Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation

 ,
,  and
and
Abstract
1. Noncanonical Sequences of the p53 Family Regulation Sites
1.1. Different Lengths of Consensus Sequence
1.2. Variance of Core Sequence
1.3. Variation in Flanking Sequence
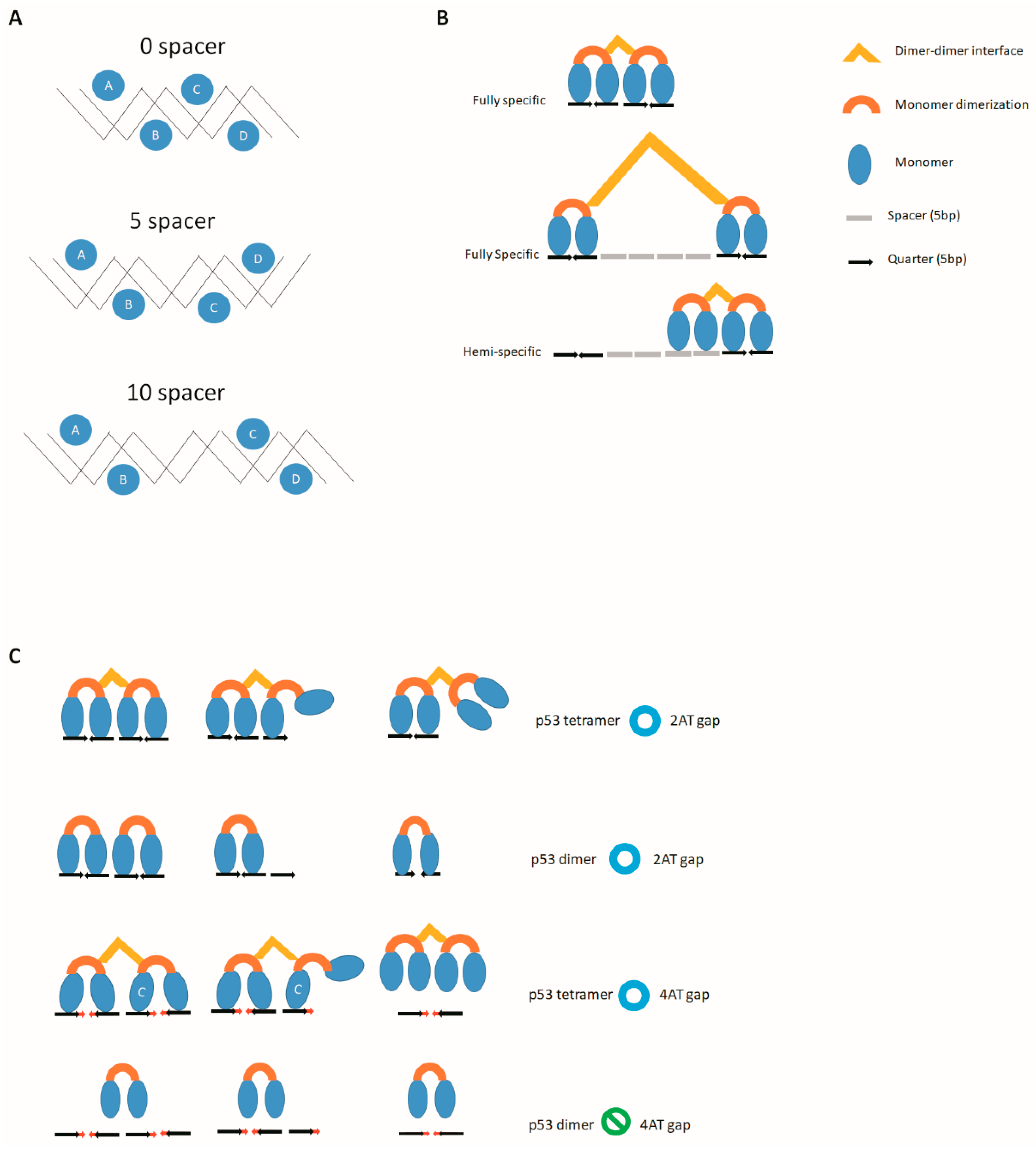
1.4. Base Number of AT Gap
2. Other Sequence Variation Also Influences p53 Family Activity
2.1. Base Number of Spacers
2.2. Cruciform DNA
3. Structure of p53 Family Members and Its Relationship to Their Target Sequences
3.1. Structure of p53
3.2. Structure of p63 and p73
3.3. The Difference in the DNA Binding Domains of p53 Family Members
3.4. Nonspecific Binding Domain in p53 and p73
3.5. The p53 Family Structure and Noncanonical Sequence Relationship
4. p53 Family-Specific Genes and Prediction
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. P63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. P73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- McLure, K.G.; Lee, P.W. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ishida, S.; Morimoto, I.; Yamashita, T.; Kojima, T.; Kihara, C.; Tanaka, T.; Imai, K.; Nakamura, Y.; Tokino, T. The p53 family member genes are involved in the notch signal pathway. J. Biol. Chem. 2002, 277, 719–724. [Google Scholar] [CrossRef]
- Tebaldi, T.; Zaccara, S.; Alessandrini, F.; Bisio, A.; Ciribilli, Y.; Inga, A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015, 16, 464. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Krysiak, O.; Inga, A.; Krysiak, B.; Resnick, M.A.; Schönfelder, G. A snp in the flt-1 promoter integrates the vegf system into the p53 transcriptional network. Proc. Natl. Acad. Sci. USA 2006, 103, 1406–1411. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Snipe, J.; Krysiak, O.; Schönfelder, G.; Resnick, M.A. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol. Cell. Biol. 2007, 27, 2590–2600. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menendez, D.; Inga, A.; Resnick, M.A. Estrogen receptor acting in cis enhances wt and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.J.; Menendez, D.; Inga, A.; Noureddine, M.; Nourredine, M.; Bell, D.A.; Bell, D.; Resnick, M.A. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008, 4, e1000104. [Google Scholar] [CrossRef]
- Cai, B.H.; Chao, C.F.; Lu, M.H.; Lin, H.C.; Chen, J.Y. A half-site of the p53-binding site on the keratin 14 promoter is specifically activated by p63. J. Biochem. 2012, 152, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Hsu, P.C.; Hsin, I.L.; Chao, C.F.; Lu, M.H.; Lin, H.C.; Chiou, S.H.; Tao, P.L.; Chen, J.Y. P53 acts as a co-repressor to regulate keratin 14 expression during epidermal cell differentiation. PLoS ONE 2012, 7, e41742. [Google Scholar] [CrossRef]
- Osada, M.; Park, H.L.; Nagakawa, Y.; Yamashita, K.; Fomenkov, A.; Kim, M.S.; Wu, G.; Nomoto, S.; Trink, B.; Sidransky, D. Differential recognition of response elements determines target gene specificity for p53 and p63. Mol. Cell. Biol. 2005, 25, 6077–6089. [Google Scholar] [CrossRef]
