BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians

 ,
,  ,
,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction to BCL-xL
1.1. The BCL-2 Protein Family
- The anti-apoptotic proteins possess the four BH regions (BH1–4), and include BCL-2, BCL-xL (BCL2-L1), BCL-W (BCL2-L2), A1 (also known as BFL-1 or BCL-2A1), and MCL-1 (Myeloid cell leukemia 1).
- The pro-apoptotic proteins, which include BAX (BCL-2 associated X protein), BAK (BCL-2 antagonist killer), and BOK/MTD (BCL-2-related ovarian killer/Matador), have three BH domains (BH1–3) and are considered the pore-forming executioners.
- BH3-only proteins generally possess one single BH region, the BH3 motif, and are also sometimes considered as a subcategory of pro-apoptotic proteins owing to their pro-cell death nature. Proteins like BID (BH3-interacting domain death antagonist), BIM/BOD (BCL2L11; BCL2-interacting mediator of cell death), BAD (BCL2 antagonist of cell death), PUMA/BBC3 (p53 up-regulated modulator of apoptosis), NOXA (phorbol-12-myristate-13-acetate-induced protein 1), BIK/BLK/NBK (BCL-2 interacting killer), HRK/DP5 and BMF (BCL-2 modifying factor) are included in this subgroup [11].
1.2. BCL-xL
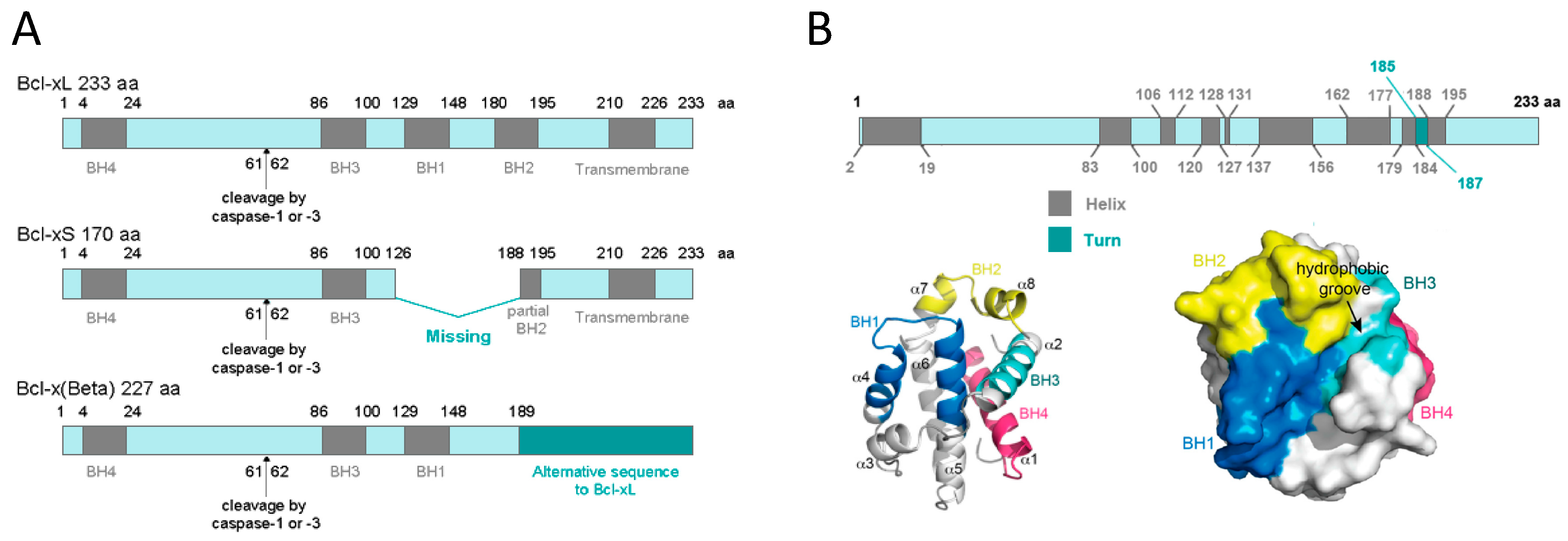
1.2.1. BCL-xL Protein Structure
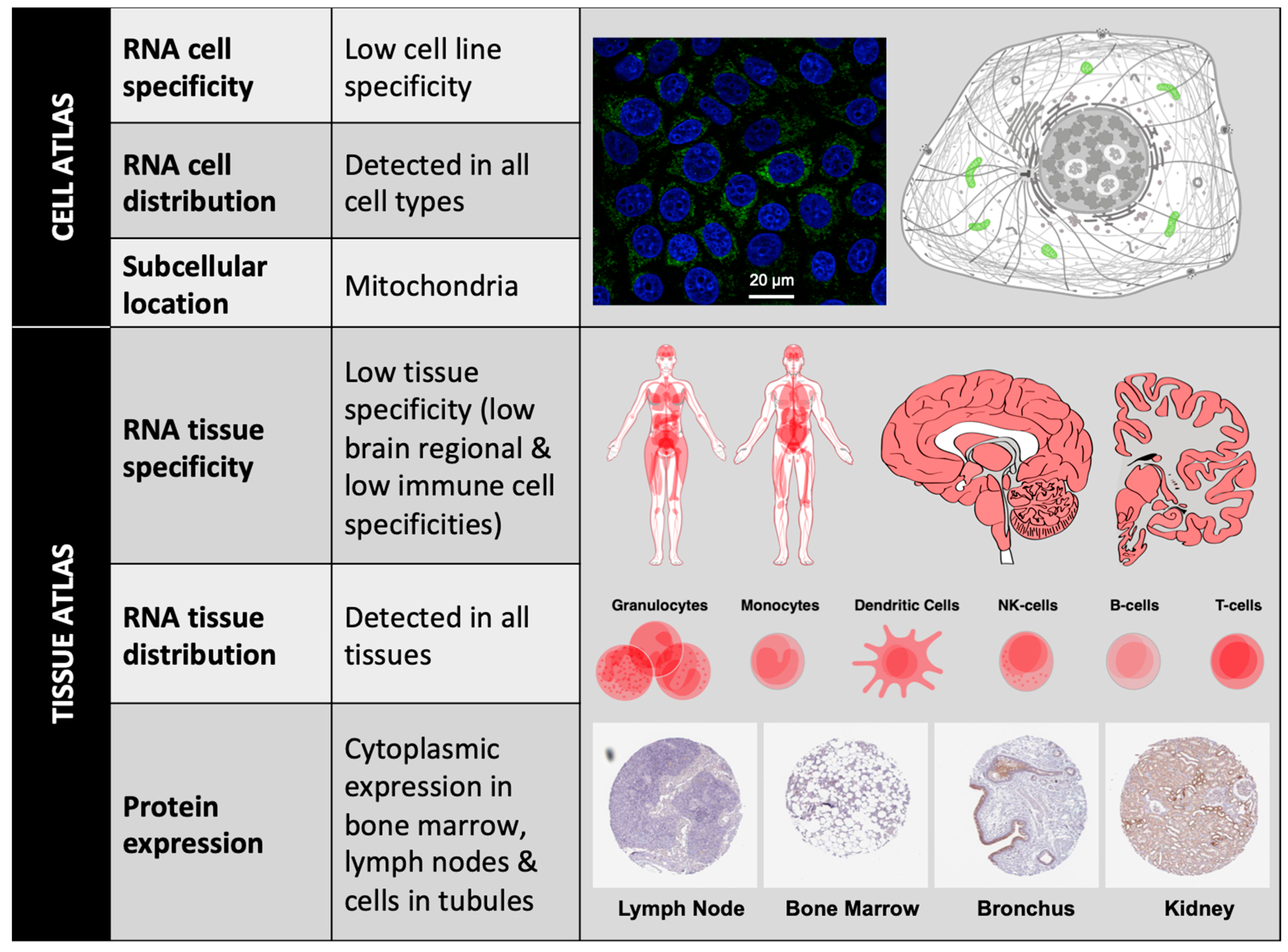
1.2.2. BCL-xL Protein Localization
2. BCL-xL Role in Mitochondrial Apoptosis
- Finally, a more recent study proposed a combination of the previous models and defined two “modes” in which anti-apoptotic proteins prevent apoptosis: by sequestering BH3-only direct activator proteins (MODE 1) or the active effectors BAX and BAK themselves (MODE 2) [48].
