the four known types of linkers, namely hydrazones, disulfides, peptides, and thioethers [57], short peptidyl linkers, such as Val-Cit dipeptide, fulfill the requirements for this critical function and even outperform the tetra-peptidyl linkers (Gly-Phe-Leu-Gly and Ala-Leu-Ala-Leu) previously used and that showed some aggregation issues upon conjugation [57,64,65]. The premature release of the payload in the case of hydrazone (due to pH changes) and disulfide linkers (due to exchange with other thiols, such as glutathione) may influence the potency of the treatment, while the delayed release of cargo in the case of thioether linkers (payload is released only after total degradation of the antibody) may cause the loss of anticancer activity [65,66]. In PadcevTM and PolivyTM, a maleimidocarpoyl moiety is added to the N-terminal of the dipeptide Val-Cit to facilitate conjugation to the antibody. The amide bond formed by the C-carboxyl group of the Cit, which is linked to a self-immolative p-amino benzyl carbamate (PABC) spacer, is stable in serum and can be rapidly hydrolyzed by lysosomal cathepsin-B releasing the PABC-MMAE moiety. This undergoes self-elimination liberating the payload MMAE [57]. The mechanism of drug release is shown in Figure 9.
5.2. Gly-Gly-Phe-Gly
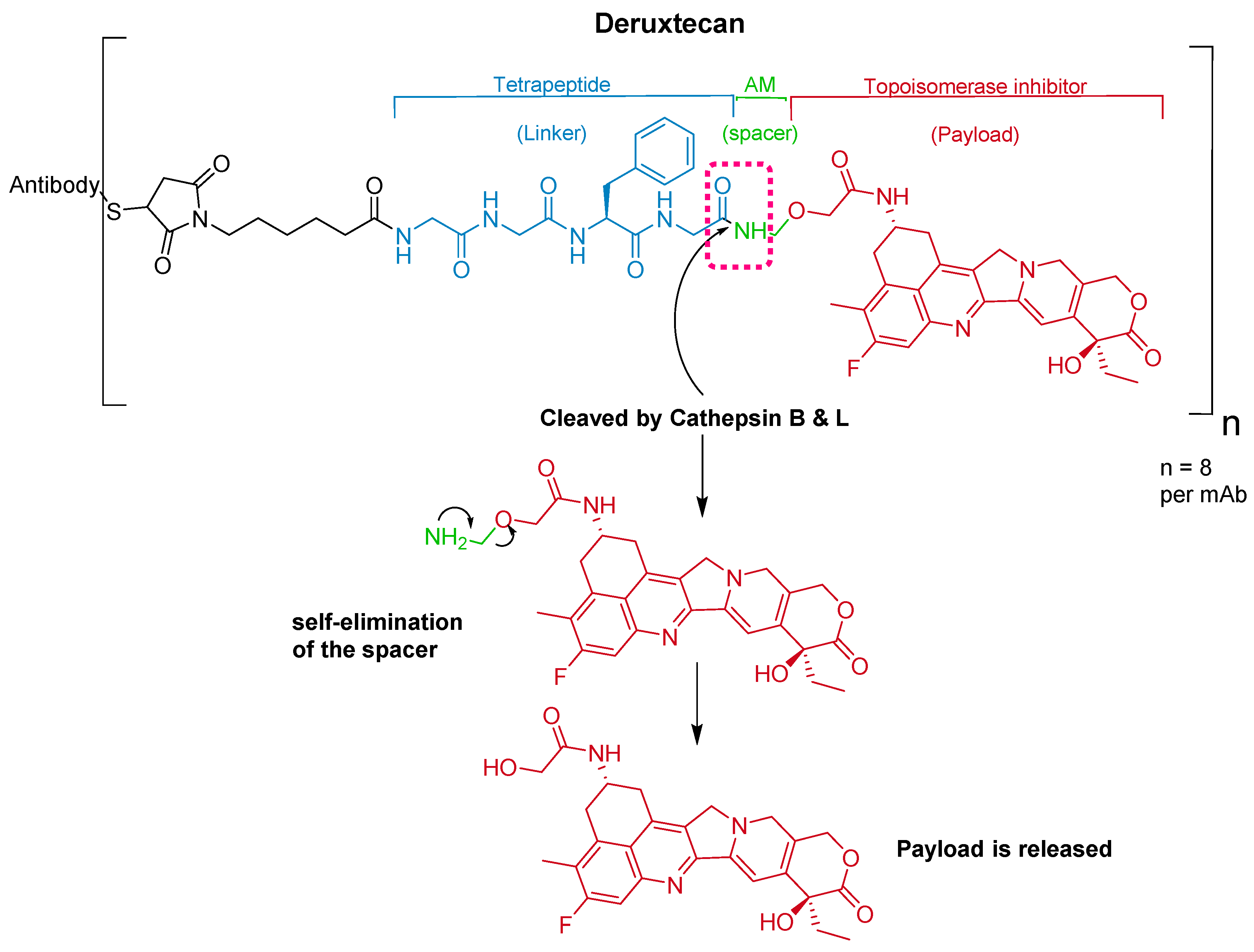
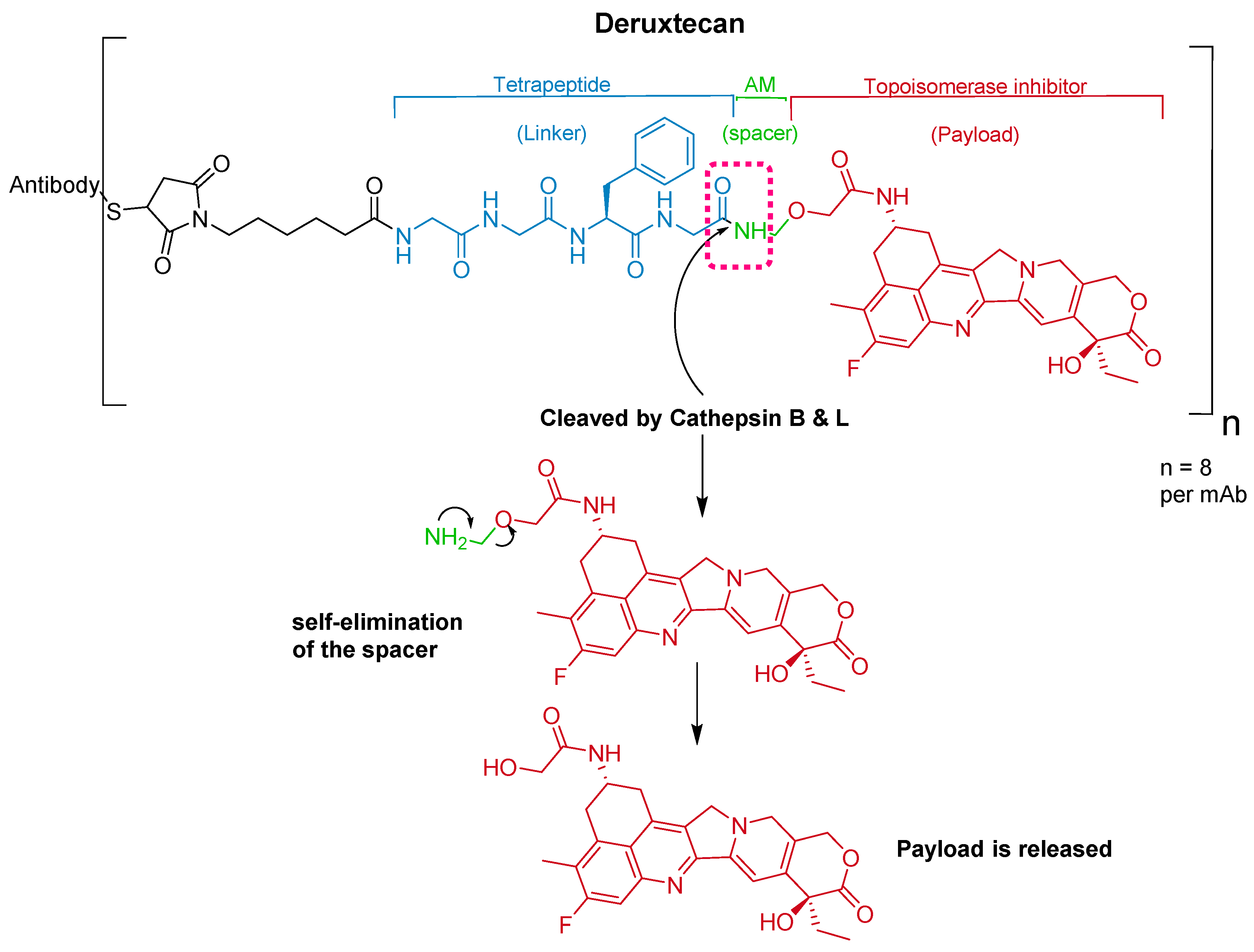
The ADC fam-trastuzumab deruxtecan-nxki (EnhertuTM) targets human epidermal growth factor receptor-2 (HER2) [66]. It is used for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer [68].
Trastuzumab is conjugated to the antibody via a tetrapeptide linker, Gly-Gly-Phe-Gly (Figure 10), which is cleavable by lysozymes [69]. The linker is connected to a Cys residue of the mAb through a maleimidocarpoyl component [66] and to a self-immolative amino methylene (AM) spacer [70].
It was developed by Daiichi Sankyo and approved by the FDA on 20 December 2019 [71].
6. Conclusions
The 2019 year has been very successful regarding the role of TIDES in the drug arena. Thus, in addition to approving two oligonucleotides and three peptide APIs, the FDA authorized two ADCs with a peptide as payload. Finally, three ADCs (including the former two) have a peptide-based linker. Therefore, eight of the 48 drugs (more than 15%) approved this year were or contained TIDES. While the peptide market is already consolidated through the large number of authorized peptides, the seven oligonucleotides recently (2017–2019) given the green light pave the way for the approval of others for diverse medical indications. Of note, is the presence of peptides as payloads and as a part of the linkers in the ADCs.
Author Contributions
All authors participated in the search for information and in writing the manuscript and have approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded in part by the following: the National Research Foundation (NRF) and the University of KwaZulu-Natal (South Africa); MINECO, (RTI2018-093831-B-100), and the Generalitat de Catalunya (2017 SGR 1439) (Spain).
Conflicts of Interest
The authors declare no conflict of interest.
References
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2016. An analysis of FDA drug approvals from a perspective of the molecule type. Molecules 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2017. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2018, 23. [Google Scholar] [CrossRef] [Green Version]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2019. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [Green Version]
