S1P/S1P Receptor Signaling in Neuromuscolar Disorders


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
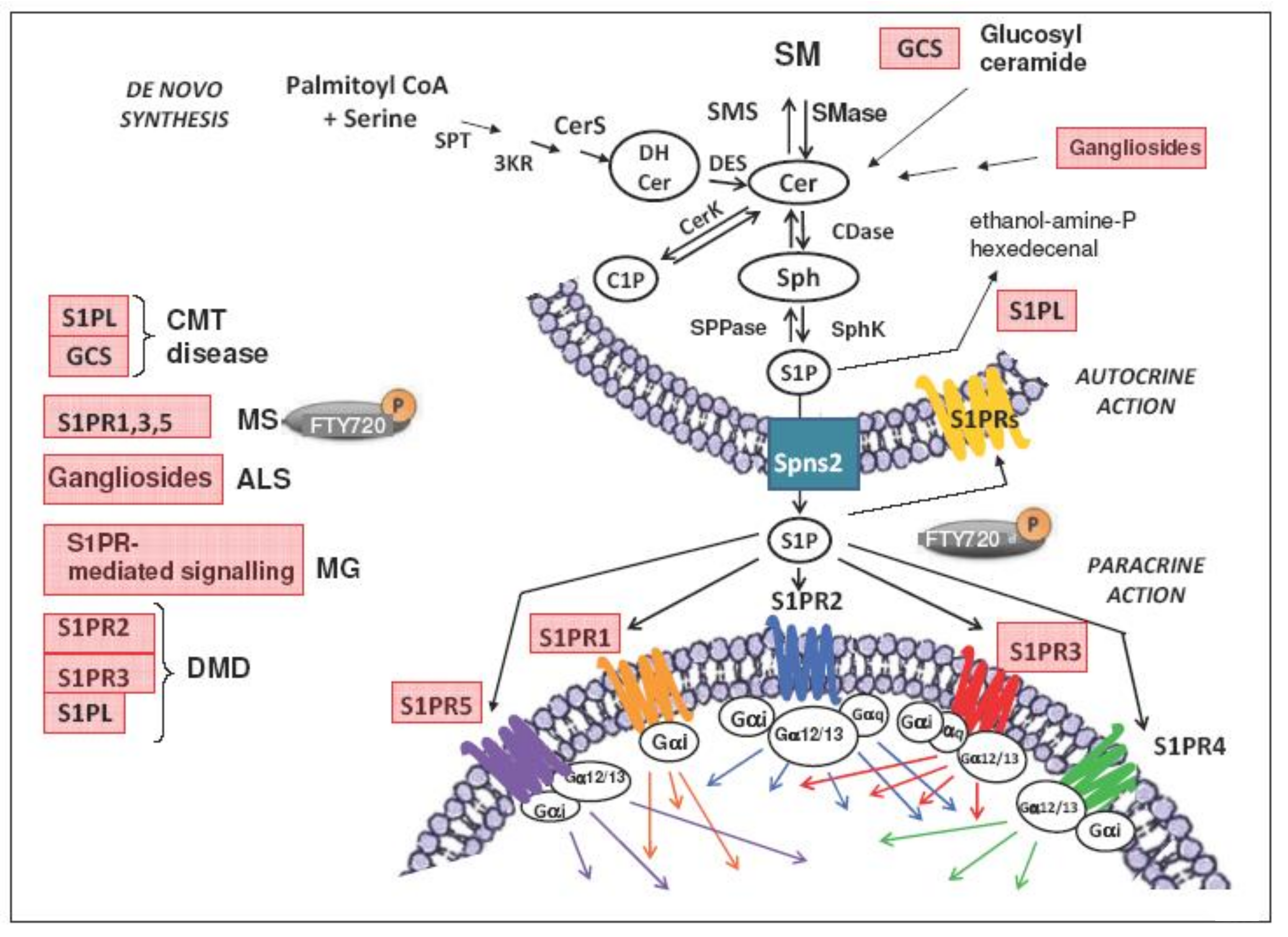
2. Sphingolipid Metabolism
3. S1P/S1PR Signaling
3.1. S1PR in Nervous System
3.2. S1PR in Skeletal Muscle System
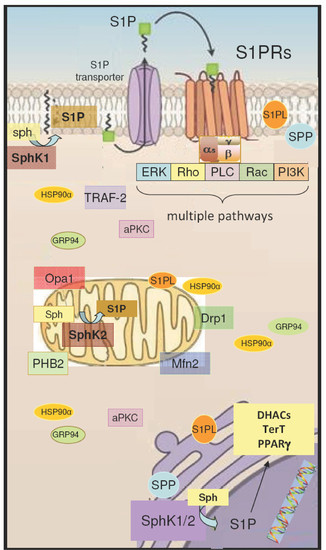
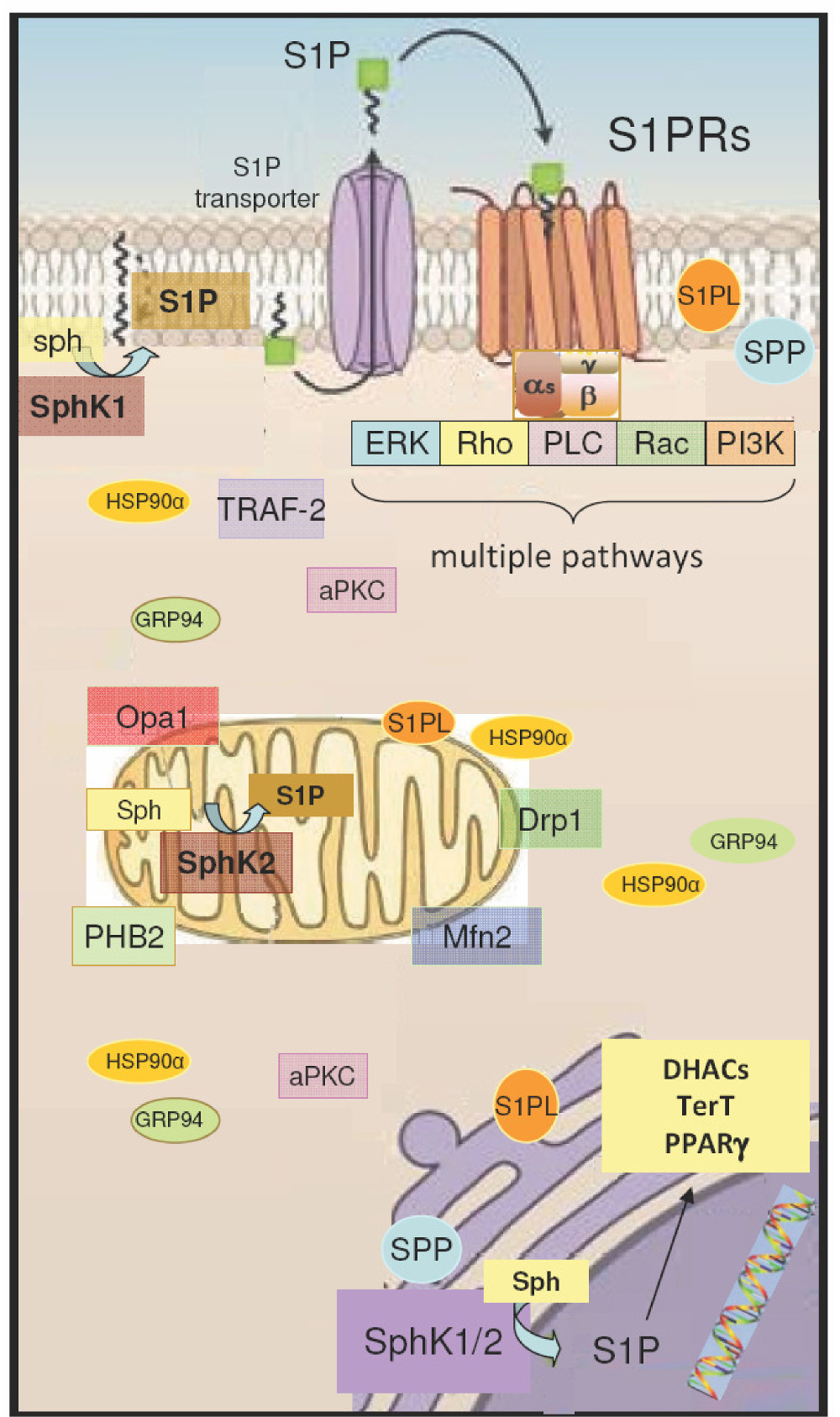
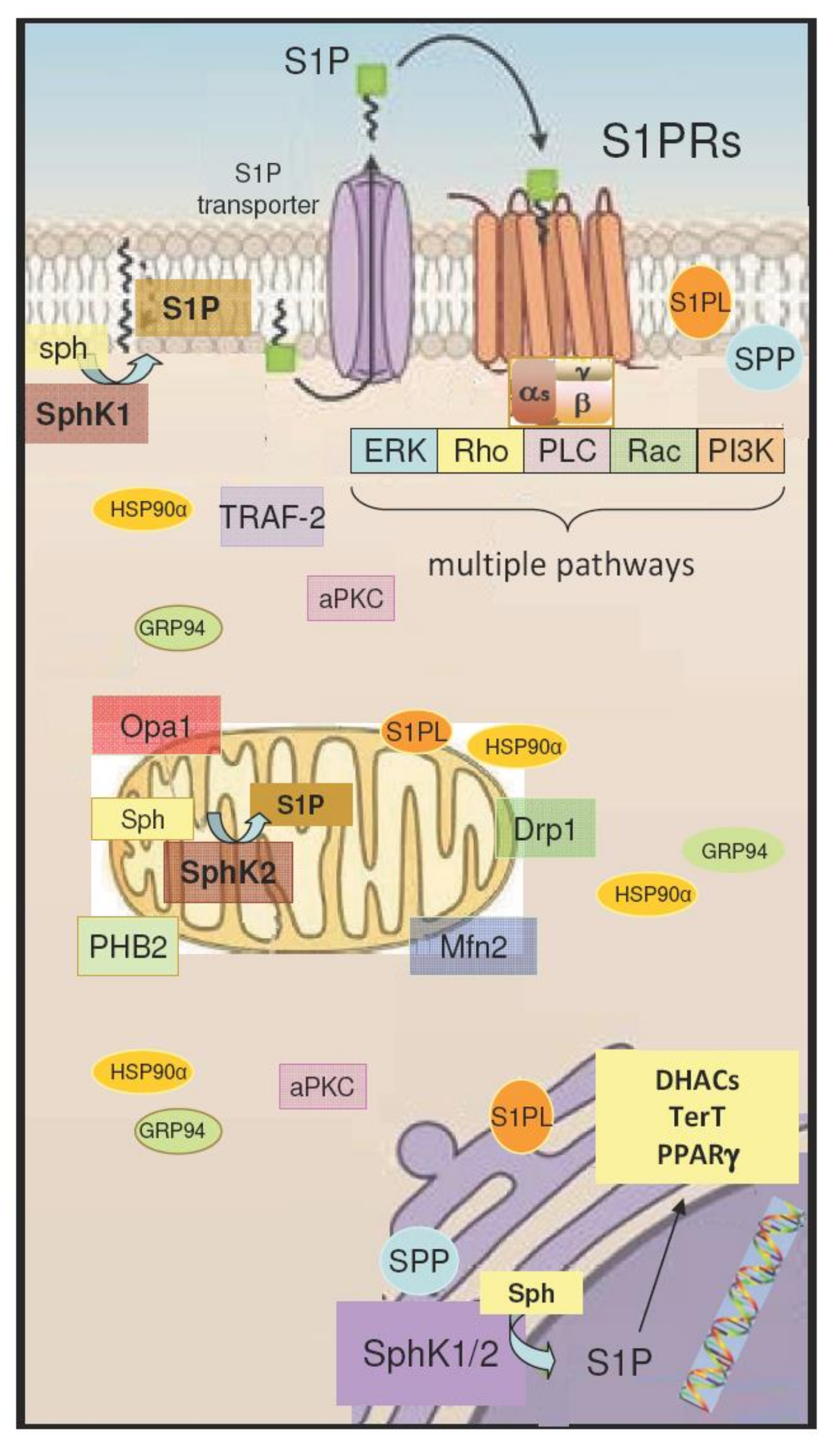
4. Intracellular Action of S1P/S1PR Signaling
4.1. S1P Signaling in Mitochondria
4.2. S1P Signaling in the Nucleus
5. S1P/S1PR Signaling in Neuromuscular Disorders
5.1. Charcot-Marie-Tooth Disease
5.2. Myasthenia Gravis
5.3. Duchenne Muscular Dystrophy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sabbadini, R.A.; Danieli-Betto, D.; Betto, R. The role of sphingolipids in the control of skeletal muscle function: a review. Ital. J. Neurol. Sci. 1999, 20, 423–430. [Google Scholar] [CrossRef]
- Danieli-Betto, D.; Peron, S.; Germinario, E.; Zanin, M.; Sorci, G.; Franzoso, S.; Sandonà, D.; Betto, R. Sphingosine 1-phosphate signaling is involved in skeletal muscleregeneration. Am. J. Physiol. Cell Physiol. 2010, 298, C550–C558. [Google Scholar] [CrossRef]
- Nikolova-Karakashian, M.N.; Reid, M.B. Sphingolipid Metabolism, Oxidant Signaling, and Contractile Function of Skeletal Muscle. Antioxid. Redox Signal. 2011, 15, 2501–2517. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.C.; Leong, W.I.; Carlson, M.E.; Oskouian, B.; Kumar, A.; Fyrst, H.; Zhang, M.; Proia, R.L.; Hoffman, E.P.; Saba, J.D. Sphingosine-1-phosphate enhances satellite cell activation in dystrophic muscles through a S1PR2/STAT3 signaling pathway. PLoS ONE 2012, 7, e37218. [Google Scholar] [CrossRef]
- Ieronimakis, N.; Pantoja, M.; Hays, A.L.; Dosey, T.L.; Qi, J.; Fischer, K.A.; Hoofnagle, A.N.; Sadilek, M.; Chamberlain, J.S.; Ruohola-Baker, H.; et al. Increased sphingosine-1-phosphate improves muscle regeneration in acutely injured mdx mice. Skelet. Muscle 2013, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Partridge, T.A.; Matsuda, R.; Zammit, P.S. Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. J. Cell Biol. 2006, 17, 245–253. [Google Scholar] [CrossRef]
- Sassoli, C.; Formigli, L.; Bini, F.; Tani, A.; Squecco, R.; Battistini, C.; Zecchi-Orlandini, S.; Francini, F.; Meacci, E. Effects of S1P on skeletal muscle repair/regeneration during eccentric contraction. J. Cell. Mol. Med. 2011, 15, 2498–2511. [Google Scholar] [CrossRef]
- Ng, M.L.; Yarla, N.S.; Menschikowski, M.; Sukocheva, O.A. Regulatory role of sphingosine kinase and sphingosine-1-phosphate receptor signaling in progenitor/stem cells. World J. Stem Cells. 2018, 10, 119–133. [Google Scholar] [CrossRef]
- Donati, C.; Meacci, E.; Nuti, F.; Becciolini, L.; Farnararo, M.; Bruni, P. Sphingosine 1-phosphate regulates myogenic differentiation: a major role for S1P2 receptor. FASEB J. 2005, 19, 449–451. [Google Scholar] [CrossRef]
- Squecco, R.; Sassoli, C.; Nuti, F.; Martinesi, M.; Chellini, F.; Nosi, D.; Zecchi-Orlandini, S.; Francini, F.; Formigli, L.; Meacci, E. Sphingosine 1-phosphate induces myoblast differentiation through Cx43 protein expression: a role for a gap junction-dependent and -independent function. Mol. Biol. Cell. 2006, 17, 4896–4910. [Google Scholar] [CrossRef]
- Meacci, E.; Nuti, F.; Donati, C.; Cencetti, F.; Farnararo, M.; Bruni, P. Sphingosine kinase activity is required for myogenic differentiation of C2C12 myoblasts. J. Cell. Physiol. 2008, 214, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Sassoli, C.; Nosi, D.; Tani, A.; Chellini, F.; Mazzanti, B.; Quercioli, F.; Zecchi-Orlandini, S.; Formigli, L. Defining the role of mesenchymal stromal cells on the regulation of matrix metalloproteinases in skeletal muscle cells. Exp. Cell Res. 2014, 323, 297–313. [Google Scholar] [CrossRef]
- De la Garza-Rodea, A.S.; Baldwin, D.M.; Oskouian, B.; Place, R.F.; Bandhuvula, P.; Kumar, A.; Saba, J.D. Sphingosine phosphate lyase regulates myogenic differentiation via S1P receptor-mediated effects on myogenic microRNA expression. FASEB J. 2014, 28, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.; Figeac, N.; White, R.B.; Knopp, P.; Zammit, P.S. Sphingosine 1-phosphate receptor 3 influences cell cycle progression in muscle satellite cells. Dev. Biol. 2013, 382, 504–551. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Wu, R.; Han, W.; Zhang, Y.; Zhu, D. miR-127 enhances myogenic cell differentiation by targeting S1PR3. Cell Death Dis. 2017, 8, e270. [Google Scholar] [CrossRef] [PubMed]