BCL-xL Role in Mitochondrial Bioenergetics
3. BCL-xL Role in Autophagy
4. BCL-xL Dual Role in Senescence
4.1. BCL-xL Role in Successful Aging
4.2. BCL-xL, a Senolytic Paradox?
5. Conclusions
Funding
Conflicts of Interest
References
- Pegoraro, L.; Palumbo, A.; Erikson, J.; Falda, M.; Giovanazzo, B.; Emanuel, B.S.; Rovera, G.; Nowell, P.C.; Croce, C.M. A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc. Natl. Acad. Sci. USA 1984, 81, 7166–7170. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef]
- Petros, A.M.; Medek, A.; Nettesheim, D.G.; Kim, D.H.; Yoon, H.S.; Swift, K.; Matayoshi, E.D.; Oltersdorf, T.; Fesik, S.W. Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. USA 2001, 98, 3012–3017. [Google Scholar] [CrossRef]
- Yin, X.M.; Oltvai, Z.N.; Korsmeyer, S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994, 369, 321–323. [Google Scholar] [CrossRef]
- Rech de Laval, V.; Deleage, G.; Aouacheria, A.; Combet, C. BCL2DB: Database of BCL-2 family members and BH3-only proteins. Database 2014, 2014. [Google Scholar] [CrossRef]
- Huska, J.D.; Lamb, H.M.; Hardwick, J.M. Overview of BCL-2 Family Proteins and Therapeutic Potentials. Methods Mol. Biol. 2019, 1877, 1–21. [Google Scholar]
- Moldoveanu, T.; Czabotar, P.E. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb. Perspect. Biol. 2019. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Knight, T.; Luedtke, D.; Edwards, H.; Taub, J.W.; Ge, Y. A delicate balance—The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics. Biochem. Pharmacol. 2019, 162, 250–261. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef]
- van Delft, M.F.; Huang, D.C. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006, 16, 203–213. [Google Scholar] [CrossRef]
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S.H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef]
- Grillot, D.A.; Gonzalez-Garcia, M.; Ekhterae, D.; Duan, L.; Inohara, N.; Ohta, S.; Seldin, M.F.; Nunez, G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J. Immunol. 1997, 158, 4750–4757. [Google Scholar]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef]
- Ban, J.; Eckhart, L.; Weninger, W.; Mildner, M.; Tschachler, E. Identification of a human cDNA encoding a novel Bcl-x isoform. Biochem. Biophys. Res. Commun. 1998, 248, 147–152. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar] [CrossRef]
- Schmitt, E.; Beauchemin, M.; Bertrand, R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene 2007, 26, 5851–5865. [Google Scholar] [CrossRef]
- Follis, A.V.; Llambi, F.; Kalkavan, H.; Yao, Y.; Phillips, A.H.; Park, C.G.; Marassi, F.M.; Green, D.R.; Kriwacki, R.W. Regulation of apoptosis by an intrinsically disordered region of Bcl-xL. Nat. Chem. Biol. 2018, 14, 458–465. [Google Scholar] [CrossRef]
- Lee, E.F.; Fairlie, W.D. The Structural Biology of Bcl-xL. Int. J. Mol. Sci. 2019, 20, 2234. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-terminal helix of Bcl-xL mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2013, 20, 333–342. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Follis, A.V.; Chipuk, J.E.; Fisher, J.C.; Yun, M.K.; Grace, C.R.; Nourse, A.; Baran, K.; Ou, L.; Min, L.; White, S.W.; et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 2013, 9, 163. [Google Scholar] [CrossRef]
- Billen, L.P.; Kokoski, C.L.; Lovell, J.F.; Leber, B.; Andrews, D.W. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008, 6, e147. [Google Scholar] [CrossRef]
- Chi, X.; Kale, J.; Leber, B.; Andrews, D.W. Regulating cell death at, on, and in membranes. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2100–2113. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar] [CrossRef]
- Vasquez-Montes, V.; Vargas-Uribe, M.; Pandey, N.K.; Rodnin, M.V.; Langen, R.; Ladokhin, A.S. Lipid-modulation of membrane insertion and refolding of the apoptotic inhibitor Bcl-xL. Biochim. Biophys. Acta. Proteins Proteom. 2019, 1867, 691–700. [Google Scholar] [CrossRef]