- New Drug Therapy Approvals 2019. 2019. Available online: https://www.fda.gov/media/134493/download (accessed on 27 February 2020).
- Heo, Y.A. Golodirsen: First approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.L.; et al. Dystrophin cardiomyopathies: Clinical management, molecular pathogenesis and evolution towards precision medicine. J. Clin. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis rescue improves cardiac function in dystrophin-deficient mice and duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Yokota, T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [PubMed]
- Vyondys 53 Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf (accessed on 27 February 2020).
- Vyondys 53 Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/211970Orig1s000ltr.pdf (accessed on 27 February 2020).
- Janas, M.M.; Harbison, C.E.; Perry, V.K.; Carito, B.; Sutherland, J.E.; Vaishnaw, A.K.; Keirstead, N.D.; Warner, G. The nonclinical safety profile of GalNAc-conjugated i therapeutics in subacute studies. Toxicol. Pathol. 2018, 46, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2’-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Vita, G.; Vita, G.L.; Stancanelli, C.; Gentile, L.; Russo, M.; Mazzeo, A. Genetic neuromuscular disorders: Living the era of a therapeutic revolution. Part 1: Peripheral neuropathies. Neurol. Sci. 2019, 40, 661–669. [Google Scholar] [CrossRef] [PubMed]
- de Paula Brandao, P.R.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Leading RNA interference therapeutics part 2: Silencing delta-aminolevulinic acid synthase 1, with a focus on givosiran. Mol. Diagn. Ther. 2020, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Givlaari Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/0212194s000lbl.pdf (accessed on 27 February 2020).
- Graham, M.M.; Gu, X.; Ginader, T.; Breheny, P.; Sunderland, J.J. 68Ga-dotatoc imaging of neuroendocrine tumors: A systematic review and metaanalysis. J. Nucl. Med. 2017, 58, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Menda, Y.; Ponto, L.L.B.; Schultz, M.K.; Zamba, G.K.D.; Watkins, G.L.; Bushnell, D.L.; Madsen, M.T.; Sunderland, J.J.; Graham, M.M.; O’Dorisio, T.M.; et al. Repeatability of gallium-68 dotatoc positron emission tomographic imaging in neuroendocrine tumors. Pancreas 2013, 42, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Le, V.S. (68)Ga generator integrated system: Elution-purification-concentration integration. Recent Results Cancer Res. 2013, 194, 43–75. [Google Scholar]
- Poeppel, T.D.; Binse, I.; Petersenn, S.; Lahner, H.; Schott, M.; Antoch, G.; Brandau, W.; Bockisch, A.; Boy, C. 68Ga-dotatoc versus 68Ga-dotatate pet/ct in functional imaging of neuroendocrine tumors. J. Nucl. Med. 2011, 52, 1864–1870. [Google Scholar] [CrossRef] [Green Version]
- Khor, L.K.; Loi, H.Y.; Sinha, A.K.; Tong, K.T.; Goh, B.C.; Loh, K.S.; Lu, S.J. 68Ga-dota-peptide: A novel molecular biomarker for nasopharyngeal carcinoma. Head Neck 2016, 38, E76–E80. [Google Scholar] [CrossRef] [PubMed]
- 68Ga-dotatoc Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210828s000lbl.pdf (accessed on 27 February 2020).
