Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome

 ,
,  ,
,  and
and 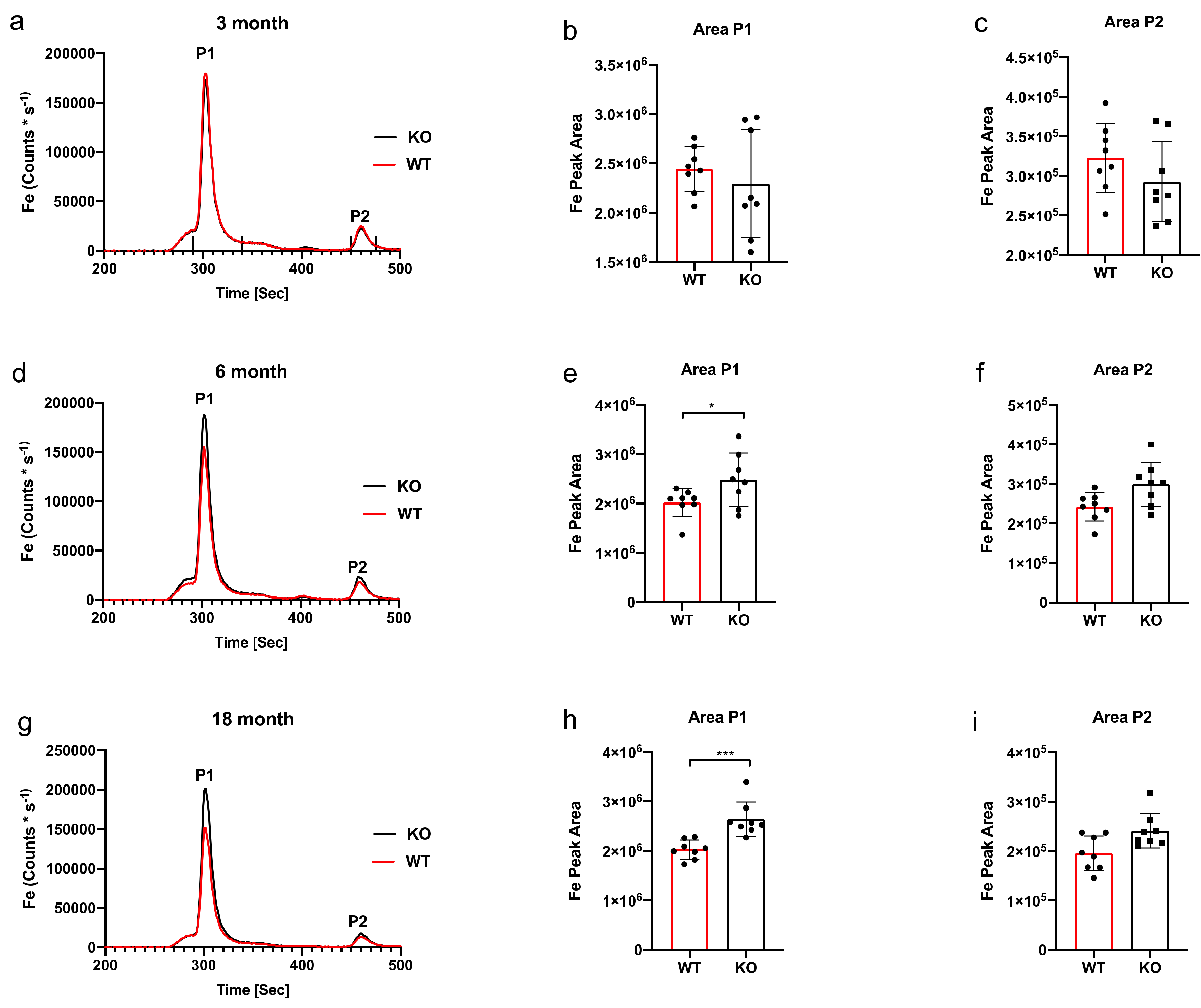{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics
2.2. Animals
2.3. SEC-ICP-MS
2.4. Statistical Analysis
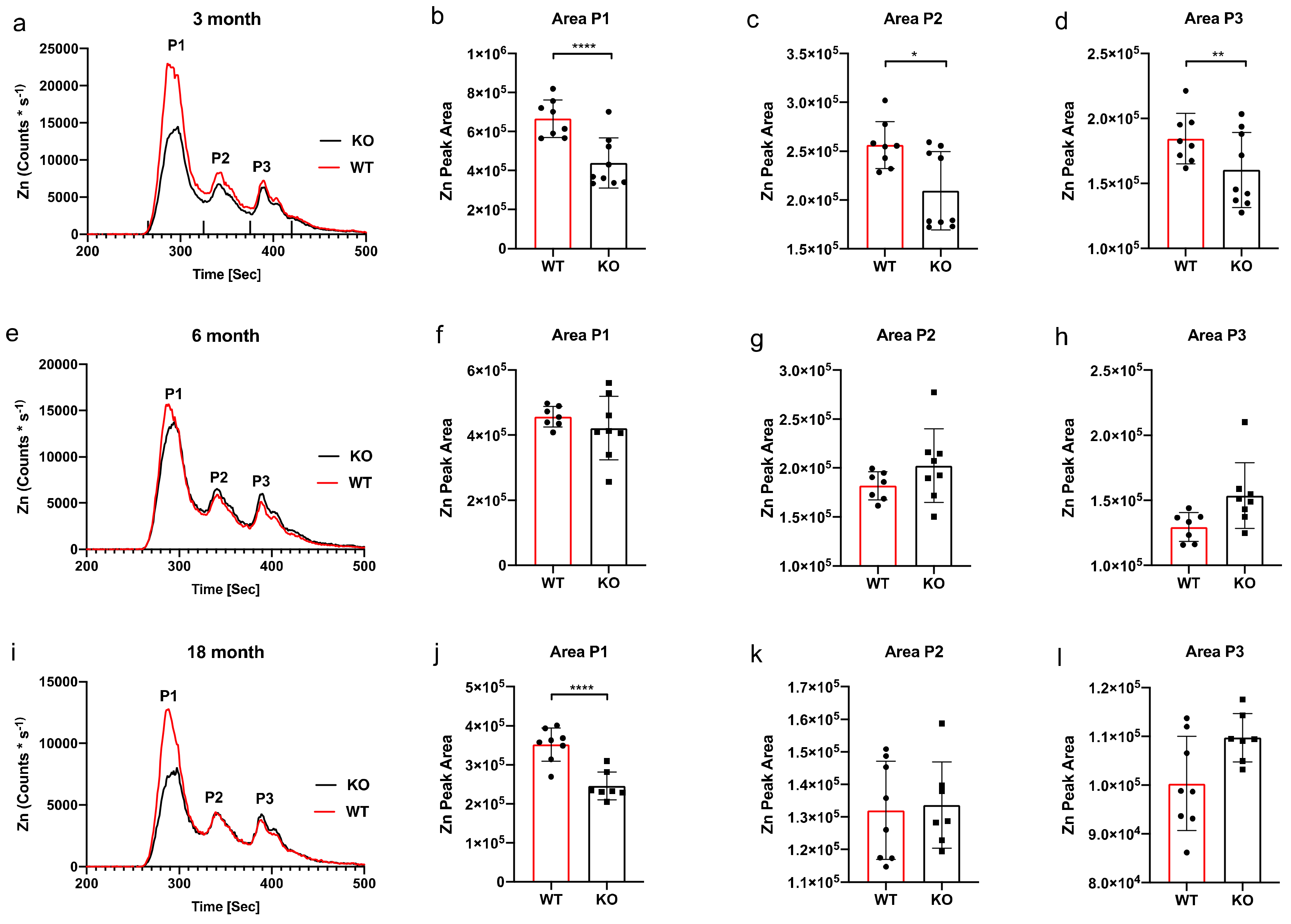
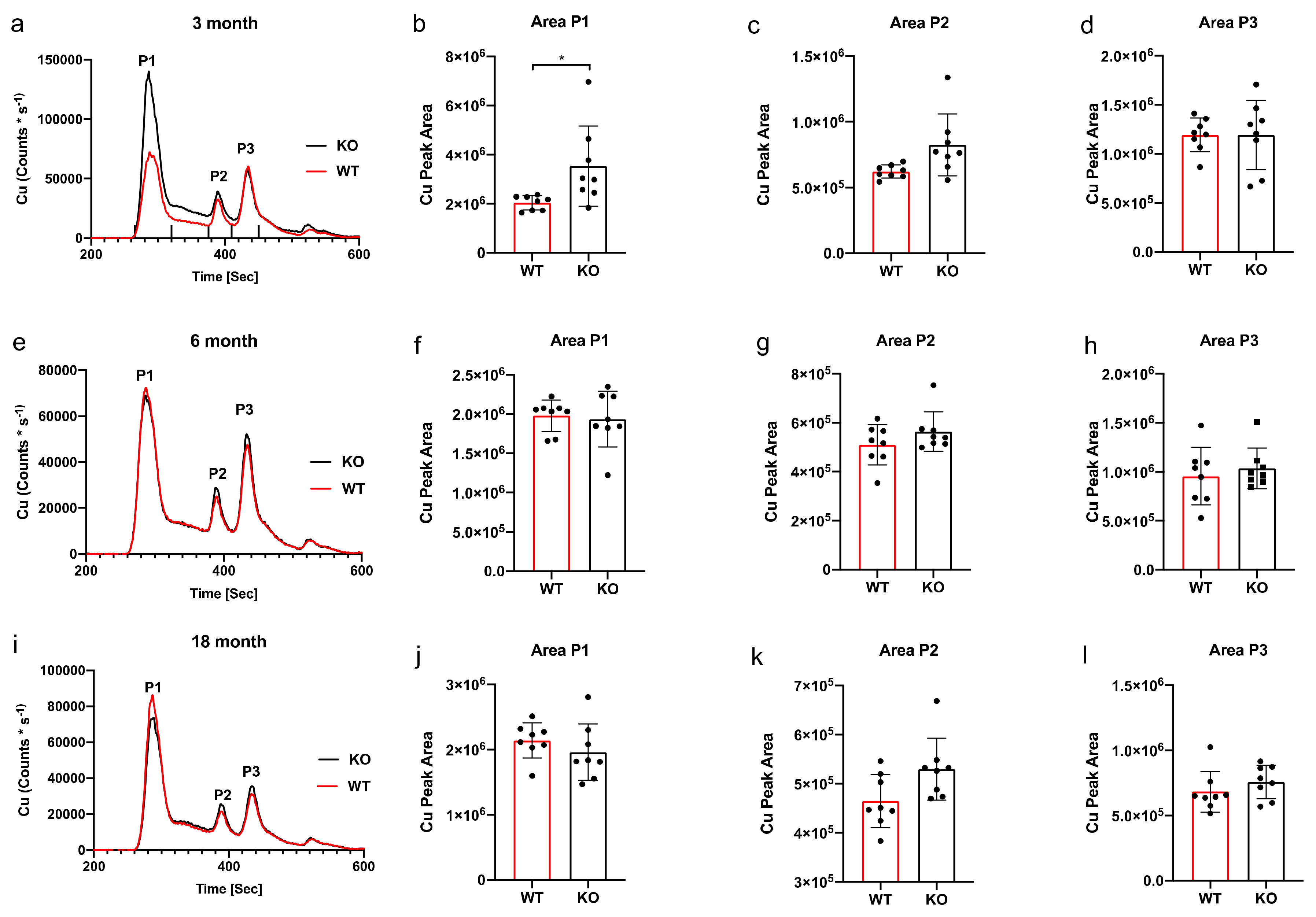
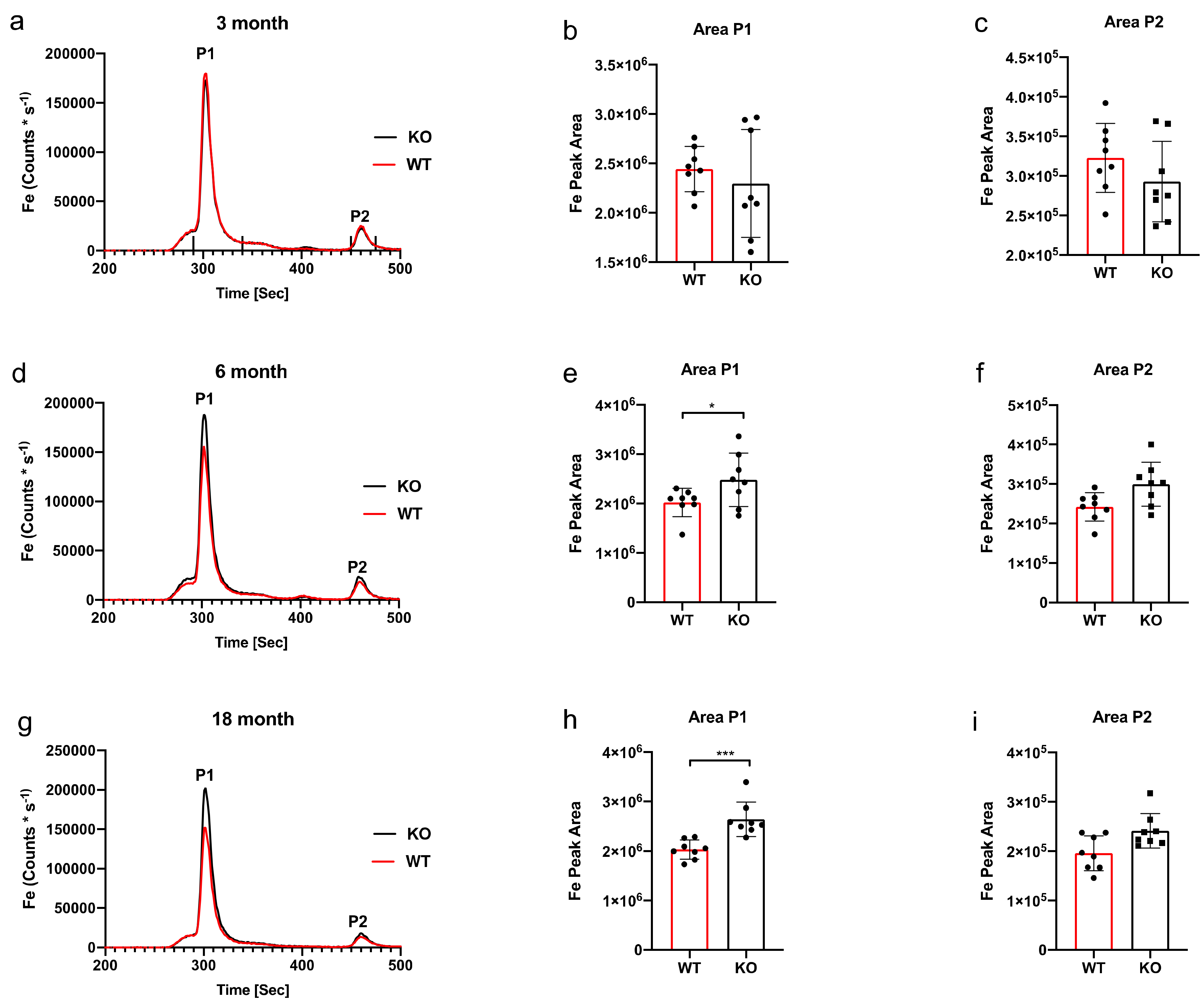
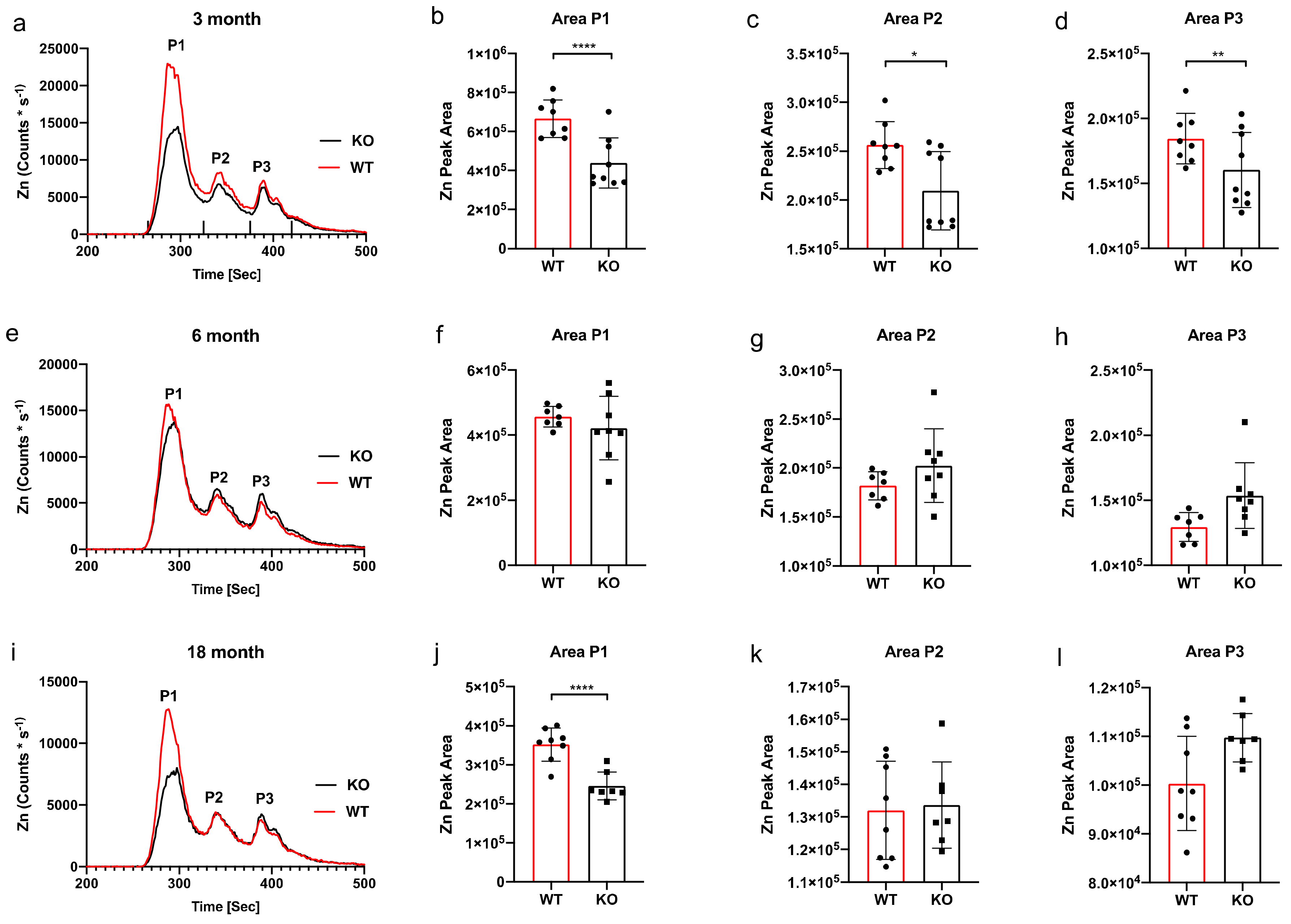
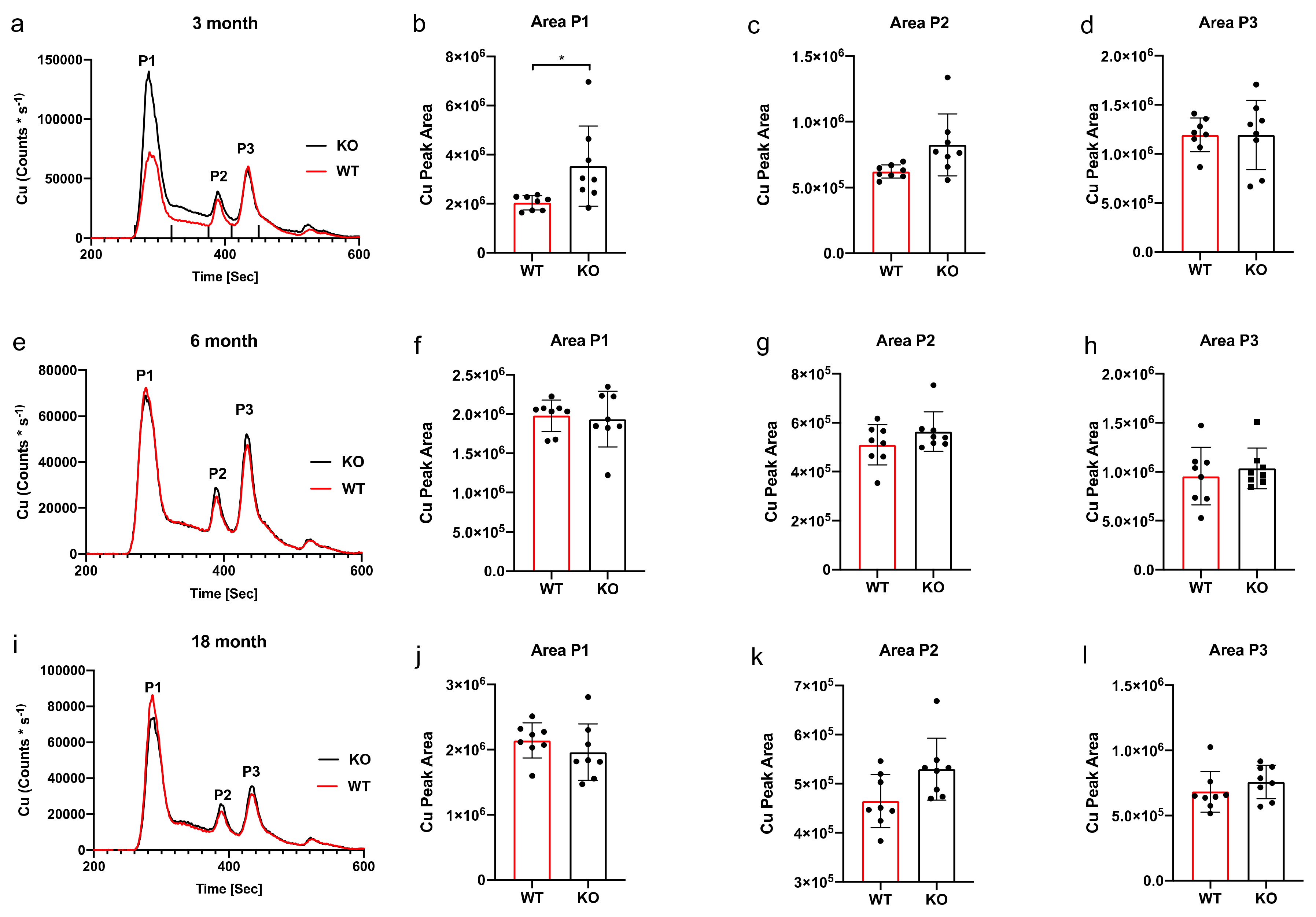
3. Results
SEC-ICP-MS
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.; Cunningham, M.G. Zinc: The brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tepaamorndech, S. The SLC30 family of zinc transporters—A review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 2013, 34, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Eide, D.J. The SLC39 family of zinc transporters. Mol. Aspects Med. 2013, 34, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlard, P.A.; Parncutt, J.; Lal, V.; James, S.; Hare, D.; Doble, P.; Finkelstein, D.I.; Bush, A.I. Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol. Dis. 2015, 81, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [Green Version]
- Adlard, P.A.; Sedjahtera, A.; Gunawan, L.; Bray, L.; Hare, D.; Lear, J.; Doble, P.; Bush, A.I.; Finkelstein, D.I.; Cherny, R.A. A novel approach to rapidly prevent age-related cognitive decline. Aging Cell 2014, 13, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linkous, D.H.; Flinn, J.M.; Koh, J.Y.; Lanzirotti, A.; Bertsch, P.M.; Jones, B.F.; Giblin, L.J.; Frederickson, C.J. Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 2008, 56, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [Green Version]
- Lothian, A.; Roberts, B.R. Standards for Quantitative Metalloproteomic Analysis Using Size Exclusion ICP-MS. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royston, J.P. Algorithm AS 181: The W-test for Normality. Appl. Stat. 1982, 31, 176–180. [Google Scholar] [CrossRef]
- Thackray, S.E.; McAllister, B.B.; Dyck, R.H. Behavioral characterization of female zinc transporter 3 (ZnT3) knockout mice. Behav. Brain Res. 2017, 321, 36–49. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kim, Y.J.; Kim, T.Y.; Koh, J.Y.; Kim, Y.H. Essential role for zinc-triggered p75NTR activation in preconditioning neuroprotection. J. Neurosci. 2008, 28, 10919–10927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.; Sonois, V.; Delaine, T.; Guilloreau, L.; Gillet, A.; Teissie, J.; Faller, P.; Vasak, M. Metal swap between Zn7-metallothionein-3 and amyloid-beta-Cu protects against amyloid-beta toxicity. Nat. Chem. Biol. 2008, 4, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, E.; Ticozzi, N.; Mandrioli, J. Psychiatric Symptoms in Amyotrophic Lateral Sclerosis: Beyond a Motor Neuron Disorder. Front. Neurosci. 