- Ponassi, R.; Terrinoni, A.; Chikh, A.; Rufini, A.; Lena, A.M.; Sayan, B.S.; Melino, G.; Candi, E. P63 and p73, members of the p53 gene family, transactivate pkcdelta. Biochem. Pharmacol. 2006, 72, 1417–1422. [Google Scholar] [CrossRef]
- Osada, M.; Park, H.L.; Park, M.J.; Liu, J.W.; Wu, G.; Trink, B.; Sidransky, D. A p53-type response element in the gdf15 promoter confers high specificity for p53 activation. Biochem. Biophys. Res. Commun. 2007, 354, 913–918. [Google Scholar] [CrossRef]
- Ortt, K.; Sinha, S. Derivation of the consensus DNA-binding sequence for p63 reveals unique requirements that are distinct from p53. FEBS Lett. 2006, 580, 4544–4550. [Google Scholar] [CrossRef] [PubMed]
- Lokshin, M.; Li, Y.; Gaiddon, C.; Prives, C. P53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic. Acids Res. 2007, 35, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chen, J.Y.; Lu, M.H.; Chang, L.T.; Lin, H.C.; Chang, Y.M.; Chao, C.F. Functional four-base a/t gap core sequence cattag of p53 response elements specifically bound tetrameric p53 differently than two-base a/t gap core sequence catg bound both dimeric and tetrameric p53. Nucleic. Acids Res. 2009, 37, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chao, C.F.; Lin, H.C.; Huang, H.Y.; Kannagi, R.; Chen, J.Y. A/t gap tolerance in the core sequence and flanking sequence requirements of non-canonical p53 response elements. J. Biochem. 2016, 159, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gorlatova, N.; Herzberg, O. Pliable DNA conformation of response elements bound to transcription factor p63. J. Biol. Chem. 2012, 287, 7477–7486. [Google Scholar] [CrossRef] [PubMed]
- Ethayathulla, A.S.; Tse, P.W.; Monti, P.; Nguyen, S.; Inga, A.; Fronza, G.; Viadiu, H. Structure of p73 DNA-binding domain tetramer modulates p73 transactivation. Proc. Natl. Acad. Sci. USA 2012, 109, 6066–6071. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Mita, H.; Toyota, M.; Ishida, S.; Morimoto, I.; Yamashita, T.; Tanaka, T.; Imai, K.; Nakamura, Y.; Tokino, T. Identification of the interleukin 4 receptor alpha gene as a direct target for p73. Cancer Res. 2003, 63, 8145–8152. [Google Scholar] [PubMed]
- Tokino, T.; Thiagalingam, S.; el-Deiry, W.S.; Waldman, T.; Kinzler, K.W.; Vogelstein, B. P53 tagged sites from human genomic DNA. Hum. Mol. Genet. 1994, 3, 1537–1542. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop l1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944. [Google Scholar] [CrossRef]
- Demir, Ö.; Ieong, P.U.; Amaro, R.E. Full-length p53 tetramer bound to DNA and its quaternary dynamics. Oncogene 2017, 36, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Vyas, P.; Beno, I.; Xi, Z.; Stein, Y.; Golovenko, D.; Kessler, N.; Rotter, V.; Shakked, Z.; Haran, T.E. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl. Acad. Sci. USA 2017, 114, 10624–10629. [Google Scholar] [CrossRef] [PubMed]