- Pierucci, F.; Frati, A.; Battistini, C.; Matteini, F.; Iachini, M.C.; Vestri, A.; Penna, F.; Costelli, P.; Meacci, E. Involvement of released sphingosine 1-phosphate/sphingosine 1-phosphate receptor axis in skeletal muscle atrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3598–3614. [Google Scholar] [CrossRef]
- Meacci, E.; Vasta, V.; Donati, C.; Farnararo, M.; Bruni, P. Receptor-mediated activation of phospholipase D by sphingosine 1-phosphate in skeletal muscle C2C12 cells. A role for protein kinase C. FEBS Lett. 1999, 457, 184–188. [Google Scholar] [CrossRef]
- Meacci, E.; Donati, C.; Cencetti, F.; Romiti, E.; Bruni, P. Permissive role of protein kinase C alpha but not protein kinase C delta in sphingosine 1-phosphate-induced Rho A activation in C2C12 myoblasts. FEBS Lett. 2000, 482, 97–101. [Google Scholar] [CrossRef]
- Meacci, E.; Cencetti, F.; Formigli, L.; Squecco, R.; Donati, C.; Tiribilli, B.; Quercioli, F.; Zecchi Orlandini, S.; Francini, F.; Bruni, P. Sphingosine 1-phosphate evokes calcium signals in C2C12 myoblasts via Edg3 and Edg5 receptors. Biochem. J. 2002, 362, 349–357. [Google Scholar] [CrossRef]
- Sbrana, F.; Sassoli, C.; Meacci, E.; Nosi, D.; Squecco, R.; Paternostro, F.; Tiribilli, B.; Zecchi-Orlandini, S.; Francini, F.; Formigli, L. Role for stress fiber contraction in surface tension development and stretch-activated channel regulation in C2C12 myoblasts. Am. J. Physiol. Cell Physiol. 2008, 295, C160–C172. [Google Scholar] [CrossRef]
- Formigli, L.; Sassoli, C.; Squecco, R.; Bini, F.; Martinesi, M.; Chellini, F.; Luciani, G.; Sbrana, F.; Zecchi-Orlandini, S.; Francini, F.; et al. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 2009, 122, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Meacci, E.; Sassoli, C.; Squecco, R.; Nosi, D.; Chellini, F.; Naro, F.; Francini, F.; Zecchi-Orlandini, S. Cytoskeleton/stretch-activated ion channel interaction regulates myogenic differentiation of skeletal myoblasts. J. Cell. Physiol. 2007, 211, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Bini, F.; Sassoli, C.; Martinesi, M.; Squecco, R.; Chellini, F.; Zecchi-Orlandini, S.; Francini, F.; Formigli, L. Functional interaction between TRPC1 channel and connexin-43 protein: a novel pathway underlying S1P action on skeletal myogenesis. Cell. Mol. Life Sci. 2010, 67, 4269–4285. [Google Scholar] [CrossRef] [PubMed]
- Gailly, P. TRP channels in normal and dystrophic skeletal muscle. Curr. Opin. Pharmacol. 2012, 12, 326–334. [Google Scholar] [CrossRef]
- Frati, A.; Ricci, B.; Pierucci, F.; Nistri, S.; Bani, D.; Meacci, E. Role of sphingosine kinase/S1P axis in ECM remodeling of cardiac cells elicited by relaxin. Mol. Endocrinol. 2015, 29, 53–67. [Google Scholar] [CrossRef]
- Vestri, A.; Pierucci, F.; Frati, A.; Monaco, L.; Meacci, E. Sphingosine 1-Phosphate Receptors: Do They Have a Therapeutic Potential in Cardiac Fibrosis? Front. Pharmacol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Ghasemi, R.; Dargahi, L.; Ahmadiani, A. Integrated sphingosine-1 phosphate signaling in the central nervous system: From physiological equilibrium to pathological damage. Pharmacol. Res. 2016, 104, 156–164. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Pierucci, F.; Vestri, A.; Meacci, E. Crosstalk between sphingolipids and vitamin D3: potential role in the nervous system. Br. J. Pharmacol. 2017, 174, 605–627. [Google Scholar] [CrossRef]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef] [PubMed]
- Langeslag, M.; Quarta, S.; Leitner, M.G.; Kress, M.; Mair, N. Sphingosine 1-phosphate to p38 signaling via S1P1 receptor and Gαi/o evokes augmentation of capsaicin-induced ionic currents in mouse sensory neurons. Mol. Pain 2014, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Sim-Selley, L.J.; Wilkerson, J.L.; Burston, J.J.; Hauser, K.F.; McLane, V.; Welch, S.P.; Lichtman, A.H.; Selley, D.E. Differential Tolerance to FTY720-Induced Antinociception in Acute Thermal and Nerve Injury Mouse Pain Models: Role of Sphingosine-1-Phosphate Receptor Adaptation. J. Pharmacol. Exp. Ther. 2018, 366, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Stockstill, K.; Doyle, T.M.; Yan, X.; Chen, Z.; Janes, K.; Little, J.W.; Braden, K.; Lauro, F.; Giancotti, L.A.; Harada, C.M.; et al. Dysregulation of sphingolipid metabolismcontributes to bortezomib-induced neuropathic pain. J. Exp. Med. 2018, 215, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Doyle, T.M.; Luongo, L.; Largent-Milnes, T.M.; Giancotti, L.A.; Kolar, G.; Squillace, S.; Boccella, S.; Walker, J.K.; Pendleton, A.; et al. Sphingosine-1-phosphate receptor 1 activation in astrocytes contributes to neuropathic pain. Proc. Natl. Acad. Sci. USA 2019, 116, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Hou, J.C.; Fang, X.M. S1PR2 deficiency enhances neuropathic pain induced bypartial sciatic nerve ligation. Turk. J. Med. Sci. 2019, 49, 412–421. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. EXPAND Clinical Investigators. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Di Pardo, A.; Castaldo, S.; Amico, E.; Pepe, G.; Marracino, F.; Capocci, L.; Giovannelli, A.; Madonna, M.; van Bergeijk, J.; Buttari, F.; et al. Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 2490–2501. [Google Scholar] [CrossRef]
