Tuning the Polymorphism of the Anti-VEGF G-rich V7t1 Aptamer by Covalent Dimeric Constructs

 ,
,  ,
,  , and
, and 
Abstract
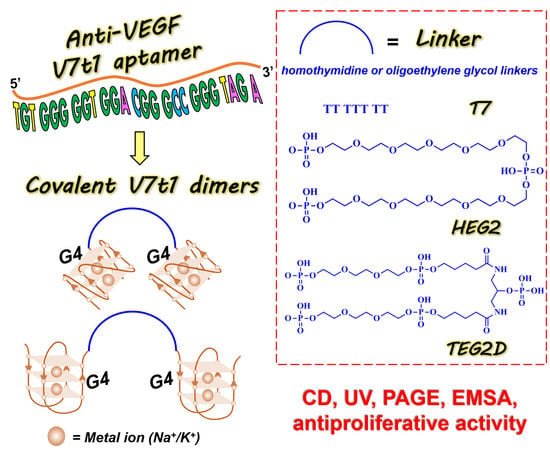
1. Introduction
2. Results and Discussion
2.1. Design and Preparation of the Covalent V7t1 Dimers
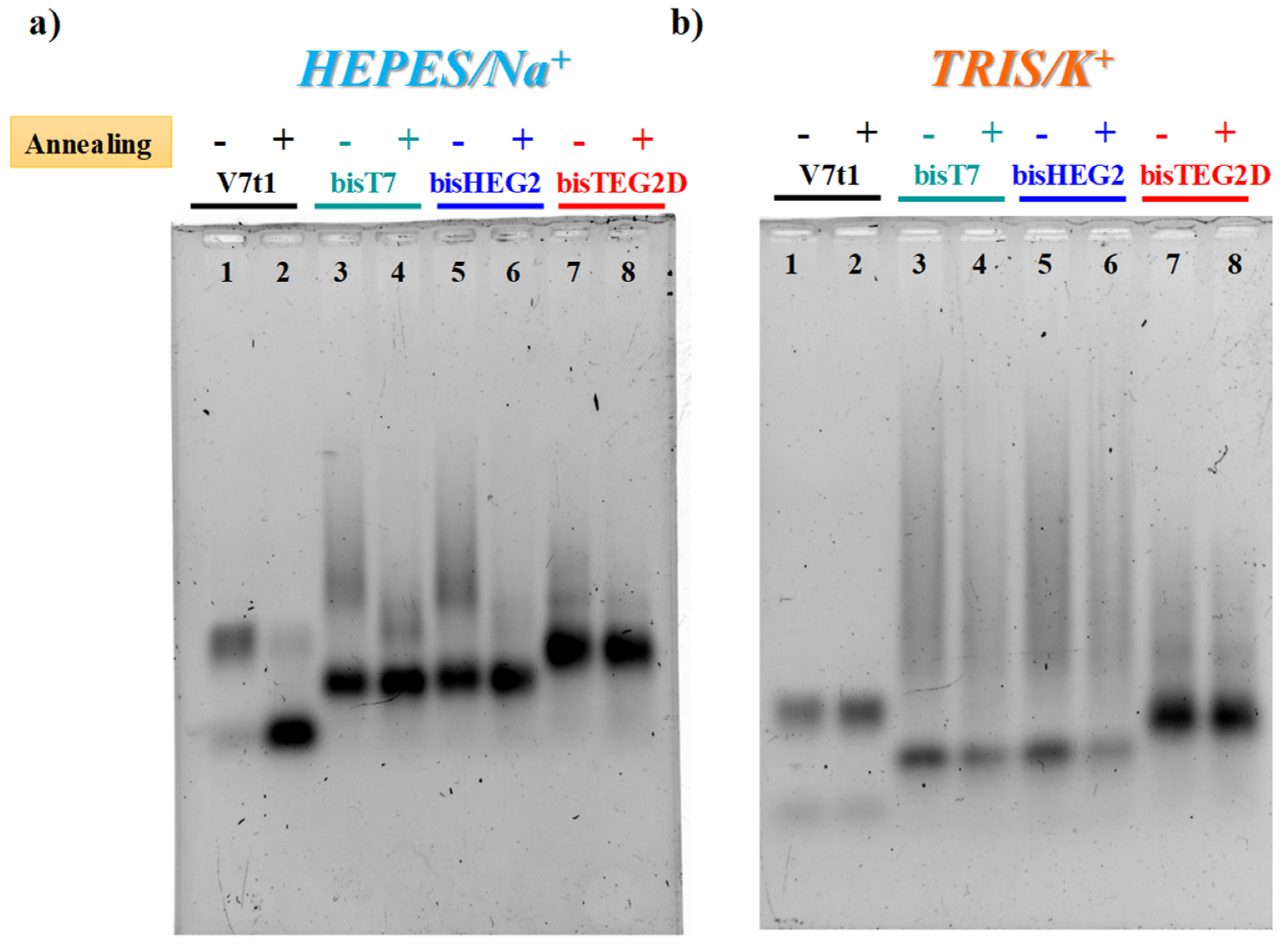
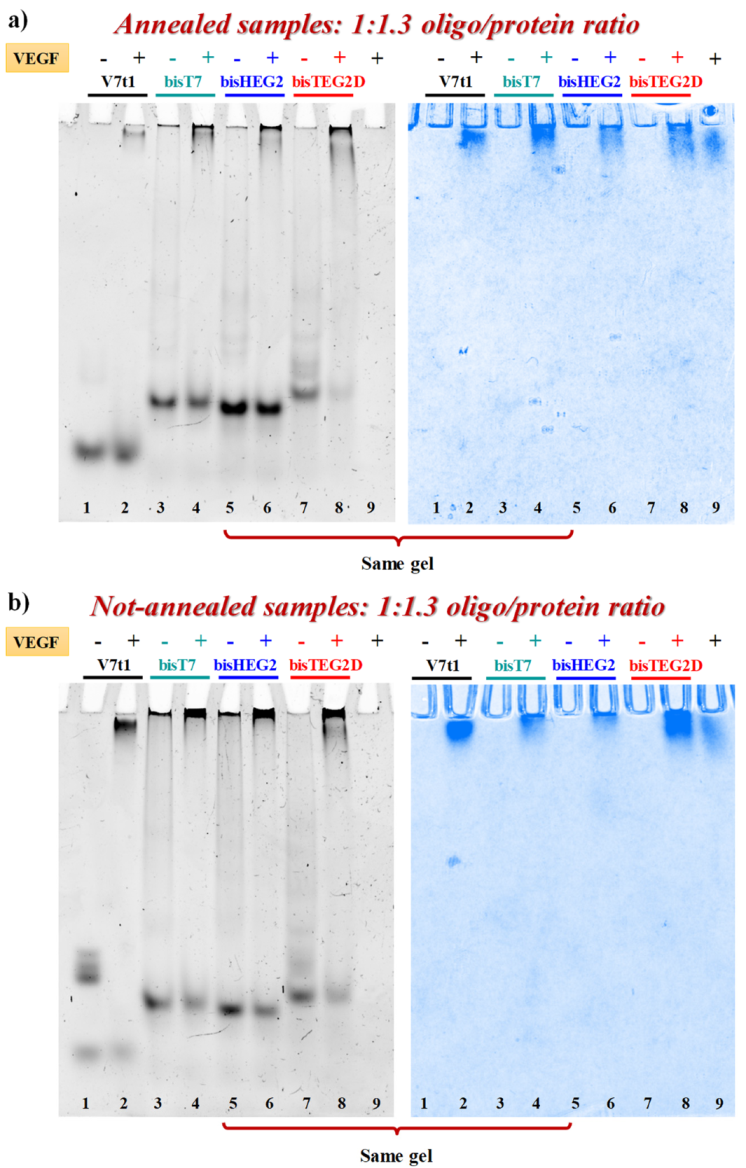
2.2. Gel Electrophoresis Analysis
2.3. Size exclusion (SE) Chromatography Analysis
2.4. Spectroscopic Characterization and Singular