- 68Ga-dotatoc Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210828Orig1s000ltr.pdf (accessed on 27 February 2020).
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2018 FDA tides harvest. Pharmaceuticals 2019, 12, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, T.K.; Sanfilippo, P.J.; Hruby, V.J.; Engel, M.H.; Heward, C.B.; Burnett, J.B.; Hadley, M.E. 4-Norleucine, 7-D-phenylalanine-a-melanocyte-stimulating hormone: A highly potent α-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. USA 1980, 77, 5754–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, A.M.; McKay, J.T.; Bonkovsky, H.L. Advances in the management of erythropoietic protoporphyria - role of afamelanotide. Appl. Clin. Genet. 2016, 9, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Spichty, R.; Balimann, M.; Barman, J.; Minder, E.I. A bioassay for the detection of neutralizing antibodies against the alpha-melanocyte stimulating hormone analog afamelanotide in patients with erythropoietic protoporphyria. J. Pharm. Biomed. Anal. 2013, 75, 192–198. [Google Scholar] [CrossRef]
- Kim, E.S.; Garnock-Jones, K.P. Afamelanotide: A review in erythropoietic protoporphyria. Am. J. Clin. Dermatol. 2016, 17, 179–185. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Harro, J.; Jaanisk, M.; Järv, A.; Podar, I.; Allik, J.; Nilsson, I.; Sakthivel, P.; Lefvert, A.K.; Hökfelt, T. Autoantibodies against neuropeptides are associated with psychological traits in eating disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 14865–14870. [Google Scholar] [CrossRef] [Green Version]
- Committee for medicinal products for human use (CHMP). Scenesse Assessment Report. 2014. Available online: https://www.ema.europa.eu/en/documents/assessment-report/scenesse-epar-public-assessment-report_en.pdf (accessed on 27 February 2020).
- Fabrikant, J.; Touloei, K.; Brown, S.M. A review and update on melanocyte stimulating hormone therapy: Afamelanotide. J. Drugs Dermatol. 2013, 12, 775–779. [Google Scholar]
- Scenesse Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210797s000lbl.pdf (accessed on 27 February 2020).
- Scenesse Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210797Orig1s000ltr.pdf (accessed on 27 February 2020).
- Molinoff, P.B.; Shadiack, A.M.; Earle, D.; Diamond, L.E.; Quon, C.Y. Pt-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann. N. Y. Acad. Sci. 2003, 994, 96–102. [Google Scholar] [CrossRef]
- Miller, M.K.; Smith, J.R.; Norman, J.J.; Clayton, A.H. Expert opinion on existing and developing drugs to treat female sexual dysfunction. Expert Opin. Emerg. Drugs 2018, 23, 223–230. [Google Scholar] [CrossRef]
- Both, S. Recent developments in psychopharmaceutical approaches to treating female sexual interest and arousal disorder. Curr. Sex Health Rep. 2017, 9, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, A.H.; Lucas, J.; DeRogatis, L.R.; Jordan, R. Phase I randomized placebo-controlled, double-blind study of the safety and tolerability of bremelanotide coadministered with ethanol in healthy male and female participants. Clin. Ther. 2017, 39, 514–526.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsberg, S.A.; Clayton, A.H.; Portman, D.; Williams, L.A.; Krop, J.; Jordan, R.; Lucas, J.; Simon, J.A. Bremelanotide for the treatment of hypoactive sexual desire disorder: Two randomized phase 3 trials. Obstet. Gynecol. 2019, 134, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Vyleesi Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210557s000lbl.pdf (accessed on 27 February 2020).
- Sohita, D.; Susan J., K. Bremalanotide: First approval. Drugs 2019, 79, 1599–1606. [Google Scholar]
- Vyleesi Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210557Orig1s000ltr.pdf (accessed on 27 February 2020).
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer. 2016, 114, 362–367. [Google Scholar] [CrossRef]
- McGregor, B.A.; Sonpavde, G. Enfortumab vedotin, a fully human monoclonal antibody against nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents 337. Synthesis of dolastatin 10 structural modifications. Anticancer Drug Des. 1995, 10, 529–544. [Google Scholar]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloyd-Williams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. Antineoplastic agents. Part 189. The absolute configuration and synthesis of natural (-)-dolastatin 10. Am. Chem. Soc. 1989, 111, 5463–5465. [Google Scholar] [CrossRef]
- Akaiwa, M.; Martin, T.; Mendelsohn, B.A. Synthesis and evaluation of linear and macrocyclic dolastatin 10 analogues containing pyrrolidine ring modifications. ACS Omega 2018, 3, 5212–5221. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.; Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 2014, 42, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Uemura, M.; Kimura, T.; Kawasaki, Y.; Takamoto, A.; Yamaguchi, A.; Melhem-Bertrandt, A.; Gartner, E.M.; Inoue, T.; Akazawa, R.; et al. A phase I study of enfortumab vedotin in japanese patients with locally advanced or metastatic urothelial carcinoma. Investig. New Drugs 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaide, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with Nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, K.S. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Padcev Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 27 February 2020).
- Padcev Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761137Orig1s000ltr.pdf (accessed on 27 February 2020).
- Sehn, L.H.; Matasar, M.J.; Flowers, C.R.; Kamdar, M.; McMillan, A.K.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab vedotin plus bendamustine with rituximab in relapsed/refractory diffuse large B-cell lymphoma: Updated results of a phase Ib/II randomized study. Blood 2019, 134, 4081. [Google Scholar] [CrossRef]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Polivy Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 27 February 2020).
- Polivy Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761121Orig1s000ltr.pdf (accessed on 27 February 2020).
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular basis of valine-citrulline-PABC linker instability in site-specific ADCs and its mitigation by linker design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Dal Corso, A.; Cazzamalli, S.; Gebleux, R.; Mattarella, M.; Neri, D. Protease-cleavable linkers modulate the anticancer activity of noninternalizing antibody-drug conjugates. Bioconjug. Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef]
- Xu, Z.; Guo, D.; Jiang, Z.; Tong, R.; Jiang, P.; Bai, L.; Chen, L.; Zhu, Y.; Guo, C.; Shi, J.; et al. Novel HER2-targeting antibody-drug conjugates of trastuzumab beyond T-DM1 in breast cancer: Trastuzumab deruxtecan(DS-8201a) and (Vic-)trastuzumab duocarmazine (SYD985). Eur. J. Med. Chem. 2019, 183, 111682. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem. Pharm. Bull. (Tokyo) 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorg. Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Enhertu Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761139Orig1s000ltr.pdf (accessed on 27 February 2020).
