2019 FDA TIDES (Peptides and Oligonucleotides) Harvest


 and
and 
Abstract
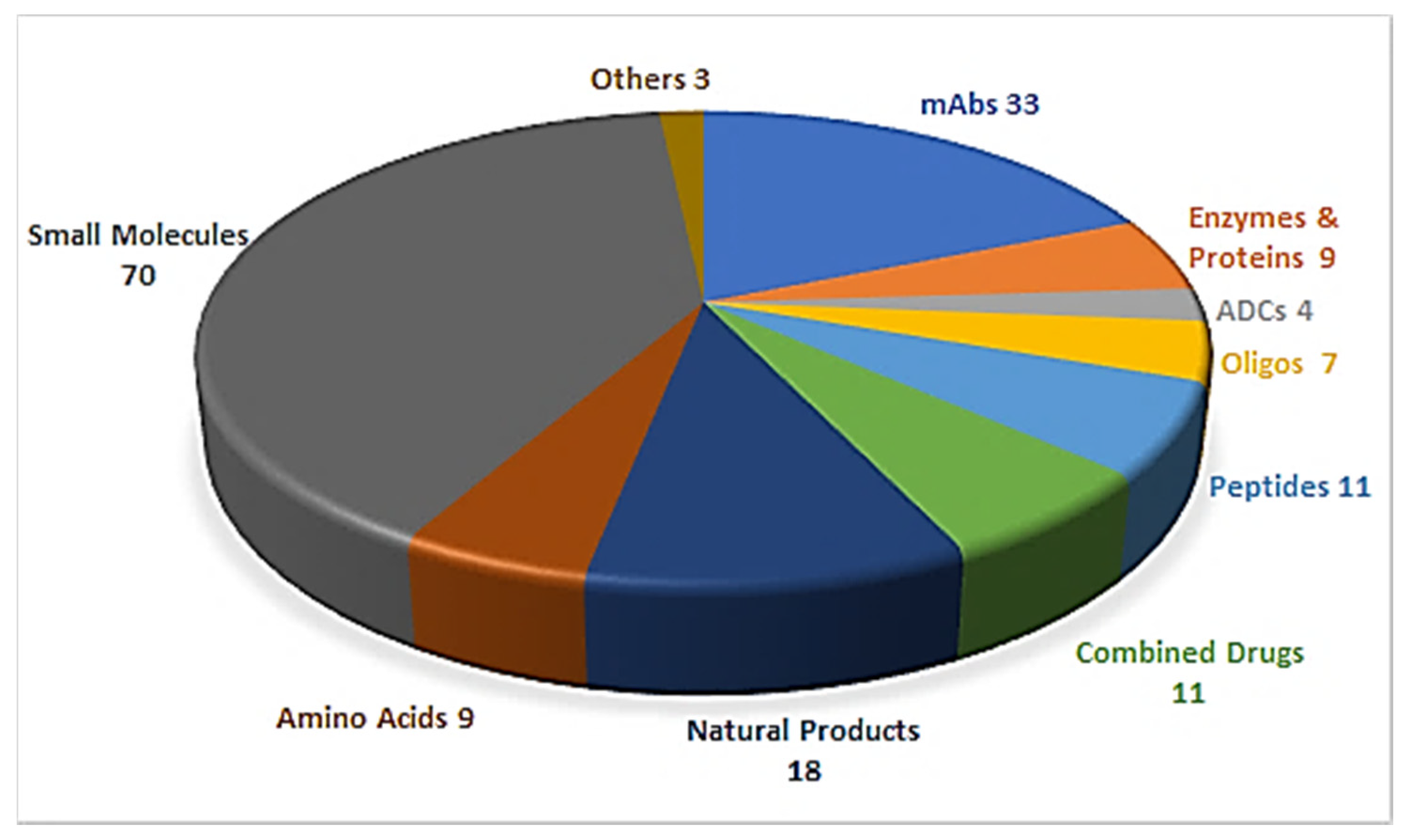
1. Introduction
2. Oligonucleotides
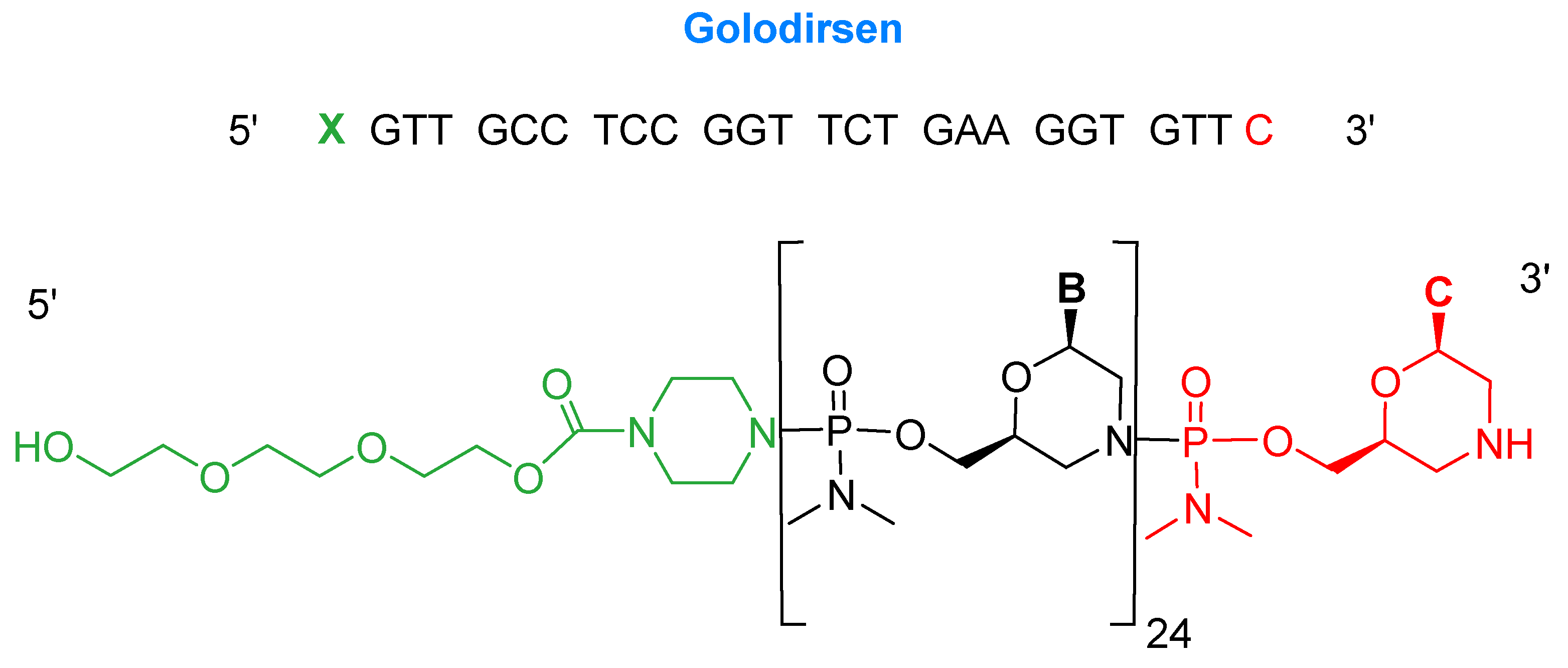
2.1. Golodirsen (Vyondys 53TM)
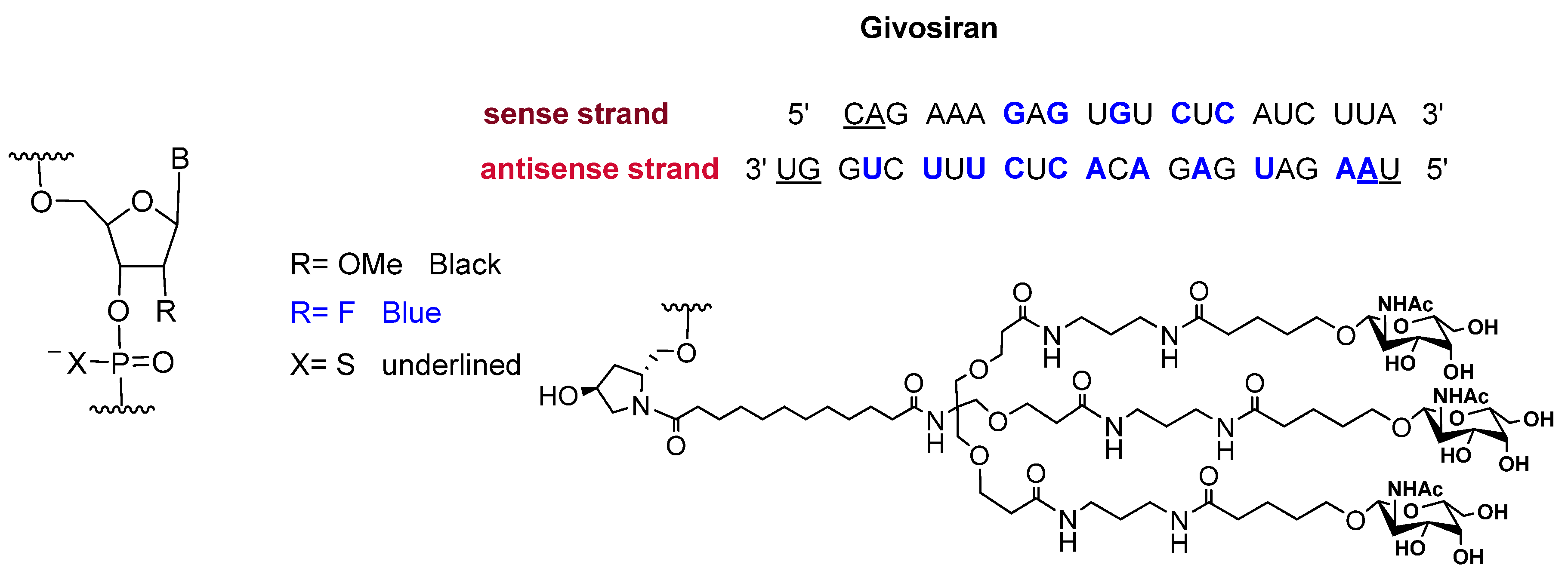
2.2. Givosiran (GivlaariTM)
3. Peptide-Based Drugs
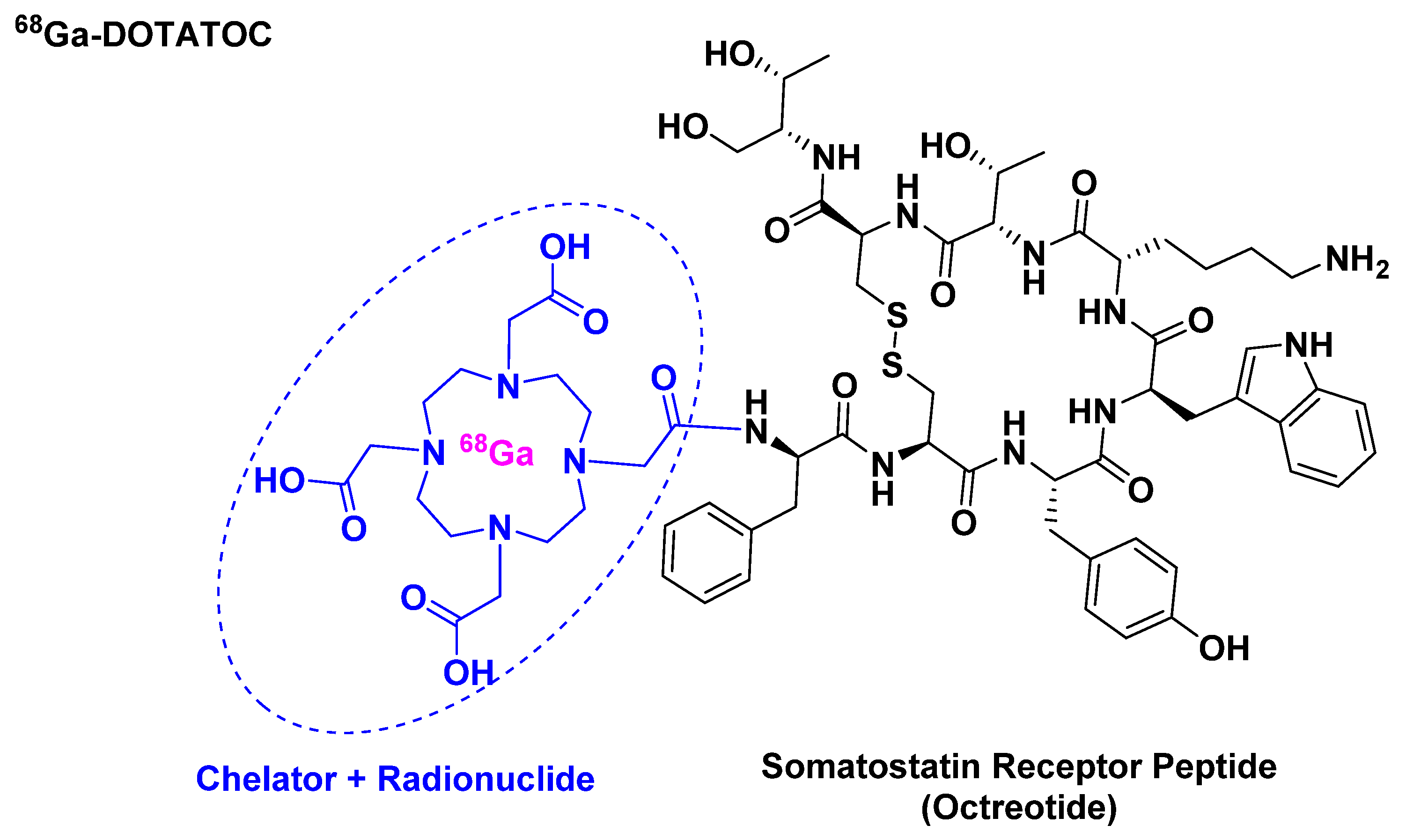
3.1. [68Ga]Ga-DOTATOC ([[68Ga]Ga-DOTA, Tyr3]-Octreotide)
3.2. Afamelanotide (Scenesse®)
3.3. Bremelanotide (VYLEESITM)
4. Peptides as Payloads in ADCs
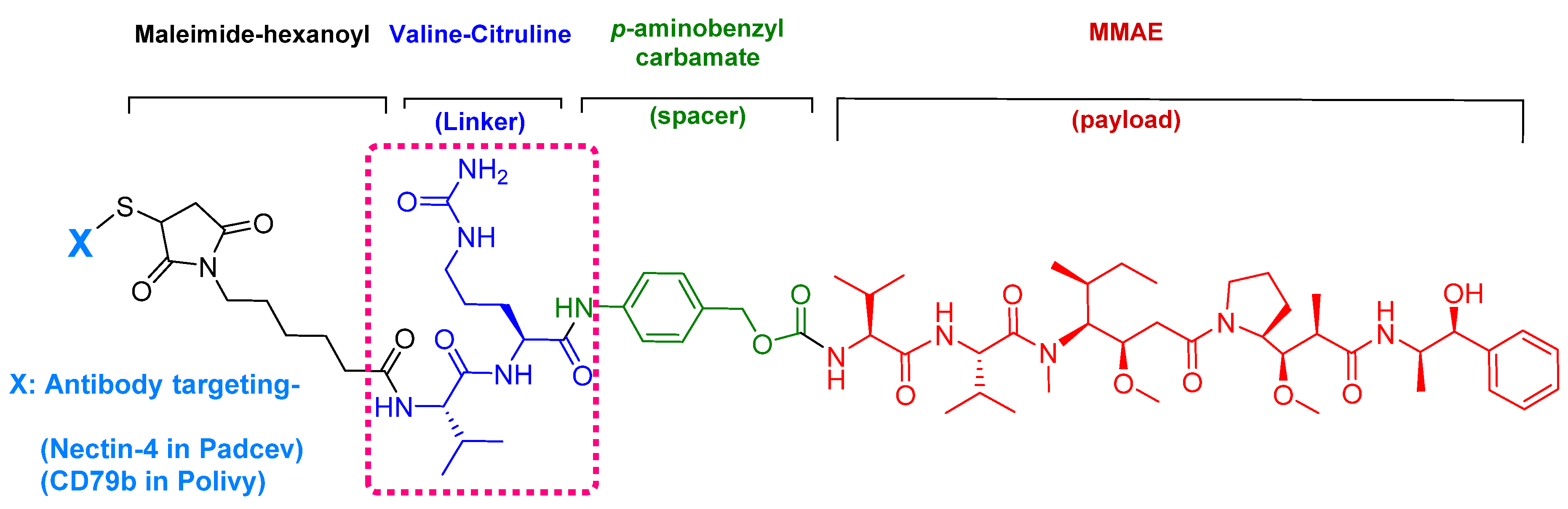