- Priya, P.; Maity, A.; Ghosh Dastidar, S. The long unstructured region of Bcl-xl modulates its structural dynamics. Proteins 2017, 85, 1567–1579. [Google Scholar] [CrossRef]
- Baruah, P.S.; Beauchemin, M.; Hebert, J.; Bertrand, R. Dynamic Bcl-xL (S49) and (S62) Phosphorylation/Dephosphorylation during Mitosis Prevents Chromosome Instability and Aneuploidy in Normal Human Diploid Fibroblasts. PLoS ONE 2016, 11, e0159091. [Google Scholar] [CrossRef]
- Wang, J.; Beauchemin, M.; Bertrand, R. Bcl-xL phosphorylation at Ser49 by polo kinase 3 during cell cycle progression and checkpoints. Cell. Signal. 2011, 23, 2030–2038. [Google Scholar] [CrossRef]
- Kharbanda, S.; Saxena, S.; Yoshida, K.; Pandey, P.; Kaneki, M.; Wang, Q.; Cheng, K.; Chen, Y.N.; Campbell, A.; Sudha, T.; et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J. Biol. Chem. 2000, 275, 322–327. [Google Scholar] [CrossRef]
- Megyesi, J.; Tarcsafalvi, A.; Seng, N.; Hodeify, R.; Price, P.M. Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak. Cell Death Discov. 2016, 2, 15066. [Google Scholar] [CrossRef]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.P.; Sadoshima, J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef]
- Dho, S.H.; Deverman, B.E.; Lapid, C.; Manson, S.R.; Gan, L.; Riehm, J.J.; Aurora, R.; Kwon, K.S.; Weintraub, S.J. Control of cellular Bcl-xL levels by deamidation-regulated degradation. PLoS Biol. 2013, 11, e1001588. [Google Scholar] [CrossRef]
- Ni, F.; Yan, C.Y.; Zhou, S.; Hui, P.Y.; Du, Y.H.; Zheng, L.; Yu, J.; Hu, X.J.; Zhang, Z.G. Repression of GRIM19 expression potentiates cisplatin chemoresistance in advanced bladder cancer cells via disrupting ubiquitination-mediated Bcl-xL degradation. Cancer Chemother Pharm. 2018, 82, 593–605. [Google Scholar] [CrossRef]
- Uren, R.T.; Dewson, G.; Chen, L.; Coyne, S.C.; Huang, D.C.; Adams, J.M.; Kluck, R.M. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J. Cell Biol. 2007, 177, 277–287. [Google Scholar] [CrossRef]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar]
- Leber, B.; Lin, J.; Andrews, D.W. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis Int. J. Program. Cell Death 2007, 12, 897–911. [Google Scholar] [CrossRef]
- Leber, B.; Lin, J.; Andrews, D.W. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene 2010, 29, 5221–5230. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef]
- Aouacheria, A.; Baghdiguian, S.; Lamb, H.M.; Huska, J.D.; Pineda, F.J.; Hardwick, J.M. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 2017, 109, 141–161. [Google Scholar] [CrossRef]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef]
- Berman, S.B.; Chen, Y.B.; Qi, B.; McCaffery, J.M.; Rucker, E.B.; Goebbels, S.; Nave, K.A.; Arnold, B.A.; Jonas, E.A.; Pineda, F.J.; et al. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 2009, 184, 707–719. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Veas-Perez de Tudela, M.; Delgado-Esteban, M.; Maestre, C.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Vecino, R.; Bolanos, J.P.; Almeida, A. Regulation of Bcl-xL-ATP Synthase Interaction by Mitochondrial Cyclin B1-Cyclin-Dependent Kinase-1 Determines Neuronal Survival. J. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef]
- Trancikova, A.; Weisova, P.; Kissova, I.; Zeman, I.; Kolarov, J. Production of reactive oxygen species and loss of viability in yeast mitochondrial mutants: Protective effect of Bcl-xL. FEMS Yeast Res. 2004, 5, 149–156. [Google Scholar] [CrossRef]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Heinlein, M.; Huang, D.C.; Vaux, D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Vaux, D.L. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy 2014, 10, 1474–1475. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.G.; Dong, J.M.; Manser, E.; Song, H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J. Biol. Chem. 2008, 283, 26274–26282. [Google Scholar] [CrossRef]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal 2014, 26, 549–555. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef]