- Jagelská, E.B.; Pivonková, H.; Fojta, M.; Brázda, V. The potential of the cruciform structure formation as an important factor influencing p53 sequence-specific binding to natural DNA targets. Biochem. Biophys. Res. Commun. 2010, 391, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Čechová, J.; Battistin, M.; Coufal, J.; Jagelská, E.B.; Raimondi, I.; Inga, A. The structure formed by inverted repeats in p53 response elements determines the transactivation activity of p53 protein. Biochem. Biophys. Res. Commun. 2017, 483, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Čechová, J.; Coufal, J.; Jagelská, E.B.; Fojta, M.; Brázda, V. P73, like its p53 homolog, shows preference for inverted repeats forming cruciforms. PLoS ONE 2018, 13, e0195835. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Reed, M.; Wang, Y.; Mayr, G.; Stenger, J.E.; Anderson, M.E.; Schwedes, J.F.; Tegtmeyer, P. P53 domains: Structure, oligomerization, and transformation. Mol. Cell. Biol. 1994, 14, 5182–5191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, B.; Pan, Y.; Gunasekaran, K.; Keskin, O.; Venkataraghavan, R.B.; Levine, A.J.; Nussinov, R. The contribution of the trp/met/phe residues to physical interactions of p53 with cellular proteins. Phys. Biol. 2005, 2, S56–S66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-resolution structure of the oligomerization domain of p53 by multidimensional nmr. Science 1994, 265, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Waterman, J.L.; Shenk, J.L.; Halazonetis, T.D. The dihedral symmetry of the p53 tetramerization domain mandates a conformational switch upon DNA binding. EMBO J. 1995, 14, 512–519. [Google Scholar] [CrossRef]
- Mateu, M.G.; Fersht, A.R. Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J. 1998, 17, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Thanos, C.D.; Bowie, J.U. P53 family members p63 and p73 are sam domain-containing proteins. Protein Sci. 1999, 8, 1708–1710. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-wells syndrome is caused by heterozygous missense mutations in the sam domain of p63. Hum. Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Ayed, A.; Arrowsmith, C.H. Solution structure of a conserved c-terminal domain of p73 with structural homology to the sam domain. EMBO J. 1999, 18, 4438–4445. [Google Scholar] [CrossRef]
- Calabrò, V.; Mansueto, G.; Parisi, T.; Vivo, M.; Calogero, R.A.; La Mantia, G. The human mdm2 oncoprotein increases the transcriptional activity and the protein level of the p53 homolog p63. J. Biol. Chem. 2002, 277, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef]
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y. Mdm2 and mdmx bind and stabilize the p53-related protein p73. Curr. Biol. 1999, 9, 829–832. [Google Scholar] [CrossRef]
- Galli, F.; Rossi, M.; D’Alessandra, Y.; De Simone, M.; Lopardo, T.; Haupt, Y.; Alsheich-Bartok, O.; Anzi, S.; Shaulian, E.; Calabrò, V.; et al. Mdm2 and fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J. Cell Sci. 2010, 123, 2423–2433. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; Pagano, M.; Melino, G. The f-box protein fbxo45 promotes the proteasome-dependent degradation of p73. Oncogene 2009, 28, 3157–3166. [Google Scholar] [CrossRef]