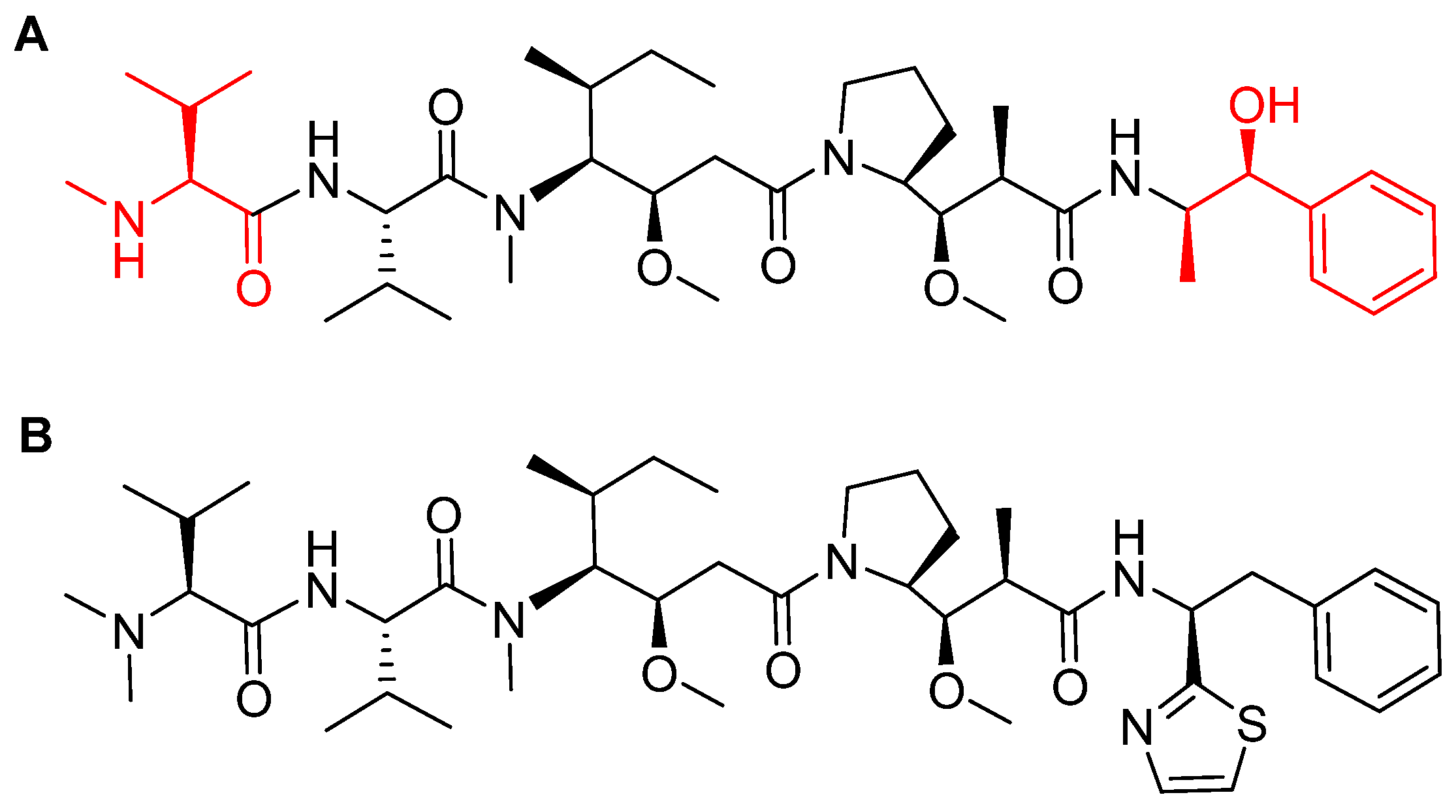
4.1. Enfortumab Vedotin-Ejfv (PADCEVTM)
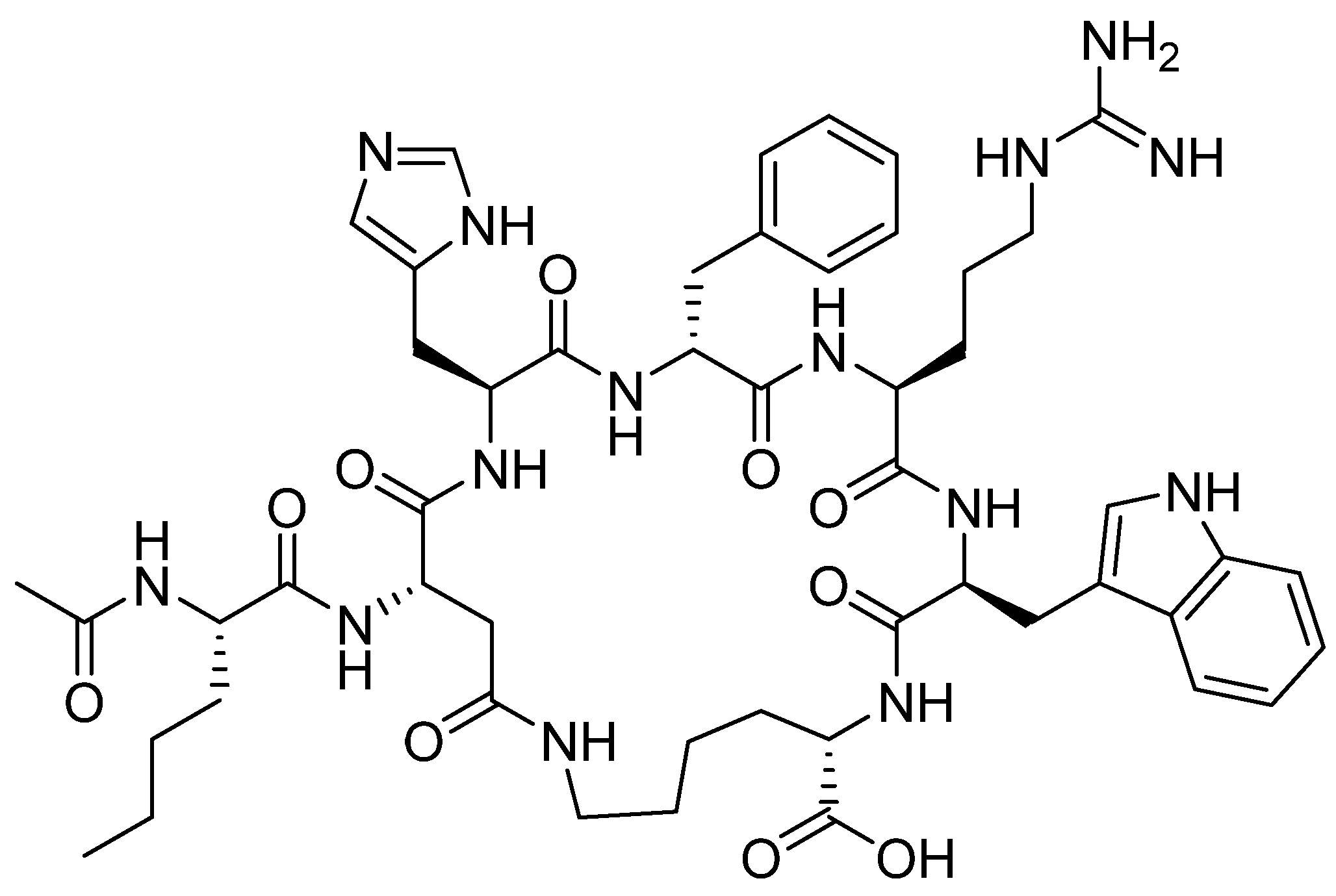
4.2. Polatuzumab Vedotin-Piiq (PolivyTM)
5. Peptides as Linkers in ADCs
5.1. Val-Cit
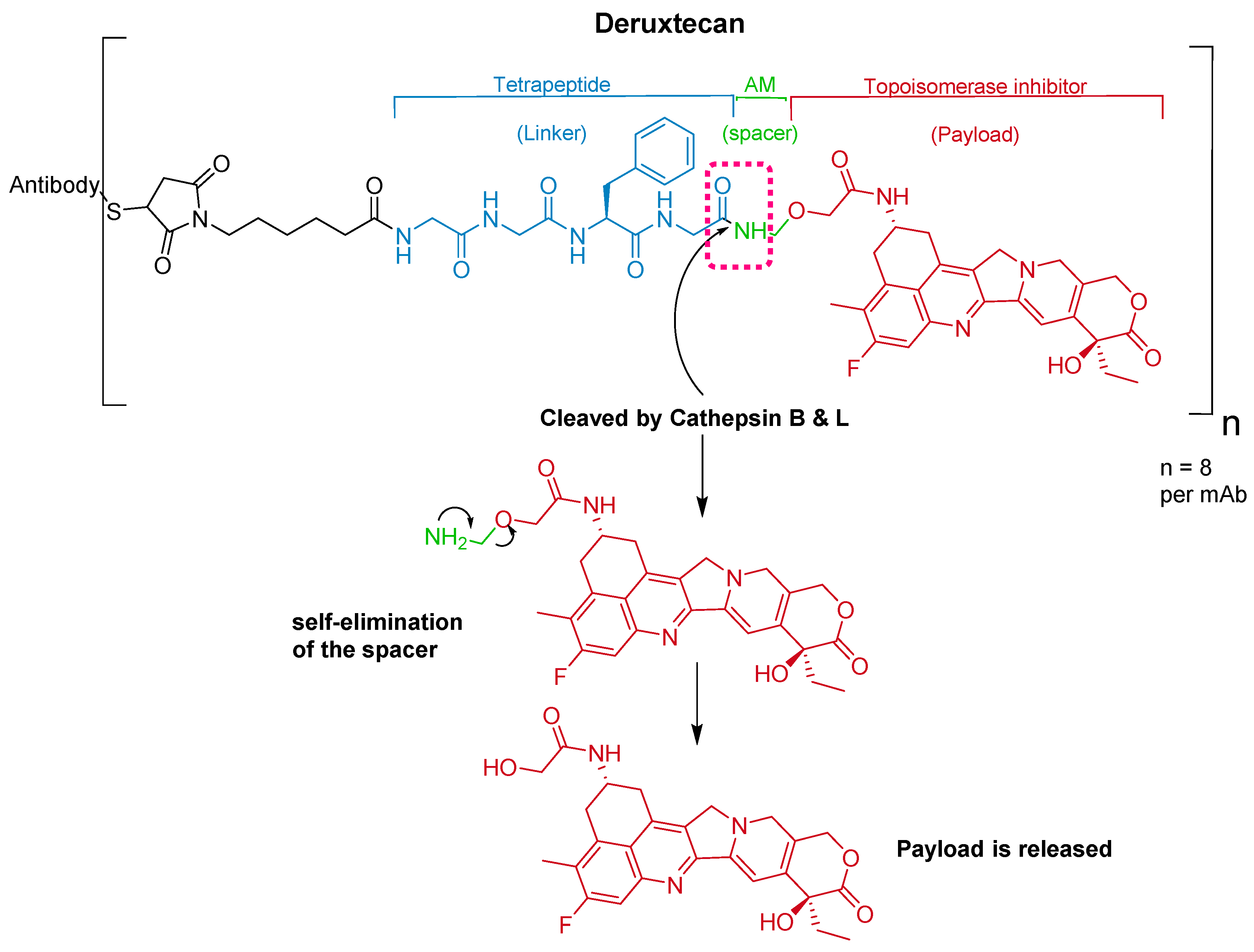
5.2. Gly-Gly-Phe-Gly
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2016. An analysis of FDA drug approvals from a perspective of the molecule type. Molecules 2017, 22. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2017. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2018, 23. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2019. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef]
- New Drug Therapy Approvals 2019. 2019. Available online: https://www.fda.gov/media/134493/download (accessed on 27 February 2020).
- Heo, Y.A. Golodirsen: First approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.L.; et al. Dystrophin cardiomyopathies: Clinical management, molecular pathogenesis and evolution towards precision medicine. J. Clin. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Gowran, A.; Bella, P.; Sitzia, C.; Scopece, A.; Castiglioni, E.; Rovina, D.; Nigro, P.; Villa, C.; Fortunato, F.; et al. Fibrosis rescue improves cardiac function in dystrophin-deficient mice and duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am. J. Pathol. 2019, 189, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Yokota, T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar] [PubMed]
- Vyondys 53 Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf (accessed on 27 February 2020).
- Vyondys 53 Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/211970Orig1s000ltr.pdf (accessed on 27 February 2020).
- Janas, M.M.; Harbison, C.E.; Perry, V.K.; Carito, B.; Sutherland, J.E.; Vaishnaw, A.K.; Keirstead, N.D.; Warner, G. The nonclinical safety profile of GalNAc-conjugated i therapeutics in subacute studies. Toxicol. Pathol. 2018, 46, 735–745. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2’-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Vita, G.; Vita, G.L.; Stancanelli, C.; Gentile, L.; Russo, M.; Mazzeo, A. Genetic neuromuscular disorders: Living the era of a therapeutic revolution. Part 1: Peripheral neuropathies. Neurol. Sci. 2019, 40, 661–669. [Google Scholar] [CrossRef] [PubMed]
- de Paula Brandao, P.R.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Leading RNA interference therapeutics part 2: Silencing delta-aminolevulinic acid synthase 1, with a focus on givosiran. Mol. Diagn. Ther. 2020, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Givlaari Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/0212194s000lbl.pdf (accessed on 27 February 2020).