2019, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estevez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [CrossRef]
- Adamo, A.M.; Liu, X.; Mathieu, P.; Nuttall, J.R.; Supasai, S.; Oteiza, P.I. Early Developmental Marginal Zinc Deficiency Affects Neurogenesis Decreasing Neuronal Number and Altering Neuronal Specification in the Adult Rat Brain. Front. Cell Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Lei, P.; Appukuttan, A.T.; Renoir, T.; Foliaki, S.; Chen, F.; Adlard, P.A.; Hannan, A.J.; Bush, A.I. Brain Zinc Deficiency Exacerbates Cognitive Decline in the R6/1 Model of Huntington’s Disease. Neurotherapeutics 2019. [Google Scholar] [CrossRef]
- Flinn, J.M.; Bozzelli, P.L.; Adlard, P.A.; Railey, A.M. Spatial memory deficits in a mouse model of late-onset Alzheimer’s disease are caused by zinc supplementation and correlate with amyloid-beta levels. Front. Aging Neurosci. 2014, 6, 174. [Google Scholar] [CrossRef] [Green Version]
- Flinn, J.M.; Hunter, D.; Linkous, D.H.; Lanzirotti, A.; Smith, L.N.; Brightwell, J.; Jones, B.F. Enhanced zinc consumption causes memory deficits and increased brain levels of zinc. Physiol. Behav. 2005, 83, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Linkous, D.H.; Adlard, P.A.; Wanschura, P.B.; Conko, K.M.; Flinn, J.M. The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers Dis. 2009, 18, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Kameo, S.; Nakai, K.; Naganuma, A.; Koyama, P.B.; Satoh, H. Simple Analysis Method for Metallothionein-1, -2 and -3 in the Brain by One-Step Size-Exclusion Column HPLC On-Line Coupling with Inductively Coupled Plasma Mass Spectrometry. J. Anal. Bioanal. Tech. 2014, 5. [Google Scholar] [CrossRef]
- Cai, S.; Gong, J.Y.; Yang, J.; Wang, J.S. Anemia following zinc treatment for Wilson’s disease: A case report and literature review. BMC Gastroenterol. 2019, 19, 120. [Google Scholar] [CrossRef]