Value Decomposition (SVD) Analyses of Covalent V7t1 Dimers Folding
2.4.1. UV Analysis
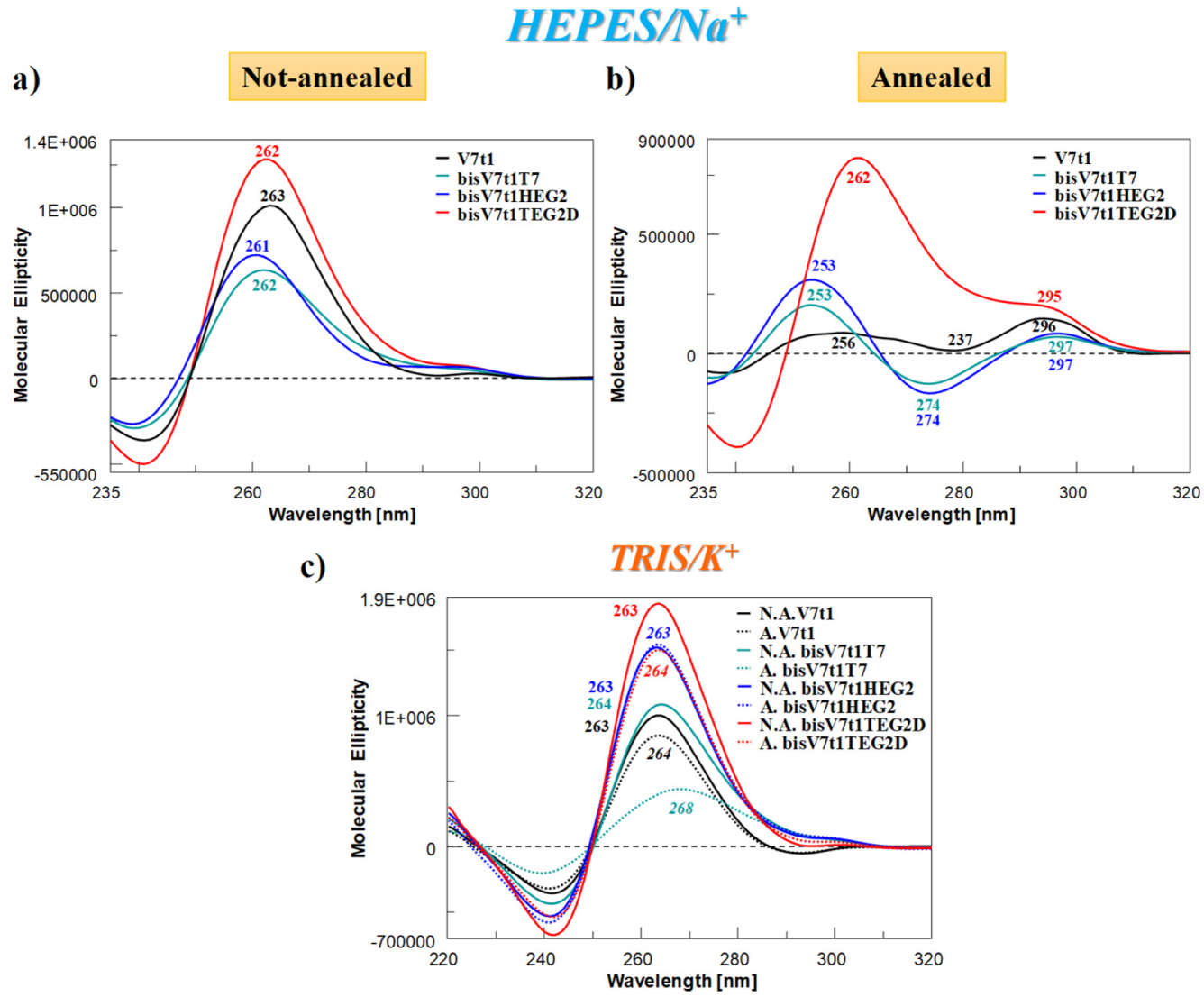
2.4.2. CD Spectra and SVD Analysis
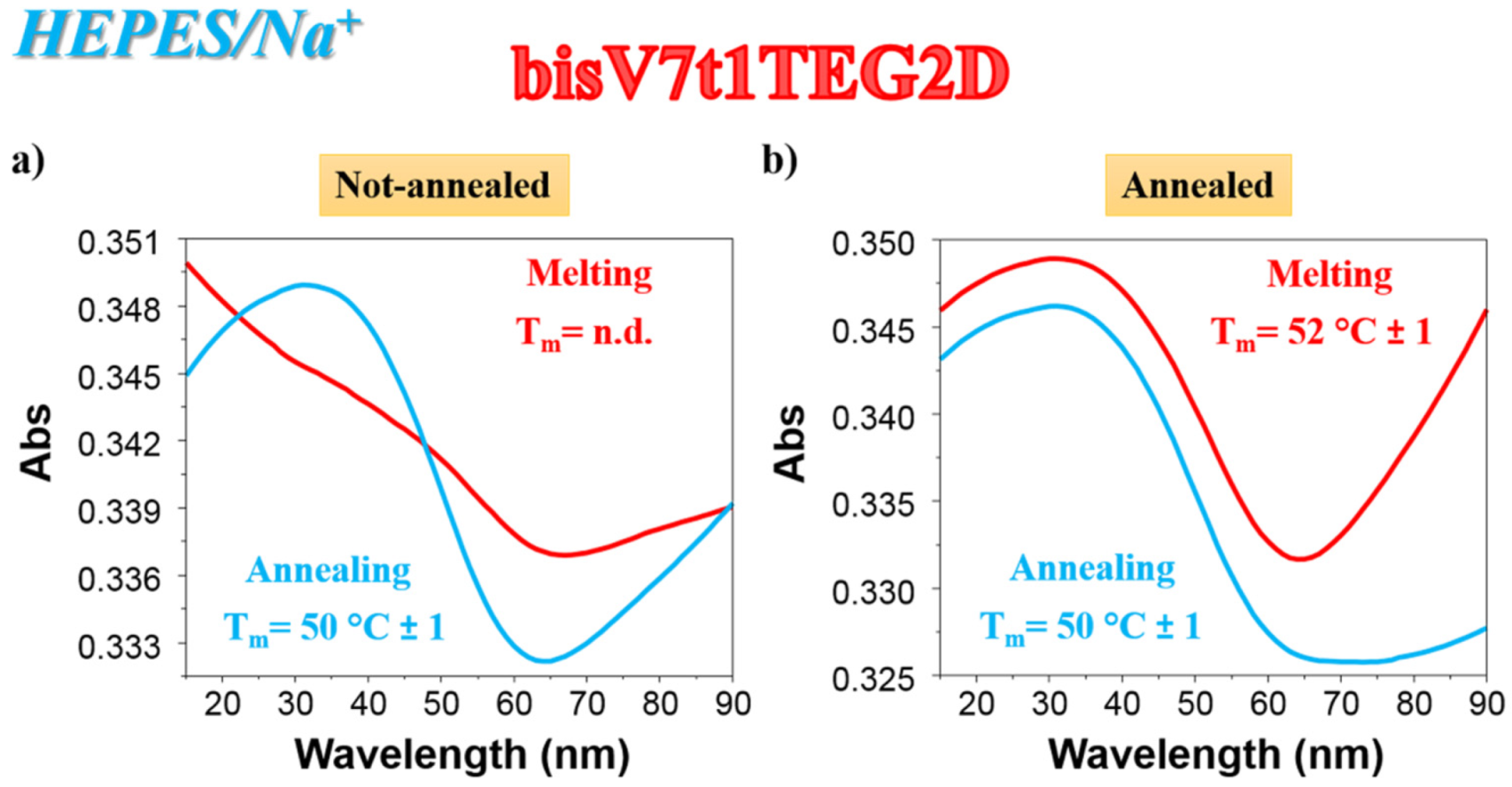
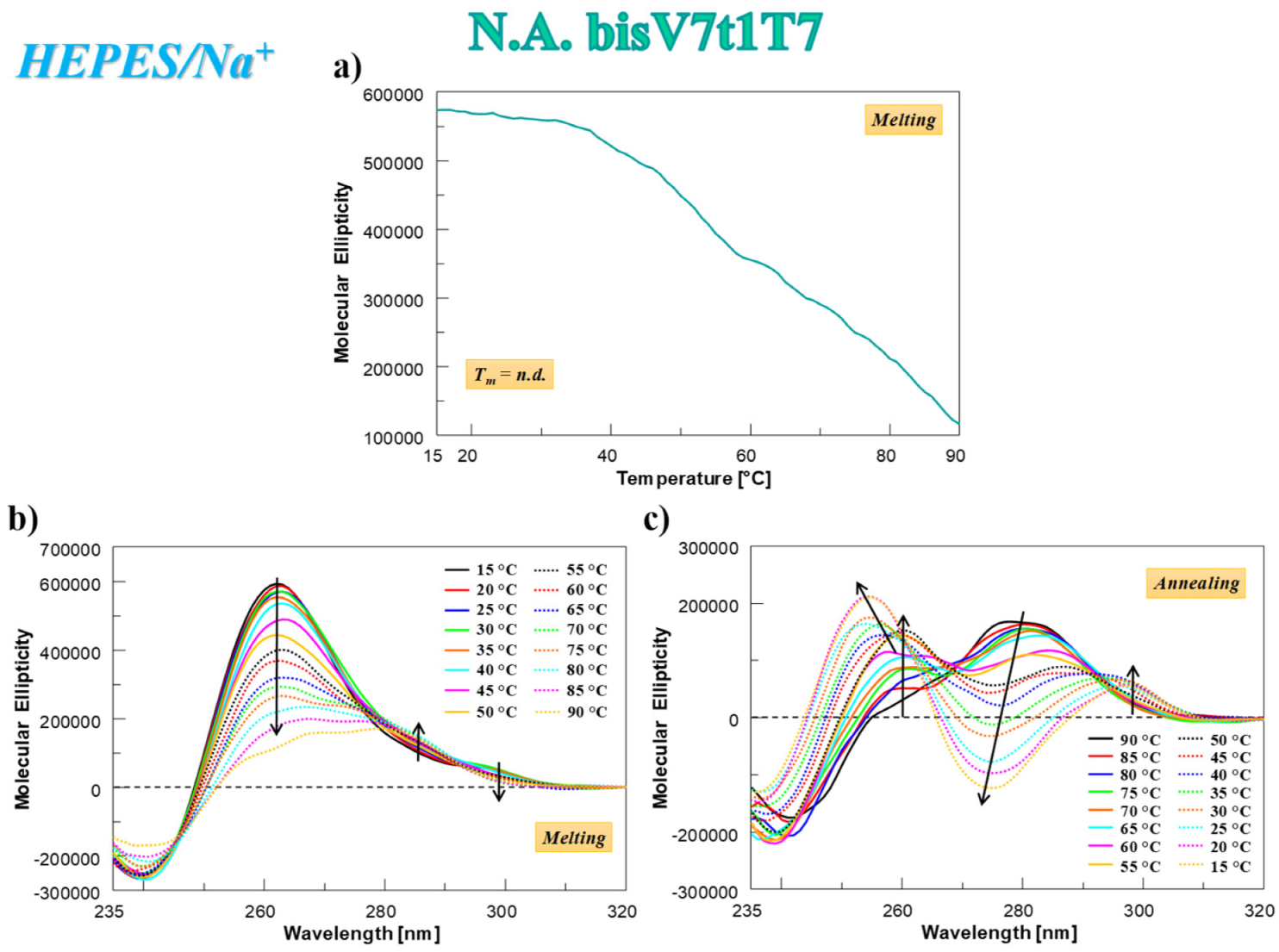