- Di Pardo, A.; Pepe, G.; Castaldo, S.; Marracino, F.; Capocci, L.; Amico, E.; Madonna, M.; Giova, S.; Jeong, S.K.; Park, B.M.; et al. Stimulation of Sphingosine Kinase 1 (SPHK1) Is Beneficial in a Huntington’s Disease Pre-clinical Model. Front. Mol. Neurosci. 2019, 12, 100. [Google Scholar] [CrossRef]
- Astudillo, L.; Sabourdy, F.; Therville, N.; Bode, H.; Ségui, B.; Andrieu-Abadie, N.; Hornemann, T.; Levade, T. Human genetic disorders of sphingolipid biosynthesis. J. Inherit. Metab. Dis. 2015, 38, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Invest. 2015, 125, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Cowart, L.A.; Hannun, Y.A. Selective substrate supply in the regulation of yeast de novo sphingolipid synthesis. J. Biol. Chem. 2007, 282, 12330–12340. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.S.; Schiffmann, S.; Parnham, M.J.; Geisslinger, G.; Grösch, S. The enigma of ceramide synthase regulation in mammalian cells. Prog. Lipid Res. 2016, 63, 93–119. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, F.; Futerman, A.H.; Casas, J. Ceramide synthases in biomedical research. Chem. Phys. Lipids 2016, 197, 25–32. [Google Scholar] [CrossRef]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef]
- Neubauer, H.A.; Pitson, S.M. Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J. 2013, 280, 5317–5336. [Google Scholar] [CrossRef]
- Saba, J.D. Fifty years of lyase and a moment of truth: sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.L.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A. Regulation of adipogenesis by ceramide 1-phosphate. Adv. Cancer Res. 2018, 140, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signaling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Maciag, T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J. Biol. Chem. 1990, 265, 9308–9313. [Google Scholar] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Molecul. Cell. Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wadham, C. Sphingosine-1-phosphate, a key mediator of the cytokine network: juxtacrine signaling. Cytokine Growth Factor Rev. 2011, 22, 45–53. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef]
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 381, ra58. [Google Scholar] [CrossRef]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M. Bonder CS Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, T.; Caliman, A.D.; Tobias, I.S.; Okada, T.; Pilo, C.A.; Van, A.N.; Andrew McCammon, J.; Nakamura, S.I.; Newton, A.C. Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter. Sci. Signal. 2019, 12, eaat6662. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Ikushiro, H.; Seo, H.S.; Shin, K.O.; Kim, Y.I.; Kim, J.Y.; Lee, Y.M.; Yano, T.; Holleran, W.M.; Elias, P.; et al. ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc. Natl. Acad Sci. USA 2016, 113, E1334–E1342. [Google Scholar] [CrossRef] [PubMed]
- Gillies, L.; Lee, S.C.; Long, J.S.; Ktistakis, N.; Pyne, N.J.; Pyne, S. The sphingosine 1-phosphate receptor 5 and sphingosine kinases 1 and 2 are localised in centrosomes: possible role in regulating cell division. Cell Signal. 2009, 21, 675–884. [Google Scholar] [CrossRef]
- Ohotski, J.; Rosen, H.; Bittman, R.; Pyne, S.; Pyne, N.J. Sphingosine kinase 2 prevents the nuclear translocation of sphingosine 1-phosphate receptor-2 and tyrosine 416 phosphorylated c-Src and increases estrogen receptor negative MDA-MB-231 breast cancer cell growth: The role of sphingosine 1-phosphate receptor-4. Cell Signal. 2014, 26, 1040–1047. [Google Scholar] [CrossRef]
- Wang, C.; Mao, J.; Redfield, S.; Mo, Y.; Lage, J.M.; Zhou, X. Systemic distribution, subcellular localization and differential expression of sphingosine-1-phosphate receptors in benign and malignant human tissues. Exp. Mol. Pathol. 2014, 97, 259–265. [Google Scholar] [CrossRef]
- Spiegel, S.; Maczis, M.A.; Maceyka, M.; Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid Res. 2019, 60, 484–489. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nishi, T.; Hirata, T.; Kihara, A.; Sano, T.; Igarashi, Y.; Yamaguchi, A. Sphingosine 1-phosphate is released from the cytosol of rat platelets in a carrier-mediated manner. J. Lipid Res. 2006, 47, 614–621. [Google Scholar] [CrossRef]