Figure 1.
A total of 175 new drugs approved by the FDA from 2016 to 2019 [1,2,3,4]. mAbs; Monoclonal antibodies, ADCs; antibody drug conjugates, Oligos; oligonucleotides.
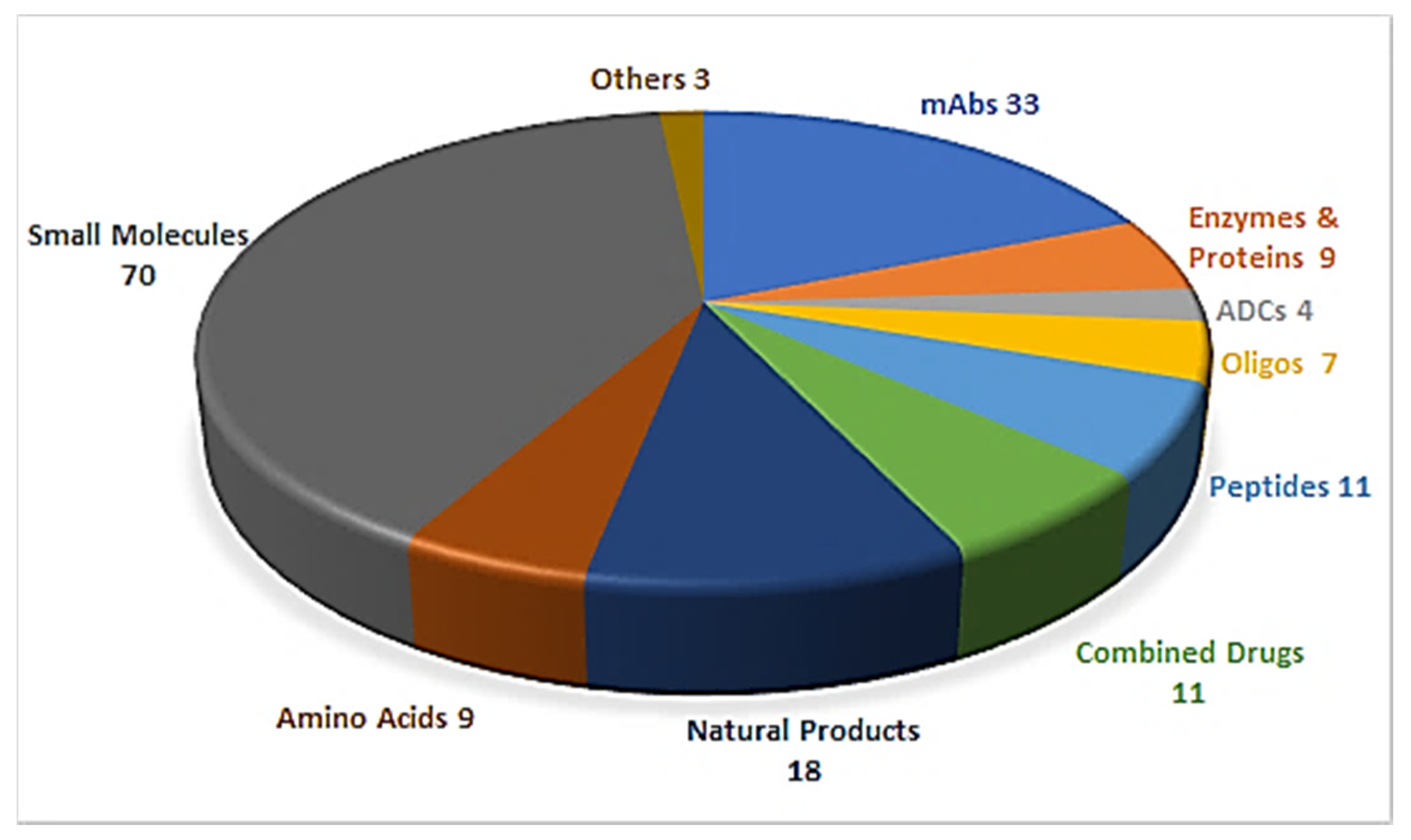
Figure 2.
Chemical structure of golodersin (Vyondys 53TM).

Figure 3.
Chemical structure of givosiran (GivlaariTM) [13].
Figure 3.
Chemical structure of givosiran (GivlaariTM) [13].
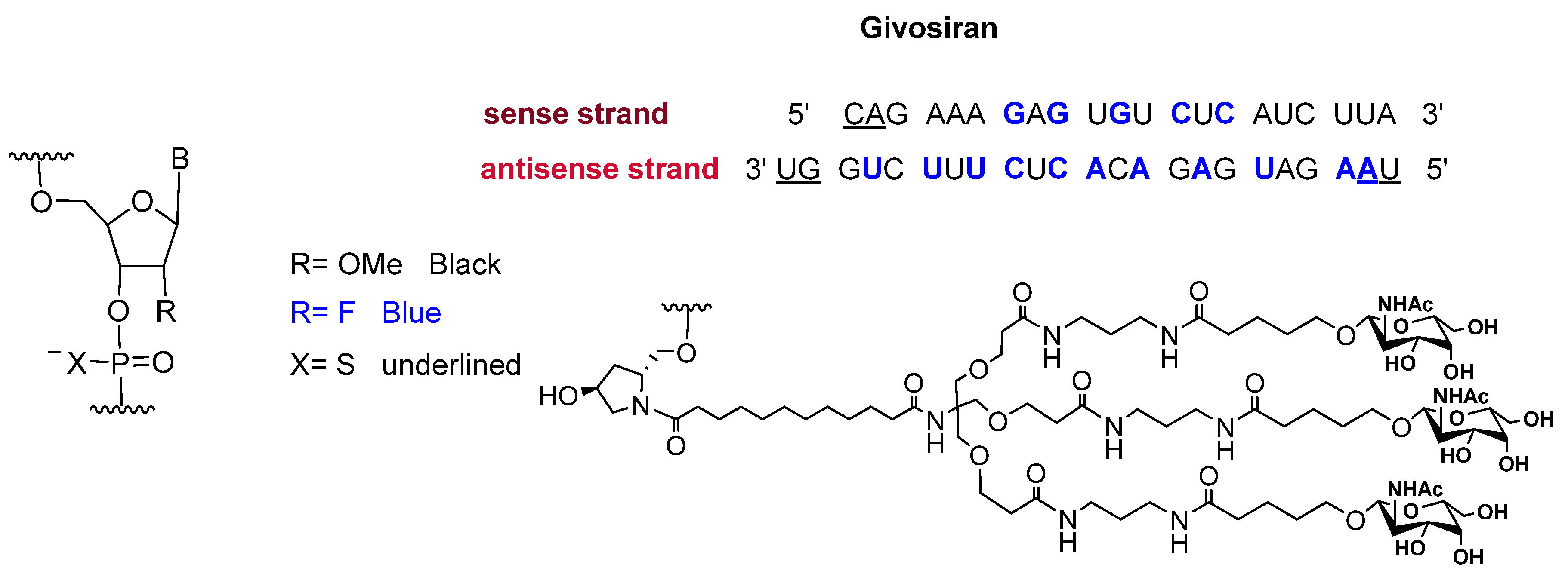
Figure 4.