- Graham, M.M.; Gu, X.; Ginader, T.; Breheny, P.; Sunderland, J.J. 68Ga-dotatoc imaging of neuroendocrine tumors: A systematic review and metaanalysis. J. Nucl. Med. 2017, 58, 1452–1458. [Google Scholar] [CrossRef]
- Menda, Y.; Ponto, L.L.B.; Schultz, M.K.; Zamba, G.K.D.; Watkins, G.L.; Bushnell, D.L.; Madsen, M.T.; Sunderland, J.J.; Graham, M.M.; O’Dorisio, T.M.; et al. Repeatability of gallium-68 dotatoc positron emission tomographic imaging in neuroendocrine tumors. Pancreas 2013, 42, 937–943. [Google Scholar] [CrossRef]
- Le, V.S. (68)Ga generator integrated system: Elution-purification-concentration integration. Recent Results Cancer Res. 2013, 194, 43–75. [Google Scholar]
- Poeppel, T.D.; Binse, I.; Petersenn, S.; Lahner, H.; Schott, M.; Antoch, G.; Brandau, W.; Bockisch, A.; Boy, C. 68Ga-dotatoc versus 68Ga-dotatate pet/ct in functional imaging of neuroendocrine tumors. J. Nucl. Med. 2011, 52, 1864–1870. [Google Scholar] [CrossRef]
- Khor, L.K.; Loi, H.Y.; Sinha, A.K.; Tong, K.T.; Goh, B.C.; Loh, K.S.; Lu, S.J. 68Ga-dota-peptide: A novel molecular biomarker for nasopharyngeal carcinoma. Head Neck 2016, 38, E76–E80. [Google Scholar] [CrossRef] [PubMed]
- 68Ga-dotatoc Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210828s000lbl.pdf (accessed on 27 February 2020).
- 68Ga-dotatoc Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210828Orig1s000ltr.pdf (accessed on 27 February 2020).
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2018 FDA tides harvest. Pharmaceuticals 2019, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K.; Sanfilippo, P.J.; Hruby, V.J.; Engel, M.H.; Heward, C.B.; Burnett, J.B.; Hadley, M.E. 4-Norleucine, 7-D-phenylalanine-a-melanocyte-stimulating hormone: A highly potent α-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. USA 1980, 77, 5754–5758. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.M.; McKay, J.T.; Bonkovsky, H.L. Advances in the management of erythropoietic protoporphyria - role of afamelanotide. Appl. Clin. Genet. 2016, 9, 179–189. [Google Scholar] [CrossRef]
- Spichty, R.; Balimann, M.; Barman, J.; Minder, E.I. A bioassay for the detection of neutralizing antibodies against the alpha-melanocyte stimulating hormone analog afamelanotide in patients with erythropoietic protoporphyria. J. Pharm. Biomed. Anal. 2013, 75, 192–198. [Google Scholar] [CrossRef]
- Kim, E.S.; Garnock-Jones, K.P. Afamelanotide: A review in erythropoietic protoporphyria. Am. J. Clin. Dermatol. 2016, 17, 179–185. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Harro, J.; Jaanisk, M.; Järv, A.; Podar, I.; Allik, J.; Nilsson, I.; Sakthivel, P.; Lefvert, A.K.; Hökfelt, T. Autoantibodies against neuropeptides are associated with psychological traits in eating disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 14865–14870. [Google Scholar] [CrossRef]
- Committee for medicinal products for human use (CHMP). Scenesse Assessment Report. 2014. Available online: https://www.ema.europa.eu/en/documents/assessment-report/scenesse-epar-public-assessment-report_en.pdf (accessed on 27 February 2020).
- Fabrikant, J.; Touloei, K.; Brown, S.M. A review and update on melanocyte stimulating hormone therapy: Afamelanotide. J. Drugs Dermatol. 2013, 12, 775–779. [Google Scholar]
- Scenesse Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210797s000lbl.pdf (accessed on 27 February 2020).
- Scenesse Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210797Orig1s000ltr.pdf (accessed on 27 February 2020).
- Molinoff, P.B.; Shadiack, A.M.; Earle, D.; Diamond, L.E.; Quon, C.Y. Pt-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann. N. Y. Acad. Sci. 2003, 994, 96–102. [Google Scholar] [CrossRef]
- Miller, M.K.; Smith, J.R.; Norman, J.J.; Clayton, A.H. Expert opinion on existing and developing drugs to treat female sexual dysfunction. Expert Opin. Emerg. Drugs 2018, 23, 223–230. [Google Scholar] [CrossRef]
- Both, S. Recent developments in psychopharmaceutical approaches to treating female sexual interest and arousal disorder. Curr. Sex Health Rep. 2017, 9, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.; Lucas, J.; DeRogatis, L.R.; Jordan, R. Phase I randomized placebo-controlled, double-blind study of the safety and tolerability of bremelanotide coadministered with ethanol in healthy male and female participants. Clin. Ther. 2017, 39, 514–526.e14. [Google Scholar] [CrossRef] [PubMed]
- Kingsberg, S.A.; Clayton, A.H.; Portman, D.; Williams, L.A.; Krop, J.; Jordan, R.; Lucas, J.; Simon, J.A. Bremelanotide for the treatment of hypoactive sexual desire disorder: Two randomized phase 3 trials. Obstet. Gynecol. 2019, 134, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Vyleesi Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210557s000lbl.pdf (accessed on 27 February 2020).
- Sohita, D.; Susan J., K. Bremalanotide: First approval. Drugs 2019, 79, 1599–1606. [Google Scholar]
- Vyleesi Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/210557Orig1s000ltr.pdf (accessed on 27 February 2020).