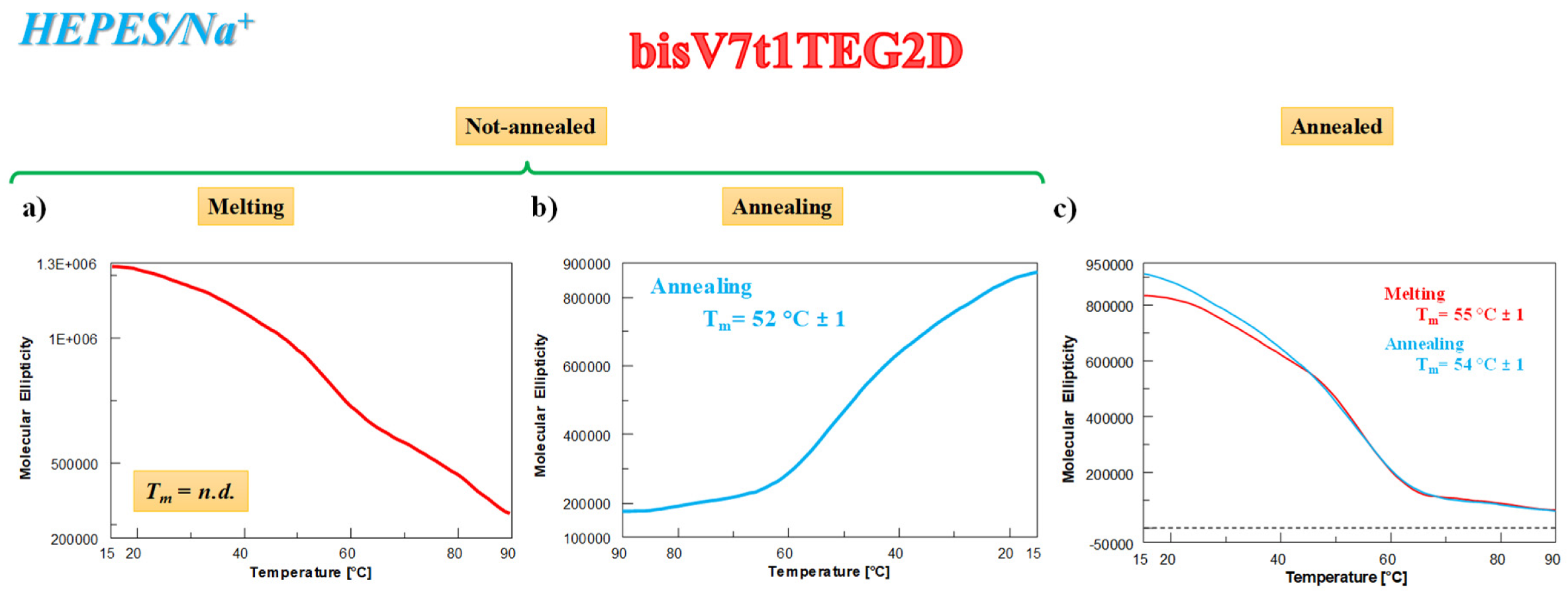
2.4.3. CD Thermal Denaturation/Renaturation Measurements
2.5. Binding Experiments with VEGF165 Protein
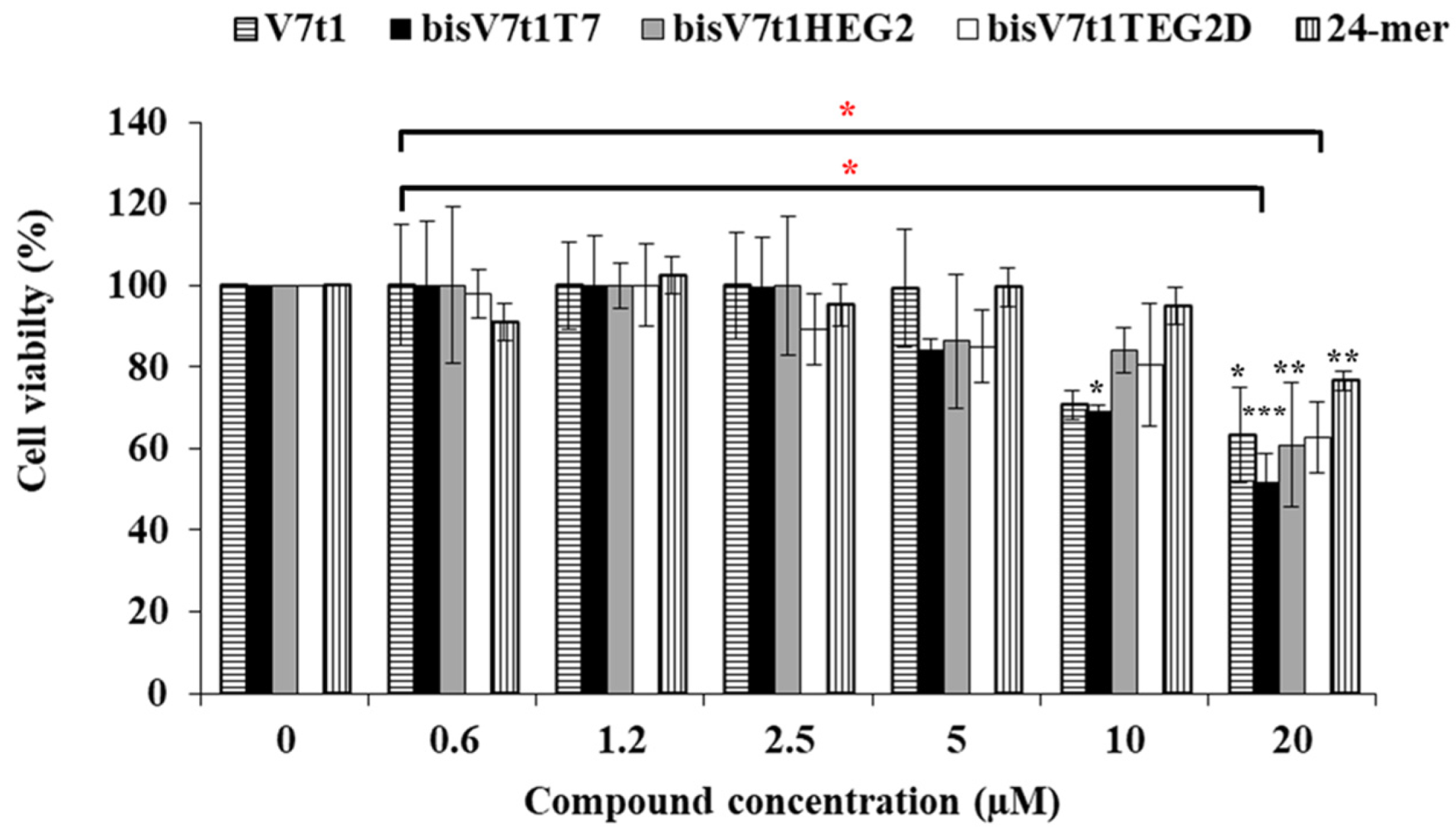
2.6. In vitro Bioactivity Studies
3. Conclusions
4. Materials and Methods
4.1. General Methods
- ▪
- V7t1 (5′TGTGGGGGTGGACGGGCCGGGTAGA3′);
- ▪
- bisV7t1T7 (5′V7t13′TTTTTTT5′V7t13′);
- ▪
- bisV7t1HEG2 (5′V7t13′C24H51O20P35′V7t13′);
- ▪
- bisV7t1TEG2D (5′V7t13′C25H53N2O24P53′V7t15′).