- Scoumanne, A.; Harms, K.L.; Chen, X. Structural basis for gene activation by p53 family members. Cancer Biol. Ther. 2005, 4, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.; Van Bokhoven, H.; McKeon, F.; et al. A c-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol. Cell. Biol. 2002, 22, 8601–8611. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Naka, M.; Takada, N.; Tada, M.; Sakiyama, S.; Nakagawara, A. Deletion of the cooh-terminal region of p73alpha enhances both its transactivation function and DNA-binding activity but inhibits induction of apoptosis in mammalian cells. Cancer Res. 1999, 59, 5902–5907. [Google Scholar] [PubMed]
- Straub, W.E.; Weber, T.A.; Schäfer, B.; Candi, E.; Durst, F.; Ou, H.D.; Rajalingam, K.; Melino, G.; Dötsch, V. The c-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis. 2010, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, G.B.; Zielonka, E.M.; Coutandin, D.; Weber, T.A.; Schäfer, B.; Hannewald, J.; Luh, L.M.; Durst, F.G.; Ibrahim, M.; Hoffmann, J.; et al. DNA damage in oocytes induces a switch of the quality control factor tap63α from dimer to tetramer. Cell 2011, 144, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Suh, E.K.; Yang, A.; Kettenbach, A.; Bamberger, C.; Michaelis, A.H.; Zhu, Z.; Elvin, J.A.; Bronson, R.T.; Crum, C.P.; McKeon, F. P63 protects the female germ line during meiotic arrest. Nature 2006, 444, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef]
- Zhu, J.; Nozell, S.; Wang, J.; Jiang, J.; Zhou, W.; Chen, X. P73 cooperates with DNA damage agents to induce apoptosis in mcf7 cells in a p53-dependent manner. Oncogene 2001, 20, 4050–4057. [Google Scholar] [CrossRef][Green Version]
- Gebel, J.; Tuppi, M.; Krauskopf, K.; Coutandin, D.; Pitzius, S.; Kehrloesser, S.; Osterburg, C.; Dötsch, V. Control mechanisms in germ cells mediated by p53 family proteins. J. Cell Sci. 2017. [Google Scholar] [CrossRef]
- Coutandin, D.; Osterburg, C.; Srivastav, R.K.; Sumyk, M.; Kehrloesser, S.; Gebel, J.; Tuppi, M.; Hannewald, J.; Schäfer, B.; Salah, E.; et al. Quality control in oocytes by p63 is based on a spring-loaded activation mechanism on the molecular and cellular level. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hötte, K.; Hoffmeister, M.; Schäfer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of chk2 and ck1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Grespi, F.; Annicchiarico-Petruzzelli, M.; Melino, G. P63 the guardian of human reproduction. Cell Cycle 2012, 11, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Coutandin, D.; Löhr, F.; Niesen, F.H.; Ikeya, T.; Weber, T.A.; Schäfer, B.; Zielonka, E.M.; Bullock, A.N.; Yang, A.; Güntert, P.; et al. Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death Differ. 2009, 16, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Luh, L.M.; Kehrloesser, S.; Deutsch, G.B.; Gebel, J.; Coutandin, D.; Schäfer, B.; Agostini, M.; Melino, G.; Dötsch, V. Analysis of the oligomeric state and transactivation potential of tap73α. Cell Death Differ. 2013, 20, 1008–1016. [Google Scholar] [CrossRef]