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer. 2016, 114, 362–367. [Google Scholar] [CrossRef]
- McGregor, B.A.; Sonpavde, G. Enfortumab vedotin, a fully human monoclonal antibody against nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents 337. Synthesis of dolastatin 10 structural modifications. Anticancer Drug Des. 1995, 10, 529–544. [Google Scholar]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloyd-Williams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. Antineoplastic agents. Part 189. The absolute configuration and synthesis of natural (-)-dolastatin 10. Am. Chem. Soc. 1989, 111, 5463–5465. [Google Scholar] [CrossRef]
- Akaiwa, M.; Martin, T.; Mendelsohn, B.A. Synthesis and evaluation of linear and macrocyclic dolastatin 10 analogues containing pyrrolidine ring modifications. ACS Omega 2018, 3, 5212–5221. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.; Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 2014, 42, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Uemura, M.; Kimura, T.; Kawasaki, Y.; Takamoto, A.; Yamaguchi, A.; Melhem-Bertrandt, A.; Gartner, E.M.; Inoue, T.; Akazawa, R.; et al. A phase I study of enfortumab vedotin in japanese patients with locally advanced or metastatic urothelial carcinoma. Investig. New Drugs 2019. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaide, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with Nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [PubMed]
- Hanna, K.S. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Padcev Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 27 February 2020).
- Padcev Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761137Orig1s000ltr.pdf (accessed on 27 February 2020).
- Sehn, L.H.; Matasar, M.J.; Flowers, C.R.; Kamdar, M.; McMillan, A.K.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab vedotin plus bendamustine with rituximab in relapsed/refractory diffuse large B-cell lymphoma: Updated results of a phase Ib/II randomized study. Blood 2019, 134, 4081. [Google Scholar] [CrossRef]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Polivy Drug Label. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 27 February 2020).
- Polivy Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761121Orig1s000ltr.pdf (accessed on 27 February 2020).
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular basis of valine-citrulline-PABC linker instability in site-specific ADCs and its mitigation by linker design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Dal Corso, A.; Cazzamalli, S.; Gebleux, R.; Mattarella, M.; Neri, D. Protease-cleavable linkers modulate the anticancer activity of noninternalizing antibody-drug conjugates. Bioconjug. Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef]
- Xu, Z.; Guo, D.; Jiang, Z.; Tong, R.; Jiang, P.; Bai, L.; Chen, L.; Zhu, Y.; Guo, C.; Shi, J.; et al. Novel HER2-targeting antibody-drug conjugates of trastuzumab beyond T-DM1 in breast cancer: Trastuzumab deruxtecan(DS-8201a) and (Vic-)trastuzumab duocarmazine (SYD985). Eur. J. Med. Chem. 2019, 183, 111682. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem. Pharm. Bull. (Tokyo) 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorg. Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Enhertu Approval Letter. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2019/761139Orig1s000ltr.pdf (accessed on 27 February 2020).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Active Ingredient Trade Name | Type | Indication | Target | Route |
|---|---|---|---|---|---|
| 1 | Golodirsen Vyondys 53TM | Antisense oligonucleotide | Duchenne’s Muscular Dystrophy (DMD) | Exon 53 in dystrophin gene | Intravenous |
| 2 | Givosiran GivlaariTM | Antisense oligonucleotide | Acute Hepatic Porphyria (AHP) | Aminolevulinate synthase 1 (ALAS1) mRNA | Subcutaneous |
| 3 | 68Ga-DOTATOC | Peptide | Scintigraphic imaging | Somatostatin receptor | Intravenous |
| 4 | Afamelanotide ScenesseTM | Peptide | Erythropoietic protoporphyria (EPP) | Melanocyte-stimulating hormone receptor | Subcutaneous |
| 5 | Bremelanotide VyleesiTM | Peptide | Hypoactive sexual desire disorder | Melanocyte-stimulating hormone receptor | Subcutaneous |
| 6 | Enfortumab vedotin-ejfv PadcevTM | ADC with peptide payload and linker | Urothelial cancers | Nectin-4 receptor | Intravenous |
| 7 | Polatuzumab vedotin-piiq PolivyTM | ADC with peptide payload and linker | Refractory diffuse large B-cell lymphoma | CD79b receptor expressed in mature B-cells | Intravenous |
| 8 | Fam-trastuzumab deruxtecan-nxki EnhertuTM | ADC with a peptide linker | Unresectable or metastatic HER2-positive breast cancer | Human epidermal growth factor receptor-2 (HER2) | Intravenous |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. https://doi.org/10.3390/ph13030040
Al Shaer D, Al Musaimi O, Albericio F, de la Torre BG. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals. 2020; 13(3):40. https://doi.org/10.3390/ph13030040
Chicago/Turabian StyleAl Shaer, Danah, Othman Al Musaimi, Fernando Albericio, and Beatriz G. de la Torre. 2020. "2019 FDA TIDES (Peptides and Oligonucleotides) Harvest" Pharmaceuticals 13, no. 3: 40. https://doi.org/10.3390/ph13030040
APA StyleAl Shaer, D., Al Musaimi, O., Albericio, F., & de la Torre, B. G. (2020). 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals, 13(3), 40. https://doi.org/10.3390/ph13030040