Chemical structure of 68Ga-DOTATOC.
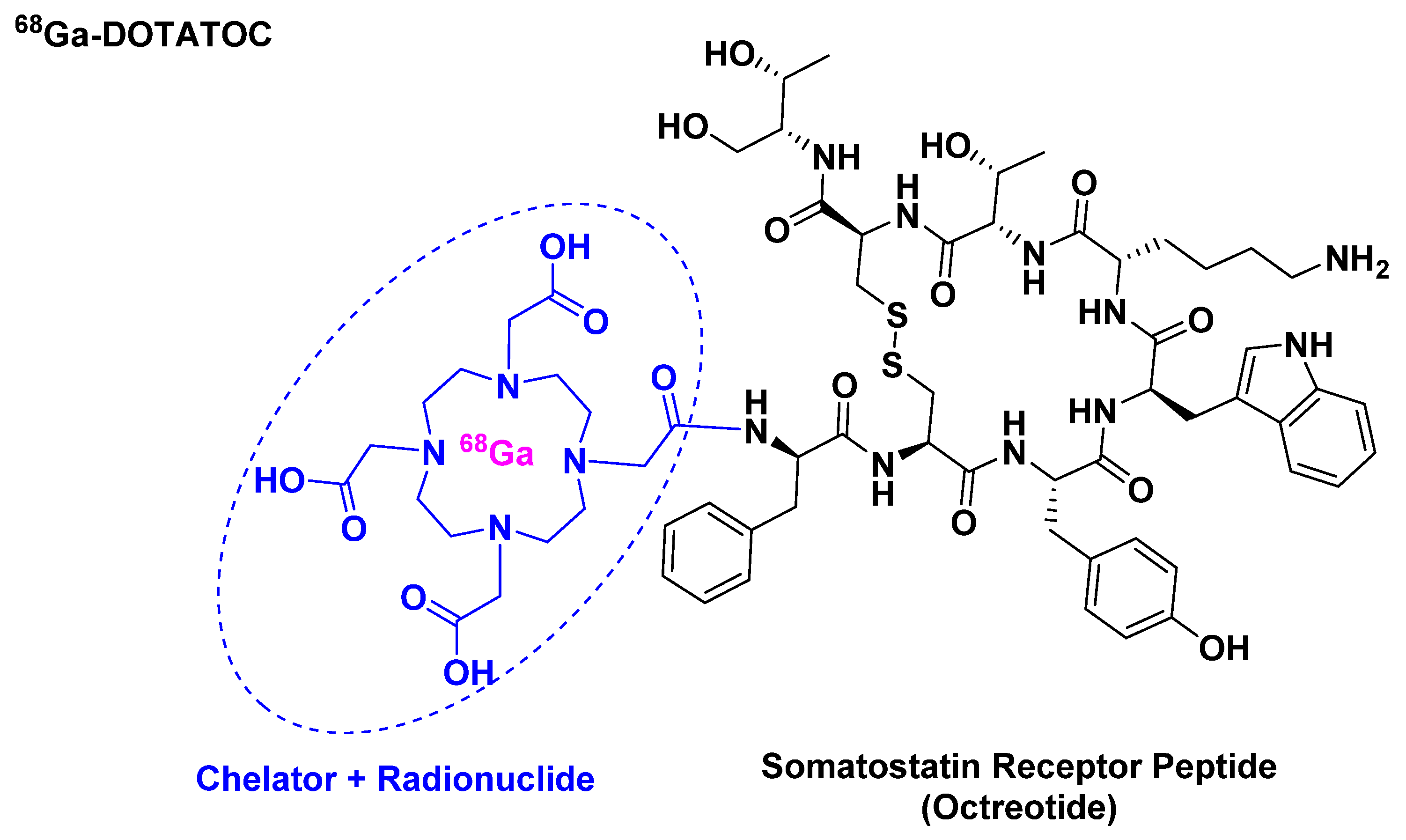
Figure 5.
Chemical structure of: (A) Afamelanotide (ScenesseTM); (B) α-melanocyte stimulating hormone (α-MSH). Differences are shown in red.
Figure 5.
Chemical structure of: (A) Afamelanotide (ScenesseTM); (B) α-melanocyte stimulating hormone (α-MSH). Differences are shown in red.