- Hoogenraad, T.U.; Dekker, A.W.; van den Hamer, C.J. Copper responsive anemia, induced by oral zinc therapy in a patient with acrodermatitis enteropathica. Sci. Total. Environ. 1985, 42, 37–43. [Google Scholar] [CrossRef]
- Porter, K.G.; McMaster, D.; Elmes, M.E.; Love, A.H. Anaemia and low serum-copper during zinc therapy. Lancet 1977, 2, 774. [Google Scholar] [CrossRef]
- Prasad, A.S.; Brewer, G.J.; Schoomaker, E.B.; Rabbani, P. Hypocupremia induced by zinc therapy in adults. JAMA 1978, 240, 2166–2168. [Google Scholar] [CrossRef]
- Arredondo, M.; Martinez, R.; Nunez, M.T.; Ruz, M.; Olivares, M. Inhibition of iron and copper uptake by iron, copper and zinc. Biol. Res. 2006, 39, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, P. Iron and zinc interactions in humans. Am. J. Clin. Nutr. 1998, 68, 442S–446S. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef]
- Weaver, B.P.; Andrews, G.K. Regulation of zinc-responsive Slc39a5 (Zip5) translation is mediated by conserved elements in the 3’-untranslated region. Biometals 2012, 25, 319–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.A.; Roberts, B.R.; Hare, D.J.; de Jonge, M.D.; Birchall, I.E.; Jenkins, N.L.; Cherny, R.A.; Bush, A.I.; McColl, G. Direct in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans. Chem. Sci. 2015, 6, 2952–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klang, I.M.; Schilling, B.; Sorensen, D.J.; Sahu, A.K.; Kapahi, P.; Andersen, J.K.; Swoboda, P.; Killilea, D.W.; Gibson, B.W.; Lithgow, G.J. Iron promotes protein insolubility and aging in C. elegans. Aging (Albany NY) 2014, 6, 975–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hancock, S.M.; Portbury, S.D.; Gunn, A.P.; Roberts, B.R.; Bush, A.I.; Adlard, P.A. Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome. Int. J. Mol. Sci. 2020, 21, 839. https://doi.org/10.3390/ijms21030839
Hancock SM, Portbury SD, Gunn AP, Roberts BR, Bush AI, Adlard PA. Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome. International Journal of Molecular Sciences. 2020; 21(3):839. https://doi.org/10.3390/ijms21030839
Chicago/Turabian StyleHancock, Sara M., Stuart D. Portbury, Adam P. Gunn, Blaine R. Roberts, Ashley I. Bush, and Paul A. Adlard. 2020. "Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome" International Journal of Molecular Sciences 21, no. 3: 839. https://doi.org/10.3390/ijms21030839
APA StyleHancock, S. M., Portbury, S. D., Gunn, A. P., Roberts, B. R., Bush, A. I., & Adlard, P. A. (2020). Zinc Transporter-3 Knockout Mice Demonstrate Age-Dependent Alterations in the Metalloproteome. International Journal of Molecular Sciences, 21(3), 839. https://doi.org/10.3390/ijms21030839