4.2. Preparation of the Oligonucleotide Samples
4.3. Gel Electrophoresis Analysis
4.4. Size Exclusion Chromatography
4.5. UV Spectroscopy
4.6. Circular Dichroism (CD) Spectroscopy
4.7. Electrophoresis Mobility Shift Assay (EMSA)
4.8. In vitro Biological Evaluation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guo, P.; Fang, Q.; Tao, H.Q.; Schafer, C.A.; Fenton, B.M.; Ding, I.; Hu, B.; Cheng, S.Y. Overexpression of vascular endothelial growth factor by MCF-7 breast cancer cells promotes estrogen-independent tumor growth in vivo. Cancer Res. 2003, 63, 4684–4691. [Google Scholar] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.R.; Zachary, I. The vascular endothelial growth factor (VEGF) family: Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular endothelial growth factor (VEGF) signaling in tumour vascularization: Potential and challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef]
- Yu, J.L.; Rak, J.W.; Klement, G.; Kerbel, R.S. Vascular endothelial growth factor isoform expression as a determinant of blood vessel patterning in human melanoma xenografts. Cancer Res. 2002, 62, 1838–1846. [Google Scholar]
- Catena, R.; Muniz-Medina, V.; Moralejo, B.; Javierre, B.; Best, C.J.M.; Emmert-Buck, M.R.; Green, J.E.; Baker, C.C.; Calvo, A. Increased expression of VEGF121/VEGF165-189 ratio results in a significant enhancement of human prostate tumor angiogenesis. Int. J. Cancer 2007, 120, 2096–2109. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef]
- Platella, C.; Riccardi, C.; Montesarchio, D.; Roviello, G.N.; Musumeci, D. G-quadruplex-based aptamers against protein targets in therapy and diagnostics. BBA-Gen. Subj. 2017, 1861, 1429–1447. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Vinores, S.A. Pegaptanib in the treatment of wet, age-related macular degeneration. Int. J. Nanomed. 2006, 3, 263–268. [Google Scholar]
- Nonaka, Y.; Sode, K.; Ikebukuro, K. Screening and improvement of an anti-VEGF DNA aptamer. Molecules 2010, 15, 215–225. [Google Scholar] [CrossRef]
- Edwards, S.L.; Poongavanam, V.; Kanwar, J.R.; Roy, K.; Hillman, K.M.; Prasad, N.; Leth-Larsen, R.; Petersen, M.; Marušič, M.; Plavec, J.; et al. Targeting VEGF with LNA-stabilized G-rich oligonucleotide for efficient breast cancer inhibition. Chem. Commun. 2015, 51, 9499–9502. [Google Scholar] [CrossRef]
- Marušič, M.; Veedu, R.N.; Wengel, J.; Plavec, J. G-rich VEGF aptamer with locked and unlocked nucleic acid modifications exhibits a unique G-quadruplex fold. Nucleic Acids Res. 2013, 41, 9524–9536. [Google Scholar] [CrossRef]
- Moccia, F.; Riccardi, C.; Musumeci, D.; Leone, S.; Oliva, R.; Petraccone, L.; Montesarchio, D. Insights into the G-rich VEGF-binding aptamer V7t1: When two G-quadruplexes are better than one! Nucleic Acids Res. 2019, 47, 8318–8331. [Google Scholar] [CrossRef]
- Nonaka, Y.; Yoshida, W.; Abe, K.; Ferri, S.; Schulze, H.; Bachmann, T.T.; Ikebukuro, K. Affinity improvement of a VEGF aptamer by in silico maturation for a sensitive VEGF-detection system. Anal. Chem. 2013, 85, 1132–1137. [Google Scholar] [CrossRef]
- Hasegawa, H.; Taira, K.I.; Sode, K.; Ikebukuro, K. Improvement of aptamer affinity by dimerization. Sensors 2008, 8, 1090–1098. [Google Scholar] [CrossRef]
- Manochehry, S.; Mcconnell, E.M.; Li, Y. Unraveling determinants of affinity enhancement in dimeric aptamers for a dimeric protein. Sci. Rep. 2019, 9, 17824. [Google Scholar] [CrossRef]
- Kaur, H.; Yung, L.Y.L. Probing high affinity sequences of DNA aptamer against VEGF165. PLoS ONE 2012, 7, e31196. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Montesarchio, D. Polyvalent nucleic acid aptamers and modulation of their activity: A focus on the thrombin binding aptamer. Pharmacol. Ther. 2012, 136, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.M.; Xiao, Y.; Tom Soh, H. Selection is more intelligent than design: Improving the affinity of a bivalent ligand through directed evolution. Nucleic Acids Res. 2012, 40, 11777–11783. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cao, Z.; Tan, W. Molecular assembly for high-performance bivalent nucleic acid inhibitor. Proc. Natl. Acad. Sci. USA 2008, 105, 5664–5669. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Wulffen, B.; Pötzsch, B.; Mayer, G. Multidomain targeting generates a high-affinity thrombin-inhibiting bivalent aptamer. ChemBioChem 2007, 8, 2223–2226. [Google Scholar] [CrossRef]
- Antipova, O.; Samoylenkova, N.; Savchenko, E.; Zavyalova, E.; Revishchin, A.; Pavlova, G.; Kopylov, A. Bimodular antiparallel G-quadruplex nanoconstruct with antiproliferative activity. Molecules 2019, 24, 3625. [Google Scholar] [CrossRef] [PubMed]
- Amato, T.; Virgilio, A.; Pirone, L.; Vellecco, V.; Bucci, M.; Pedone, E.; Esposito, V.; Galeone, A. Investigating the properties of TBA variants with twin thrombin binding domains. Sci. Rep. 2019, 9, 9184. [Google Scholar] [CrossRef] [PubMed]
- Batool, S.; Argyropoulos, K.V.; Azad, R.; Okeoma, P.; Zumrut, H.; Bhandari, S.; Dekhang, R.; Mallikaratchy, P.R. Dimerization of an aptamer generated from ligand-guided selection (LIGS) yields a high affinity scaffold against B-cells. Biochim. Biophys. Acta 2019, 1863, 232–240. [Google Scholar] [CrossRef]