- Reitsema, V.; Bouma, H.; Kok, J.W. Sphingosine-1-phosphate transport and its role in immunology. AIMS Mol. Sci. 2014, 1, 183–201. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Natarajan, V.; Dudek, S.M.; Jacobson, J.R.; Moreno-Vinasco, L.; Huang, L.S.; Abassi, T.; Mathew, B.; Zhao, Y.; Wang, L.; Bittman, R.; et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Li, P.Y.; Wada, A.; Mitsutake, S.; Igarashi, Y. Identifi1cation of functional nuclear export sequences in human sphingosine kinase 1. Biochem. Biophys. Res. Commun. 2003, 311, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Erwin, P.A.; Dantas, A.P.; Chen, H.; Michel, T. VEGF induces S1P1 receptors in endothelial cells: Implications for cross-talk between sphingolipid and growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 10664–10669. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Powell, J.A.; Bonder, C.S. Regulation of sphingosine kinase in hematological malignancies and other cancers. Anticancer Agents Med. Chem. 2011, 11, 799–809. [Google Scholar] [CrossRef]
- Spiegel, S.; Kolesnick, R. Sphingosine 1-phosphate as a therapeutic agent. Leukemia 2002, 16, 1596–1602. [Google Scholar] [CrossRef]
- Xia, P.; Gamble, J.R.; Wang, L.; Pitson, S.M.; Moretti, P.A.; Wattenberg, B.W.; D’Andrea, R.J.; Vadas, M.A. An oncogenic role of sphingosine kinase. Curr. Biol. 2000, 10, 1527–1530. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signaling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Pitman, M.R.; Pitson, S.M. Inhibitors of the sphingosine kinase pathway as potential therapeutics. Curr. Cancer Drug Targets 2010, 10, 354–367. [Google Scholar] [CrossRef]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Ding, G.; Sonoda, H.; Kajimoto, T.; Haga, Y.; Khosrowbeygi, A.; Gao, S.; Miwa, N.; Jahangeer, S.; Nakamura, S. Involvement of N-terminal-extended form of sphingosine kinase 2 in serum-dependent regulation of cell proliferation and apoptosis. J. Biol. Chem. 2005, 280, 36318–36325. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, H.A.; Pham, D.H.; Zebol, J.R.; Moretti, P.A.; Peterson, A.L.; Leclercq, T.M.; Chan, H.; Powell, J.A.; Pitman, M.R.; Samuel, M.S.; et al. An oncogenic role for sphingosine kinase 2. Oncotarget 2016, 7, 64886–64899. [Google Scholar] [CrossRef]
- Wallington-Beddoe, C.T.; Powell, J.A.; Tong, D.; Pitson, S.M.; Bradstock, K.F.; Bendall, L.J. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res. 2014, 74, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Schiffmann, S.; Sekar, D.; Ley, S.; Menrad, H.; Werno, C.; Grosch, S.; Geisslinger, G.; Brüne, B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int. J. Cancer 2009, 125, 2114–2121. [Google Scholar] [CrossRef]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lepine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef]
- Gao, P.; Smith, C.D. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol. Cancer Res. 2011, 9, 1509–1519. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Prince, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Olivera, A.; Rivera, J. Sphingolipids and the balancing of immune cell function: lessons from the mast cell. J. Immunol. 2005, 174, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Kawasaki-Nishi, S.; Otsuka, M.; Hisano, Y.; Yamaguchi, A.; Nishi, T. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 2018, 8, 4969. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Anada, Y.; Tani, M.; Ikeda, M.; Sano, T.; Kihara, A.; Igarashi, Y. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem. Biophys. Res. Commun. 2007, 357, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.; Gonzalez-Cabrera, P.J.; Sanna, M.G.; Brown, S. Sphingosine 1-phosphate receptor signaling. Annual Rev. Biochem. 2009, 78, 743–768. [Google Scholar] [CrossRef]
- Hla, T.; Venkataraman, K.; Michaud, J. The vascular SIP gradient-cellular sources and biological significance. Biochim. Biophys. Acta 2008, 1781, 477–482. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar] [CrossRef]
- Rao, T.S.; Lariosa-Willingham, K.D.; Lin, F.F.; Yu, N.; Tham, C.S.; Chun, J.; Webb, M. Growth factor pre-treatment differentially regulates phosphoinositide turnover downstream of lysophospholipid receptor and metabotropic glutamate receptors in cultured rat cerebrocortical astrocytes. Int. J. Dev. Neurosci. 2004, 22, 131–135. [Google Scholar] [CrossRef]