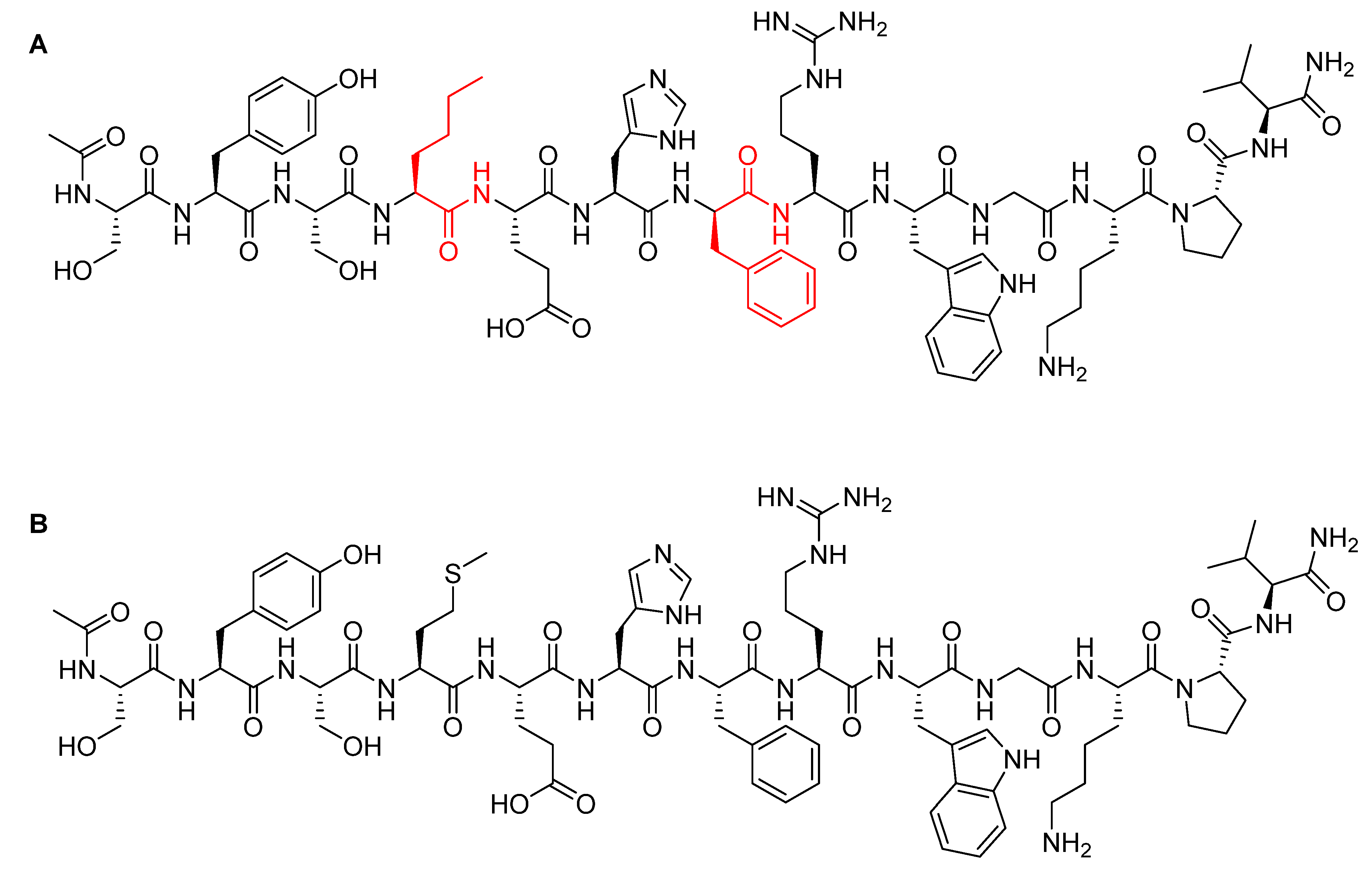
Figure 6.
Chemical structure of bremelanotide (VyleesiTM).
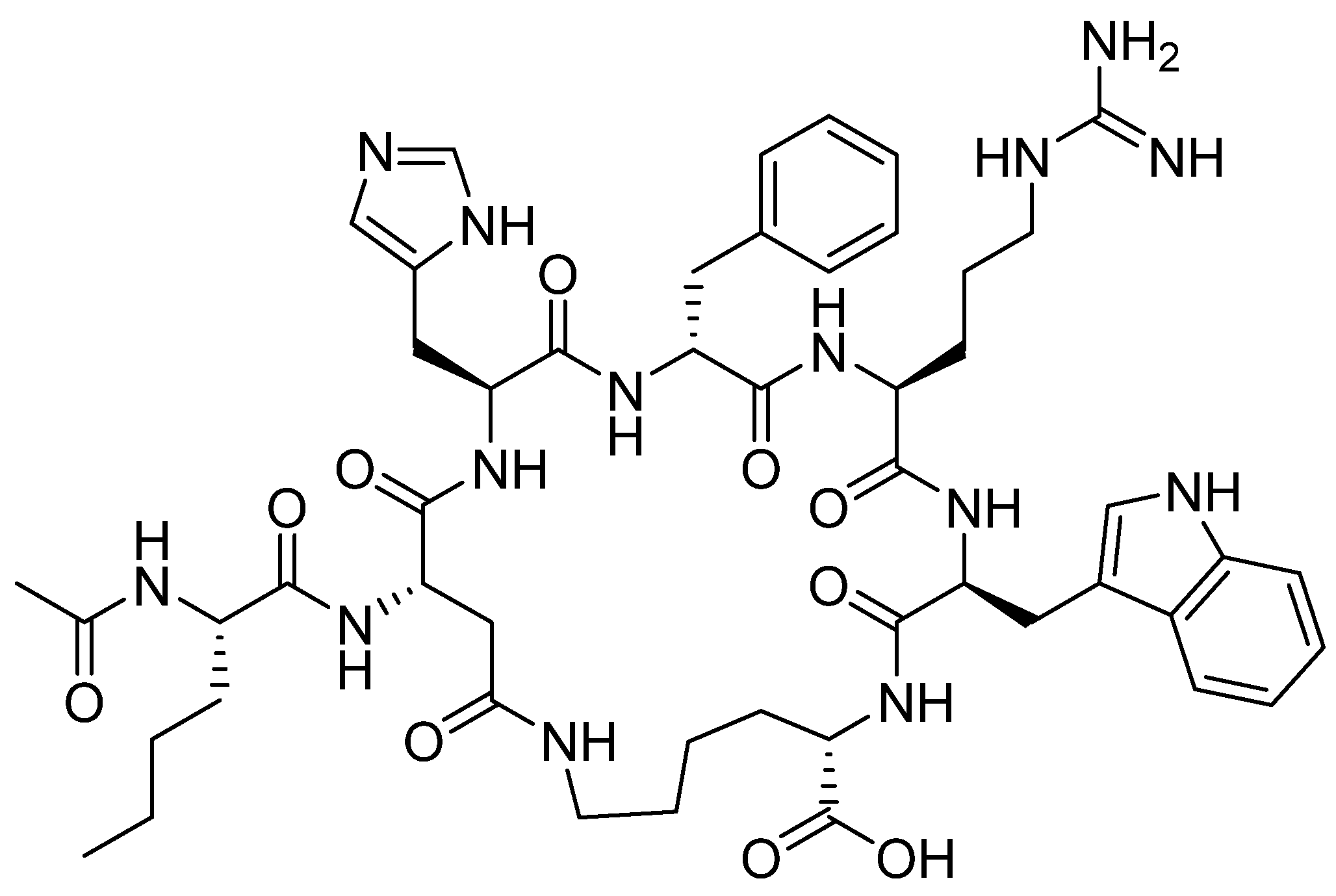
Figure 7.
Chemical structure of enfortumab vedotin-ejfv (PadcevTM) and polatuzumab vedotin-piiq (PolivyTM). MMAE; monomethyl auristatin E.
Figure 7.
Chemical structure of enfortumab vedotin-ejfv (PadcevTM) and polatuzumab vedotin-piiq (PolivyTM). MMAE; monomethyl auristatin E.