- Mallikaratchy, P.R.; Ruggiero, A.; Gardner, J.R.; Kuryavyi, V.; Maguire, W.F.; Heaney, M.L.; McDevitt, M.R.; Patel, D.J.; Scheinberg, D.A. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2011, 39, 2458–2469. [Google Scholar] [CrossRef]
- Stuart, C.H.; Riley, K.R.; Boyacioglu, O.; Herpai, D.M.; Debinski, W.; Qasem, S.; Marini, F.C.; Colyer, C.L.; Gmeiner, W.H. Selection of a novel aptamer against vitronectin using capillary electrophoresis and next generation sequencing. Mol. Ther.-Nucleic Acids 2016, 5, e386. [Google Scholar] [CrossRef]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; De Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 aptamers as powerful immune response modulators. Mol. Ther.-Nucleic Acids 2013, 2, e98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lis, J.T.; Shi, H. A systematic study of the features critical for designing a high avidity multivalent aptamer. Nucleic Acid Ther. 2013, 23, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Kuai, H.; Zhao, Z.; Mo, L.; Liu, H.; Hu, X.; Fu, T.; Zhang, X.; Tan, W. Circular bivalent aptamers enable in vivo stability and recognition. J. Am. Chem. Soc. 2017, 139, 9128–9131. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for improving aptamer binding affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent aptamers: Versatile tools for diagnostic and therapeutic applications. Molecules 2016, 21, 613. [Google Scholar] [CrossRef] [PubMed]
- Hianik, T.; Grman, I.; Karpisova, I. The effect of DNA aptamer configuration on the sensitivity of detection thrombin at surface by acoustic method. Chem. Commun. 2009, 6303–6305. [Google Scholar] [CrossRef]
- Tsukakoshi, K.; Ikuta, Y.; Abe, K.; Yoshida, W.; Iida, K.; Ma, Y.; Nagasawa, K.; Sode, K.; Ikebukuro, K. Structural regulation by a G-quadruplex ligand increases binding abilities of G-quadruplex-forming aptamers. Chem. Commun. 2016, 52, 12646–12649. [Google Scholar] [CrossRef]
- De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Musumeci, D.; Piccialli, G. Synthesis and characterization of new 3′-3′ linked oligodeoxyribonucleotides for alternate strand triple helix formation. Tetrahedron 1999, 55, 9899–9914. [Google Scholar] [CrossRef]
- Hardin, C.C.; Watson, T.; Corregan, M.; Bailey, C. Cation-dependent transition between the quadruplex and Watson-Crick hairpin forms of d(CGCG3GCG). Biochemistry 1992, 31, 833–841. [Google Scholar] [CrossRef]
- Hud, N.V.; Smith, F.W.; Anet, F.A.L.; Feigon, J. The selectivity for K+ versus Na+ in DNA quadruplexes is dominated by relative free energies of hydration: A thermodynamic analysis by 1H NMR. Biochemistry 1996, 35, 15383–15390. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Arachchilage, G.M.; Basu, S. Metal cations in G-quadruplex folding and stability. Front. Chem. 2016, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Mergny, J.L.; Gabelica, V. Role of alkali metal ions in G-quadruplex nucleic acid structure and stability. Metal Ions Life Sci. 2016, 16, 203–258. [Google Scholar] [CrossRef]
- Riccardi, C.; Meyer, A.; Vasseur, J.J.; Russo Krauss, I.; Paduano, L.; Oliva, R.; Petraccone, L.; Morvan, F.; Montesarchio, D. Stability is not everything: The case of the cyclization of the thrombin binding aptamer. ChemBioChem 2019, 20, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Meyer, A.; Vasseur, J.-J.; Russo Krauss, I.; Paduano, L.; Morvan, F.; Montesarchio, D. Fine-tuning the properties of the thrombin binding aptamer through cyclization: Effect of the 5′-3′ connecting linker on the aptamer stability and anticoagulant activity. Bioorg. Chem. 2020, 94, 103379. [Google Scholar] [CrossRef]
- Stellwagen, N.C.; Bossi, A.; Gelfi, C.; Righetti, P.G. DNA and buffers: Are there any non interacting, neutral pH buffers? Anal. Biochem. 2000, 287, 167–175. [Google Scholar] [CrossRef]
- Dailey, M.M.; Clarke Miller, M.; Bates, P.J.; Lane, A.N.; Trent, J.O. Resolution and characterization of the structural polymorphism of a single quadruplex-forming sequence. Nucleic Acids Res. 2010, 38, 4877–4888. [Google Scholar] [CrossRef]
- Largy, E.; Mergny, J.-L. Shape matters: Size-exclusion HPLC for the study of nucleic acid structural polymorphism. Nucleic Acids Res. 2014, 42, e149. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Russo Krauss, I.; Piccolo, M.; Irace, C.; Paduano, L.; Montesarchio, D. Exploring the conformational behaviour and aggregation properties of lipid-conjugated AS1411 aptamers. Int. J. Biol. Macromol. 2018, 118, 1384–1399. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Li, J.; Lacroix, L.; Amrane, S.; Chaires, J.B. Thermal difference spectra: A specific signature for nucleic acid structures. Nucleic Acids Res. 2005, 33, 1–6. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Lacroix, L. UV Melting of G-Quadruplexes. Curr. Protoc. Nucleic Acid Chem. 2009, 37, 17.1.1–17.1.15. [Google Scholar] [CrossRef]
- Malgowska, M.; Gudanis, D.; Teubert, A.; Dominiak, G.; Gdaniec, Z. How to study G-quadruplex structures. J. Biotechnol. Comput. Biol. Bionanotechnol. 2012, 93, 381–390. [Google Scholar] [CrossRef]
- Poniková, S.; Tlučková, K.; Antalík, M.; Víglaský, V.; Hianik, T. The circular dichroism and differential scanning calorimetry study of the properties of DNA aptamer dimers. Biophys. Chem. 2011, 155, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Paramasivan, S.; Rujan, I.; Bolton, P.H. Circular dichroism of quadruplex DNAs: Applications to structure, cation effects and ligand binding. Methods 2007, 43, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef]