- Ulfig, N.; Briese, M. Evidence for the presence of the sphingosine-1-phosphate receptor Edg-8 in human radial glial fibers. Acta Histochem. 2004, 106, 373–378. [Google Scholar] [CrossRef]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Van Het Hof, B.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef] [PubMed]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Akiyama, T.; Irei, I.; Hamazaki, S.; Sadahira, Y. Cellular Localization of Sphingosine-1-phosphate Receptor 1 Expression in the Human Central Nervous System. J. Histochem. Cytochem. 2010, 2010 58, 847–856. [Google Scholar] [CrossRef]
- Tham, C.S.; Lin, F.F.; Rao, T.S.; Yu, N.; Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 2003, 21, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Bae, Y.J.; Choi, J.W. S1P1 Regulates M1/M2 polarization toward brain injury after transient focal cerebral ischemia. Biomol. Ther. 2019, 27, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Terai, K.; Soga, T.; Takahashi, M.; Kamohara, M.; Ohno, K.; Yatsugi, S.; Okada, M.; Yamaguchi, T. Edg-8 receptors are preferentially expressed in oligodendrocyte lineage cells of the rat CNS. Neuroscience 2003, 116, 1053–1062. [Google Scholar] [CrossRef]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef]
- Kempf, A.; Tews, B.; Arzt, M.E.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS. Biol. 2014, 12, e1001763. [Google Scholar] [CrossRef]
- Harada, J.; Foley, M.; Moskowitz, M.A.; Waeber, C. Sphingosine-1- phosphate induces proliferation and morphological changes of neural progenitor cells. J. Neurochem. 2004, 88, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.H.; Mumaw, J.; Machacek, D.W.; Sturkie, C.; Callihan, P.; Stice, S.L.; Hooks, S.B. Human neural progenitors express functional lysophospholipid receptors that regulate cell growth and morphology. BMC Neurosci. 2008, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Ohmori, R.; Ohkawa, R.; Madoiwa, S.; Mimuro, J.; Murakami, T.; Kobayashi, E.; Hoshino, Y.; Yatomi, Y.; Sakata, Y. Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem. Cells 2007, 25, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ohmori, T.; Kashiwakura, Y.; Ohkawa, R.; Madoiwa, S.; Mimuro, J.; Shimazaki, K.; Hoshino, Y.; Yatomi, Y.; Sakata, Y. Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke 2008, 39, 3411–3417. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhao, Z.; Xu, H.; Zhang, X.; Su, X.; Yang, Y.; Yu, X.; He, X. Activation of Sphingosine 1-Phosphate Receptor 1 Enhances Hippocampus Neurogenesis in a Rat Model of Traumatic Brain Injury: An Involvement of MEK/Erk Signaling Pathway. Neural. Plast. 2016, 2016, 8072156. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hong, F.; Zhang, L.; Feng, L. The sphingosine-1-phosphate analogue, FTY-720, promotes the proliferation of embryonic neural stem cells, enhances hippocampal neurogenesis and learning and memory abilities in adult mice. Br. J. Pharmacol. 2016, 173, 2793–2807. [Google Scholar] [CrossRef]
- Stessin, A.M.; Banu, M.A.; Clausi, M.G.; Berry, N.; Boockvar, J.A.; Ryu, S. FTY720/fingolimod, an oral S1PR modulator, mit.gates radiation induced cognitive deficits. Neurosci. Lett. 2017, 658, 1–5. [Google Scholar] [CrossRef]
- Metzdorf, J.; Hobloss, Z.; Schlevogt, S.; Ayzenberg, I.; Stahlke, S.; Pedreiturria, X.; Haupeltshofer, S.; Gold, R.; Tönges, L.; Kleiter, I. Fingolimod for Irradiation-Induced Neurodegeneration. Front. Neurosci. 2019, 13, 699. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Ciric, B.; Ma, C.G.; Gran, B.; Rostami, A.; Zhang, G.X. Effect of Fingolimod on Neural Stem Cells: A Novel Mechanism and Broadened Application for Neural Repair. Mol. Ther. 2017, 25, 401–415. [Google Scholar] [CrossRef]
- Cohen, J.A.; Tenenbaum, N.; Bhatt, A.; Zhang, Y.; Kappos, L. Extended treatment with fingolimod for relapsing multiple sclerosis: the 14-year LONGTERMS study results. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419878324. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Paganoni, S.; Atassi, N.; Macklin, E.A.; Goyal, N.; Rivner, M.; Simpson, E.; Appel, S.; Grasso, D.L.; Mejia, N.I.; et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve 2017, 56, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, V. Phase III SUNBEAM and RADIANCE PART B trials for Ozanimod in relapsing multiple sclerosis demonstrate superiority versus interferon-β-1a (Avonex®) in reducing annualized relapse rates and MRI brain lesions. Neurodegener. Dis. Manag. 2018, 8, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Comi, G.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.P.; Montalban, X.; Kubala Havrdová, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019, 18, 1021–1033. [Google Scholar] [CrossRef]