- Yang, J.; Yao, S. JNK-Bcl-2/Bcl-xL-Bax/Bak Pathway Mediates the Crosstalk between Matrine-Induced Autophagy and Apoptosis via Interplay with Beclin 1. Int. J. Mol. Sci. 2015, 16, 25744–25758. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path—A mini-review. Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef]
- Sanz, C.; Benet, I.; Richard, C.; Badia, B.; Andreu, E.J.; Prosper, F.; Fernandez-Luna, J.L. Antiapoptotic protein Bcl-x(L) is up-regulated during megakaryocytic differentiation of CD34+ progenitors but is absent from senescent megakaryocytes. Exp. Hematol. 2001, 29, 728–735. [Google Scholar] [CrossRef]
- Borras, C.; Abdelaziz, K.M.; Gambini, J.; Serna, E.; Ingles, M.; de la Fuente, M.; Garcia, I.; Matheu, A.; Sanchis, P.; Belenguer, A.; et al. Human exceptional longevity: Transcriptome from centenarians is distinct from septuagenarians and reveals a role of Bcl-xL in successful aging. Aging 2016, 8, 3185–3208. [Google Scholar] [CrossRef]
- Jung, M.S.; Jin, D.H.; Chae, H.D.; Kang, S.; Kim, S.C.; Bang, Y.J.; Choi, T.S.; Choi, K.S.; Shin, D.Y. Bcl-xL and E1B-19K proteins inhibit p53-induced irreversible growth arrest and senescence by preventing reactive oxygen species-dependent p38 activation. J. Biol. Chem. 2004, 279, 17765–17771. [Google Scholar] [CrossRef]
- Gayle, S.S.; Sahni, J.M.; Webb, B.M.; Weber-Bonk, K.L.; Shively, M.S.; Spina, R.; Bar, E.E.; Summers, M.K.; Keri, R.A. Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells. J. Biol. Chem. 2019, 294, 875–886. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354. [Google Scholar] [CrossRef]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35. [Google Scholar] [CrossRef]
- Krajewska, M.; Mai, J.K.; Zapata, J.M.; Ashwell, K.W.; Schendel, S.L.; Reed, J.C.; Krajewski, S. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002, 9, 145–157. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, M.; Garcia, I.; Ding, L.; O’Shea, S.; Boise, L.H.; Thompson, C.B.; Nunez, G. Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc. Natl. Acad. Sci. USA 1995, 92, 4304–4308. [Google Scholar] [CrossRef]
- Li, H.; Alavian, K.N.; Lazrove, E.; Mehta, N.; Jones, A.; Zhang, P.; Licznerski, P.; Graham, M.; Uo, T.; Guo, J.; et al. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat. Cell Biol. 2013, 15, 773–785. [Google Scholar] [CrossRef]
- Hickman, J.A.; Hardwick, J.M.; Kaczmarek, L.K.; Jonas, E.A. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J. Neurophysiol. 2008, 99, 1515–1522. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M. Understanding Aging. N. Engl. J. Med. 2017, 376, 1083–1085. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrás, C.; Mas-Bargues, C.; Román-Domínguez, A.; Sanz-Ros, J.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians. Int. J. Mol. Sci. 2020, 21, 418. https://doi.org/10.3390/ijms21020418
Borrás C, Mas-Bargues C, Román-Domínguez A, Sanz-Ros J, Gimeno-Mallench L, Inglés M, Gambini J, Viña J. BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians. International Journal of Molecular Sciences. 2020; 21(2):418. https://doi.org/10.3390/ijms21020418
Chicago/Turabian StyleBorrás, Consuelo, Cristina Mas-Bargues, Aurora Román-Domínguez, Jorge Sanz-Ros, Lucia Gimeno-Mallench, Marta Inglés, Juan Gambini, and José Viña. 2020. "BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians" International Journal of Molecular Sciences 21, no. 2: 418. https://doi.org/10.3390/ijms21020418
APA StyleBorrás, C., Mas-Bargues, C., Román-Domínguez, A., Sanz-Ros, J., Gimeno-Mallench, L., Inglés, M., Gambini, J., & Viña, J. (2020). BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians. International Journal of Molecular Sciences, 21(2), 418. https://doi.org/10.3390/ijms21020418