- Masiero, S.; Trotta, R.; Pieraccini, S.; De Tito, S.; Perone, R.; Randazzo, A.; Spada, G.P. A non-empirical chromophoric interpretation of CD spectra of DNA G-quadruplex structures. Org. Biomol. Chem. 2010, 8, 2683–2692. [Google Scholar] [CrossRef]
- Karsisiotis, A.I.; Hessari, N.M.A.; Novellino, E.; Spada, G.P.; Randazzo, A.; Webba da Silva, M. Topological characterization of nucleic acid G-Quadruplexes by UV absorption and circular dichroism. Angew. Chem. Int. Ed. Eng. 2011, 50, 10645–10648. [Google Scholar] [CrossRef]
- Vorlíčková, M.; Kejnovská, I.; Sagi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Circular dichroism and guanine quadruplexes. Methods 2012, 57, 64–75. [Google Scholar] [CrossRef]
- Randazzo, A.; Spada, G.P.; Webba da Silva, M. Circular dichroism of quadruplex structures. Top. Curr. Chem. 2013, 330, 67–86. [Google Scholar] [CrossRef]
- Del Villar-Guerra, R.; Gray, R.D.; Chaires, J.B. Characterization of quadruplex DNA structure by circular dichroism. Curr. Protoc. Nucleic Acid Chem. 2017, 68, 17.8.1–17.8.16. [Google Scholar] [CrossRef]
- Del Villar-Guerra, R.; Trent, J.O.; Chaires, J.B. G-quadruplex secondary structure obtained from circular dichroism spectroscopy. Angew. Chem. Int. Ed. Eng. 2018, 57, 7171–7175. [Google Scholar] [CrossRef]
- Hellman, L.M.; Fried, M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Lönne, M.; Bolten, S.; Lavrentieva, A.; Stahl, F.; Scheper, T.; Walter, J.G. Development of an aptamer-based affinity purification method for vascular endothelial growth factor. Biotechnol. Rep. 2015, 8, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Roviello, G.N.; Rigione, G.; Capasso, D.; Di Gaetano, S.; Riccardi, C.; Roviello, V.; Montesarchio, D. Benzodifuran derivatives as potential antiproliferative agents: Possible correlation between their bioactivity and aggregation properties. ChemPlusChem 2017, 82, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Cioffi, F.; Broersen, K.; Roviello, V.; Riccardi, C.; Montesarchio, D.; Capasso, D.; Di Gaetano, S.; Roviello, G.N. Benzodifurans for biomedical applications: BZ4, a selective anti-proliferative and anti-amyloid lead compound. Future Med. Chem. 2019, 11, 285–302. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, N.; Tang, Y.; Yang, Y.; Chu, X.; Zhao, Y. SL2B aptamer and folic acid dual-targeting DNA nanostructures for synergic biological effect with chemotherapy to combat colorectal cancer. Int. J. Nanomedicine 2017, 12, 2657–2672. [Google Scholar] [CrossRef]
- Kaur, H.; Li, J.J.; Bay, B.H.; Yung, L.Y.L. Investigating the antiproliferative activity of high affinity DNA aptamer on cancer cells. Plos ONE 2013, 8, e50964. [Google Scholar] [CrossRef]
- Petraccone, L.; Pagano, B.; Giancola, C. Studying the effect of crowding and dehydration on DNA G-quadruplexes. Methods 2012, 57, 76–83. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Phan, A.; Lacroix, L. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998, 435, 74–78. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Linker Length |
|---|---|---|
| bisV7t1T7 |  | 42 |
| bisV7t1HEG2 |  | 40 |
| bisV7t1TEG2D |  | 40 |
| Prediction of G4 Topologies Relative Abundance from SVD Analysis of CD Spectra | |||
|---|---|---|---|
| HEPES/Na+ | |||
| Parallel (%) | Hybrid (%) | Antiparallel (%) | |
| N.A. V7t1 | 100 | 1 | 0 |
| A. V7t1 | 36 | 0 | 63 |
| N.A. bisV7t1T7 | 72 | 7 | 20 |
| A. bisV7t1T7 | 32 | 0 | 67 |
| N.A. bisV7t1HEG2 | 78 | 0 | 23 |
| A. bisV7t1HEG2 | 34 | 0 | 65 |
| N.A. bisV7t1TEG2D | 100 | 1 | 0 |
| A. bisV7t1TEG2D | 75 | 26 | 0 |
| TRIS/K+ | |||
| N.A. V7t1 | 100 | 1 | 0 |
| A. V7t1 | 92 | 2 | 6 |
| N.A. bisV7t1T7 | 97 | 4 | 0 |
| A. bisV7t1T7 | 45 | 38 | 16 |
| N.A. bisV7t1HEG2 | 100 | 1 | 0 |
| A. bisV7t1HEG2 | 100 | 1 | 0 |
| N.A. bisV7t1TEG2D | 100 | 1 | 0 |
| A. bisV7t1TEG2D | 100 | 1 | 0 |
| Molecule | IC50 (µM) |
|---|---|
| V7t1 | >20 µM |
| bisV7t1T7 | 19 µM |
| bisV7t1HEG2 | >20 µM |
| bisV7t1TEG2D | >20 µM |
| control 24-mer | >20 µM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riccardi, C.; Musumeci, D.; Platella, C.; Gaglione, R.; Arciello, A.; Montesarchio, D. Tuning the Polymorphism of the Anti-VEGF G-rich V7t1 Aptamer by Covalent Dimeric Constructs. Int. J. Mol. Sci. 2020, 21, 1963. https://doi.org/10.3390/ijms21061963
Riccardi C, Musumeci D, Platella C, Gaglione R, Arciello A, Montesarchio D. Tuning the Polymorphism of the Anti-VEGF G-rich V7t1 Aptamer by Covalent Dimeric Constructs. International Journal of Molecular Sciences. 2020; 21(6):1963. https://doi.org/10.3390/ijms21061963
Chicago/Turabian StyleRiccardi, Claudia, Domenica Musumeci, Chiara Platella, Rosa Gaglione, Angela Arciello, and Daniela Montesarchio. 2020. "Tuning the Polymorphism of the Anti-VEGF G-rich V7t1 Aptamer by Covalent Dimeric Constructs" International Journal of Molecular Sciences 21, no. 6: 1963. https://doi.org/10.3390/ijms21061963
APA StyleRiccardi, C., Musumeci, D., Platella, C., Gaglione, R., Arciello, A., & Montesarchio, D. (2020). Tuning the Polymorphism of the Anti-VEGF G-rich V7t1 Aptamer by Covalent Dimeric Constructs. International Journal of Molecular Sciences, 21(6), 1963. https://doi.org/10.3390/ijms21061963