- Meacci, E.; Cencetti, F.; Donati, C.; Nuti, F.; Farnararo, M.; Kohno, T.; Igarashi, Y.; Bruni, P. Down-regulation of EDG5/S1P2 during myogenic differentiation results in the specific uncoupling of sphingosine 1-phosphate signaling to phospholipase D. Biochim. Biophys. Acta 2003, 1633, 133–142. [Google Scholar] [CrossRef]
- Zanin, M.; Germinario, E.; Dalla Libera, L.; Sandonà, D.; Sabbadini, R.A.; Betto, R.; Danieli-Betto, D. Trophic action of sphingosine 1-phosphate in denervated rat soleus skeletal muscle. Am. J. Physiol. Cell. Physiol. 2008, 294, C36–C46. [Google Scholar] [CrossRef]
- Sassoli, C.; Pierucci, F.; Zecchi-Orlandini, S.; Meacci, E. Sphingosine 1-Phosphate (S1P)/ S1P Receptor Signaling and Mechanotransduction: Implications for Intrinsic Tissue Repair/Regeneration. Int. J. Mol. Sci. 2019, 20, 5545. [Google Scholar] [CrossRef]
- Bondì, M.; Germinario, E.; Pirazzini, M.; Zanetti, G.; Cencetti, F.; Donati, C.; Gorza, L.; Betto, R.; Bruni, P.; Danieli-Betto, D. Ablation of S1P3 receptor protects mouse soleus from age-related drop in muscle mass, force, and regenerative capacity. Am. J. Physiol. Cell. Physiol. 2017, 313, C54–C67. [Google Scholar] [CrossRef]
- Danieli-Betto, D.; Germinario, E.; Esposito, A.; Megighian, A.; Midrio, M.; Ravara, B.; Damiani, E.; Libera, L.D.; Sabbadini, R.A.; Betto, R. Sphingosine 1-phosphate protects mouse extensor digitorum longus skeletal muscle during fatigue. Am. J. Physiol. Cell. Physiol. 2005, 288, C1367–C1373. [Google Scholar] [CrossRef]
- Bencini, C.; Squecco, R.; Piperio, C.; Formigli, L.; Meacci, E.; Nosi, D.; Tiribilli, B.; Vassalli, M.; Quercioli, F.; Bruni, P.; et al. Effects of sphingosine 1-phosphate on excitation-contraction coupling in mammalian skeletal muscle. J. Muscle Res. Cell Motil. 2003, 24, 539–554. [Google Scholar] [CrossRef]
- Germinario, E.; Peron, S.; Toniolo, L.; Betto, R.; Cencetti, F.; Donati, C.; Bruni, P.; Danieli-Betto, D. S1P2 receptor promotes mouse skeletal muscle regeneration. J. Appl. Physiol. 2012, 113, 707–713. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baranowski, M.; Błachnio-Zabielska, A.U.; Charmas, M.; Helge, J.W.; Dela, F.; Książek, M.; Długołęcka, B.; Klusiewicz, A.; Chabowski, A.; Górski, J. Exercise increases sphingoid base-1-phosphate levels in human blood and skeletal muscle in a time- and intensity-dependent manner. Eur. J. Appl. Physiol. 2015, 115, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.R.; Katashima, C.K.; Bueno Silva, C.G.; Lenhare, L.; Micheletti, T.O.; Camargo, R.L.; Ghezzi, A.C.; Camargo, J.A.; Assis, A.M.; Tobar, N.; et al. Hypothalamic S1P/S1PR1 axis controls energy homeostasis in Middle-Aged Rodents: the reversal effects of physical exercise. Aging 2016, 9, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, V.R.; Gaspar, R.C.; Kuga, G.K.; Pavan, I.C.B.; Simabuco, F.M.; da Silva, A.S.R.; de Moura, L.P.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R. The effects of aging on rho kinase and insulin signaling in skeletal muscle and white adipose tissue of rats. J. Gerontol. A Biol. Sci. Med. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Michel, V. and Bakovic, M. Lipid rafts in health and disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Santos, A.L.; Preta, G. Lipids in the cell: organisation regulates function. Cell. Mol. Life Sci. 2018, 75, 1909–1927. [Google Scholar] [CrossRef]
- Whitley, B.N.; Engelhart, E.A.; Hoppins, S. Mitochondrial dynamics and their potential as a therapeutic target. Mitochondrion 2019, 269–283. [Google Scholar] [CrossRef]
- Cao, Y.-P. and Zheng, M. Mitochondrial dynamics and inter-communication in the heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Perry, C.G.; Lally, J.; Holloway, G.P.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 2010, 588, 4795–4810. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 2010, 1800, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, R.; Léger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Dériaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport. Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef]
- Gomez, M.; Paillard, M.; Price, Q.; Chen, G.; Teixeira, S.; Spiegel, E.J.; Lesnefsky, E.J. A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res. Cardiol. 2011, 106, 1341–1353. [Google Scholar] [CrossRef]
- Fang, R.; Zhang, L.; Zhang, L.; Li, W.; Li, M.; Wen, K. Sphingosine 1-phosphate postconditioning protects against myocardial ischemia/reperfusion injury in rats via mitochondrial signaling and Akt-Gsk3β phosphorylation. Arch. Med. Res. 2017, 48, 147–155. [Google Scholar] [CrossRef]