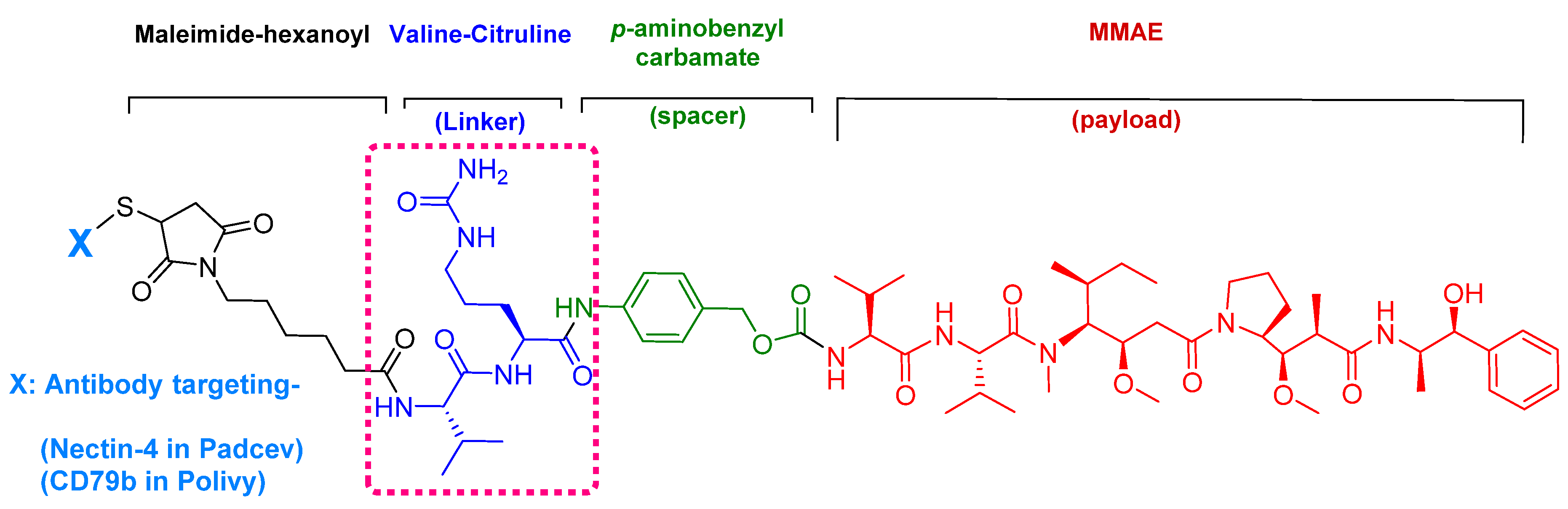
Figure 8.
Chemical structure of: (A) Synthetic monomethyl auristatin E (MMAE) analogue; (B) natural dolastatin 10. Differences are shown in red [50].
Figure 8.
Chemical structure of: (A) Synthetic monomethyl auristatin E (MMAE) analogue; (B) natural dolastatin 10. Differences are shown in red [50].
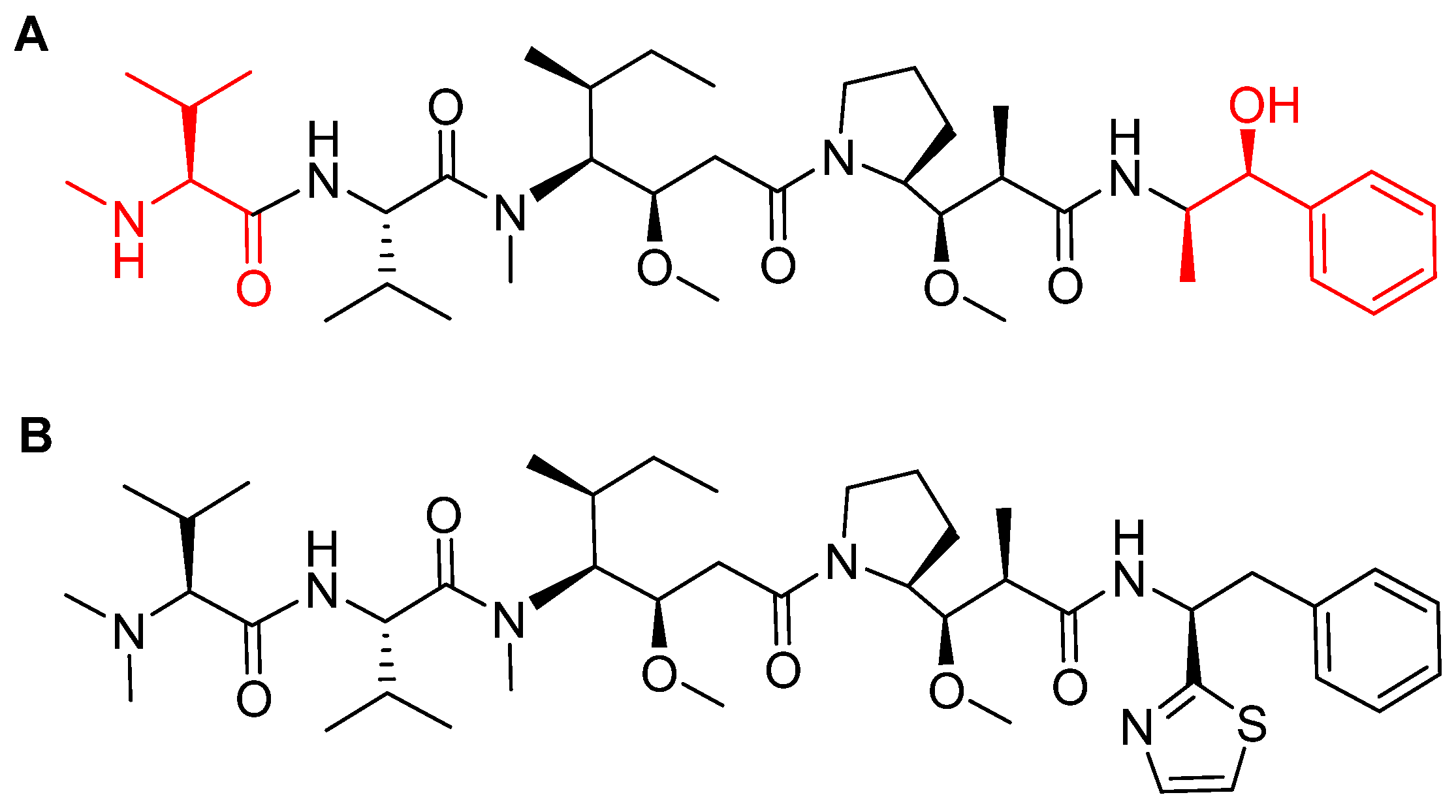
Figure 9.
Mechanism of payload release in ADCs with Val-Cit linker and p-aminobenzyl carbamate as a spacer [67]. MMAE; monomethyl auristatin E.
Figure 9.
Mechanism of payload release in ADCs with Val-Cit linker and p-aminobenzyl carbamate as a spacer [67]. MMAE; monomethyl auristatin E.