- Ke, M.; Tang, Q.; Pan, Z.; Yin, Y.; Zhang, L.; Wen, K. Sphingosine-1-phosphate attenuates hypoxia/reoxygenation-induced cardiomyocyte injury via a mitochondrial pathway. Biochem. Biophys. Res. Commun. 2019, 510, 142–148. [Google Scholar] [CrossRef]
- Pulli, I.; Löf, C.; Blom, T.; Asghar, M.Y.; Lassila, T.; Bäck, N.; Lin, K.L.; Nyström, J.H.; Kemppainen, K.; Toivola, D.M.; et al. Sphingosine kinase 1 overexpression induces MFN2 fragmentation and alters mitochondrial matrix Ca(2+) handling in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lee, H.; Kwon, S.E.; Park, E.-J.; Rhee, C.-J.; Park, K.-W.; Toivola, D.M.; Oh, S.-W.; Park, W.-Y. LeeDeficiency of sphingosine-1-phosphate reduces the expression of prohibitin and causes â-cell impairment via mitochondrial dysregulation. Endocrinol. Metab. 2018, 33, 403. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sieburth, D. Sphingosine kinase activates the mitochondrial unfolded protein response and is targeted to mitochondria by stress. Cell. Rep. 2018, 24, 2932–2945. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Bennett, M.K.; Wallington-Beddoe, C.T.; Pitson, S.M. Sphingolipids and the unfolded protein response. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1483–1494. [Google Scholar] [CrossRef]
- Pierucci, F.; Frati, A.; Squecco, R.; Lenci, E.; Vicenti, C.; Slavik, J.; Francini, F.; Machala, M.; Meacci, E. Non-dioxin-like organic toxicant PCB153 modulates sphingolipid metabolism in liver progenitor cells: its role in Cx43-formed gap junction impairment. Arch. Toxicol. 2017, 91, 749–760. [Google Scholar] [CrossRef]
- Ho, N.; Xu, C.; Thibault, G. From the unfolded protein response to metabolic diseases—Lipids under the spotlight. J. Cell Sci. 2018, 131, jcs199307. [Google Scholar] [CrossRef]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite atrophy and apoptosis mediated by PERK signaling after accumulation of GM2-ganglioside. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 225–239. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Young, M.M.; Kester, M.; Wang, H.G. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: its implication in cancer therapeutics. Cell. Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Song, D.D.; Zhang, T.T.; Chen, J.L.; Xia, Y.F.; Qin, Z.H.; Waeber, C.; Sheng, R. Sphingosine kinase 2 activates autophagy and protects neurons against ischemic injury through interaction with Bcl-2 via its putative BH3 domain. Cell Death Dis. 2017, 8, e2912. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Karunakaran, I.; Graler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 2017, 13, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hashimoto, M.; Lin, Q.X.; Tan, D.Q.; Suda, T. Sphingosine-1-phosphate signaling modulates terminal erythroid differentiation through the regulation of mitophagy. Exp. Hematol. 2019, 72, 47–59. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Nuclear sphingolipids: metabolism and signaling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef]
- Lucki, N.C.; Sewer, M.B. Nuclear sphingolipid metabolism. Annu. Rev. Physiol. 2012, 74, 131–151. [Google Scholar] [CrossRef]
- Scassellati, C.; Albi, E.; Cmarko, D.; Tiberi, C.; Cmarkova, J.; Bouchet-Marquis, C.; Verschure, P.J.; Driel, R.; Magni, M.V.; Fakan, S. Intranuclear sphingomyelin is associated with transcriptionally active chromatin and plays a role in nuclear integrity. Bio. Cell 2010, 102, 361–375. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Albi, E. Nuclear Lipids in the Nervous System: What they do in Health and Disease. Neurochem. Res. 2017, 42, 321–336. [Google Scholar] [CrossRef]
- Fu, P.; Ebenezer, D.L.; Ha, A.W.; Suryadevara, V.; Harijith, A.; Natarajan, V. Nuclear lipid mediators: Role of nuclear sphingolipids and sphingosine-1-phosphate signaling in epigenetic regulation of inflammation and gene expression. J. Cell. Biochem. 2018, 119, 6337–6353. [Google Scholar] [CrossRef]
- Ihlefeld, K.; Claas, R.F.; Koch, A.; Pfeilschifter, J.M.; Meyer Zu Heringdorf, D. Evidence for a link between histone deacetylation and Ca²+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 2012, 447, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Tran, D.H.; Hait, N.C.; Sperber, H.; Qi, J.; Fischer, K.; Ieronimakis, N.; Pantoja, M.; Hays, A.; Allegood, J.; Reyes, M.; et al. Molecular mechanism of sphingosine-1-phosphate action in Duchenne muscular dystrophy. Dis. Models Mech. 2014, 7, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Suryadevara, V.; Zhao, Y.; Natarajan, V. Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: Role of S1P lyase. Adv. Biol. Regul. 2017, 63, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Gardner, N.M.; Riley, R.T.; Showker, J.L.; Voss, K.A.; Sachs, A.J.; Maddox, J.R.; Gelineau-van Waes, J.B. Elevated Nuclear and Cytoplasmic FTY720-Phosphate in Mouse Embryonic Fibroblasts Suggests the Potential for Multiple Mechanisms in FTY720-Induced Neural Tube Defects. Toxicol. Sci. 2016, 150, 161–168. [Google Scholar] [CrossRef]
- Hait, N.C.; Wise, L.E.; Allegood, J.C.; O’Brien, M.; Avni, D.; Reeves, T.M.; Knapp, P.E.; Lu, J.; Luo, C.; Miles, M.F.; et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 2014, 17, 971–980. [Google Scholar] [CrossRef]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef]
- Vermeulen, M.; Walter, W.; Le Guezennec, X.; Kim, J.; Edayathumangalam, R.S.; Lasonder, E.; Luger, K.; Roeder, R.G.; Logie, C.; Berger, S.; et al. A feed-forward repression mechanism anchors the Sin3/histone deacetylase and N-CoR/SMRT corepressors on chromatin. Mol. Cell. Biol. 2006, 26, 5226–5236. [Google Scholar] [CrossRef]
- Baymaz, H.I.; Karemaker, I.D.; Vermeulen, M. Perspective on unraveling the versatility of ‘co-repressor’ complexes. Biochim. Biophys. Acta 2015, 1849, 1051–1056. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Berdyshev, E.V.; Bronova, I.A.; Liu, Y.; Tiruppathi, C.; Komarova, Y.; Benevolenskaya, E.V.; Suryadevara, V.; Ha, A.W.; Harijith, A.; et al. Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax 2019, 74, 579–591. [Google Scholar] [CrossRef]
- Pareyson, D.; Stojkovic, T.; Reilly, M.M.; Leonard-Louis, S.; Laurà, M.; Blake, J.; Parman, Y.; Battaloglu, E.; Tazir, M.; Bellatache, M.; et al. A multicenter retrospective study of charcot-marie-tooth disease type 4B (CMT4B) associated with mutations in myotubularin-related proteins (MTMRs). Ann. Neurol. 2019, 86, 55–67. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: the next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.; Nikodinovic Glumac, J.; Asselbergh, B.; Ermanoska, B.; Blocquel, D.; Steiner, R.; Estrada-Cuzcano, A.; Peeters, K.; Ooms, T.; De Vriendt, E.; et al. Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology 2017, 88, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.U.; Linzer, R.W.; Truman, J.P.; Gurevich, M.; Hannun, Y.A.; Senkal, C.E.; Obeid, L.M. Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F. FASEB J. 2018, 32, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Eisner, V.; Chiong, M.; Criollo, A.; Moraga, F.; Garcia, A.; Härtel, S.; Jaimovich, E.; Zorzano, A.; Hidalgo, C.; et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 2008, 77, 387–397. [Google Scholar] [CrossRef]
- Peragallo, J.H. Pediatric Myasthenia Gravis. Semin. Pediatr. Neurol. 2017, 24, 116–121. [Google Scholar] [CrossRef]
- Kohno, T.; Tsuji, T.; Hirayama, K.; Iwatsuki, R.; Hirose, M.; Watabe, K.; Yoshikawa, H. A novel immunomodulator, FTY720, prevents development of experimental autoimmune myasthenia gravis in C57BL/6 mice. Biol. Pharm. Bull. 2005, 28, 736–739. [Google Scholar] [CrossRef]
- Pelz, A.; Schaffert, H.; Diallo, R.; Hiepe, F.; Meisel, A.; Kohler, S. S1P receptor antagonists fingolimod and siponimod do not improve the outcome of experimental autoimmune myasthenia gravis mice after disease onset. Eur. J. Immunol. 2018, 48, 498–508. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, T.; Wang, H.; Zhao, Y. Treatment of experimental autoimmune myasthenia gravis rats with FTY720 and its effect on Th1/Th2 cells. Mol. Med. Rep. 2018, 17, 7409–7414. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef]
- Pantoja, M.; Ruohola-Baker, H. Drosophila as a starting point for developingtherapeutics for the rare disease Duchenne Muscular Dystrophy. Rare Dis. 2013, 1, e24995. [Google Scholar] [CrossRef]
- Heydemann, A. Severe murine limb-girdle muscular dystrophy type 2C pathology is diminished by FTY720 treatment. Muscle Nerve 2017, 56, 486–494. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meacci, E.; Garcia-Gil, M. S1P/S1P Receptor Signaling in Neuromuscolar Disorders. Int. J. Mol. Sci. 2019, 20, 6364. https://doi.org/10.3390/ijms20246364
Meacci E, Garcia-Gil M. S1P/S1P Receptor Signaling in Neuromuscolar Disorders. International Journal of Molecular Sciences. 2019; 20(24):6364. https://doi.org/10.3390/ijms20246364
Chicago/Turabian StyleMeacci, Elisabetta, and Mercedes Garcia-Gil. 2019. "S1P/S1P Receptor Signaling in Neuromuscolar Disorders" International Journal of Molecular Sciences 20, no. 24: 6364. https://doi.org/10.3390/ijms20246364
APA StyleMeacci, E., & Garcia-Gil, M. (2019). S1P/S1P Receptor Signaling in Neuromuscolar Disorders. International Journal of Molecular Sciences, 20(24), 6364. https://doi.org/10.3390/ijms20246364