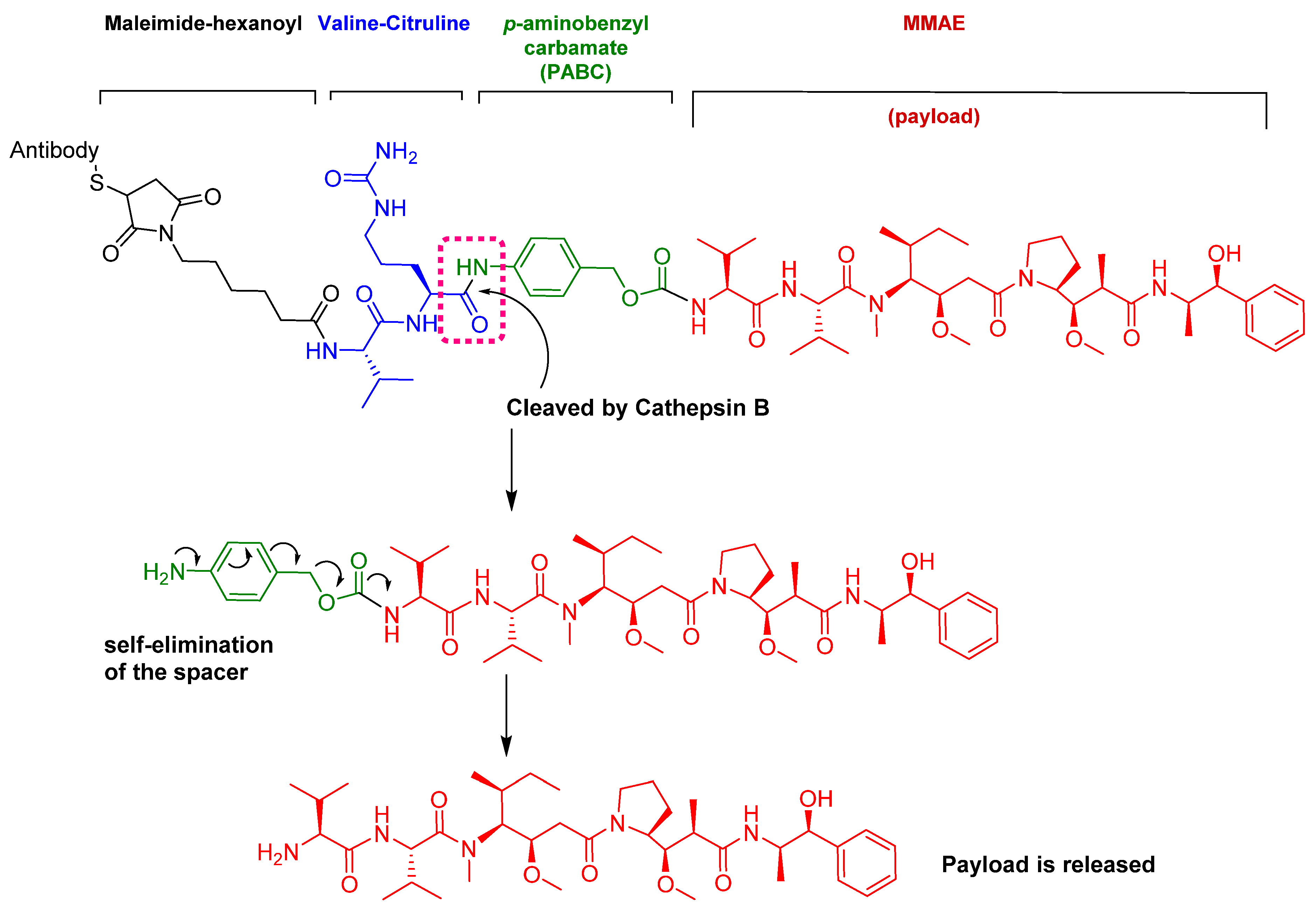
Figure 10.
Chemical structure of fam-trastuzumab deruxtecan-nxki (EnhertuTM) showing the tetrapeptide linker and amino methylene cleavage in the payload-release process. AM; aminomethyl.
Figure 10.
Chemical structure of fam-trastuzumab deruxtecan-nxki (EnhertuTM) showing the tetrapeptide linker and amino methylene cleavage in the payload-release process. AM; aminomethyl.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the 2019 FDA peptides & oligonucleotides (TIDES) harvest [5].
Table 1.
Summary of the 2019 FDA peptides & oligonucleotides (TIDES) harvest [5].
| # | Active Ingredient Trade Name | Type | Indication | Target | Route |
|---|---|---|---|---|---|
| 1 | Golodirsen Vyondys 53TM | Antisense oligonucleotide | Duchenne’s Muscular Dystrophy (DMD) | Exon 53 in dystrophin gene | Intravenous |
| 2 | Givosiran GivlaariTM | Antisense oligonucleotide | Acute Hepatic Porphyria (AHP) | Aminolevulinate synthase 1 (ALAS1) mRNA | Subcutaneous |
| 3 | 68Ga-DOTATOC | Peptide | Scintigraphic imaging | Somatostatin receptor | Intravenous |
| 4 | Afamelanotide ScenesseTM | Peptide | Erythropoietic protoporphyria (EPP) | Melanocyte-stimulating hormone receptor | Subcutaneous |
| 5 | Bremelanotide VyleesiTM | Peptide | Hypoactive sexual desire disorder | Melanocyte-stimulating hormone receptor | Subcutaneous |
| 6 | Enfortumab vedotin-ejfv PadcevTM | ADC with peptide payload and linker | Urothelial cancers | Nectin-4 receptor | Intravenous |
| 7 | Polatuzumab vedotin-piiq PolivyTM | ADC with peptide payload and linker | Refractory diffuse large B-cell lymphoma | CD79b receptor expressed in mature B-cells | Intravenous |
| 8 | Fam-trastuzumab deruxtecan-nxki EnhertuTM | ADC with a peptide linker | Unresectable or metastatic HER2-positive breast cancer | Human epidermal growth factor receptor-2 (HER2) | Intravenous |
ADC; antibody drug conjugate.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. https://doi.org/10.3390/ph13030040
AMA Style
Al Shaer D, Al Musaimi O, Albericio F, de la Torre BG. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals. 2020; 13(3):40. https://doi.org/10.3390/ph13030040
Chicago/Turabian StyleAl Shaer, Danah, Othman Al Musaimi, Fernando Albericio, and Beatriz G. de la Torre. 2020. "2019 FDA TIDES (Peptides and Oligonucleotides) Harvest" Pharmaceuticals 13, no. 3: 40. https://doi.org/10.3390/ph13030040
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.