- Seeliger, M.A.; Moll, U.M. P73-constitutively open for business. Cell Death Differ. 2013, 20, 972–973. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef]
- Chen, C.; Gorlatova, N.; Kelman, Z.; Herzberg, O. Structures of p63 DNA binding domain in complexes with half-site and with spacer-containing full response elements. Proc. Natl. Acad. Sci. USA 2011, 108, 6456–6461. [Google Scholar] [CrossRef]
- D’Abramo, M.; Bešker, N.; Desideri, A.; Levine, A.J.; Melino, G.; Chillemi, G. The p53 tetramer shows an induced-fit interaction of the c-terminal domain with the DNA-binding domain. Oncogene 2016, 35, 3272–3281. [Google Scholar] [CrossRef]
- Perez, C.A.; Ott, J.; Mays, D.J.; Pietenpol, J.A. P63 consensus DNA-binding site: Identification, analysis and application into a p63mh algorithm. Oncogene 2007, 26, 7363–7370. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.A.; Shepard, E.M.; Scotto, K.W. Differential regulation of mdr1 transcription by the p53 family members. Role of the DNA binding domain. J. Biol. Chem. 2005, 280, 13213–13219. [Google Scholar] [CrossRef] [PubMed]
- Dehner, A.; Klein, C.; Hansen, S.; Müller, L.; Buchner, J.; Schwaiger, M.; Kessler, H. Cooperative binding of p53 to DNA: Regulation by protein-protein interactions through a double salt bridge. Angew. Chem. Int. Ed. 2005, 44, 5247–5251. [Google Scholar] [CrossRef] [PubMed]
- Enthart, A.; Klein, C.; Dehner, A.; Coles, M.; Gemmecker, G.; Kessler, H.; Hagn, F. Solution structure and binding specificity of the p63 DNA binding domain. Sci. Rep. 2016, 6, 26707. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.L.; Freund, S.M.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Regulation of DNA binding of p53 by its c-terminal domain. J. Mol. Biol. 2004, 342, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, T.; Pramanik, A.; Kawasaki, T.; Tan-No, K.; Gileva, I.; Lindegren, H.; Langel, U.; Ekstrom, T.J.; Rigler, R.; Terenius, L.; et al. P53 latency. C-terminal domain prevents binding of p53 core to target but not to nonspecific DNA sequences. J. Biol. Chem. 2001, 276, 15650–15658. [Google Scholar] [CrossRef]
- Sauer, M.; Bretz, A.C.; Beinoraviciute-Kellner, R.; Beitzinger, M.; Burek, C.; Rosenwald, A.; Harms, G.S.; Stiewe, T. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic. Acids Res. 2008, 36, 1900–1912. [Google Scholar] [CrossRef]
- McKinney, K.; Prives, C. Efficient specific DNA binding by p53 requires both its central and c-terminal domains as revealed by studies with high-mobility group 1 protein. Mol. Cell. Biol. 2002, 22, 6797–6808. [Google Scholar] [CrossRef]
- Nie, Y.; Li, H.H.; Bula, C.M.; Liu, X. Stimulation of p53 DNA binding by c-abl requires the p53 c terminus and tetramerization. Mol. Cell. Biol. 2000, 20, 741–748. [Google Scholar] [CrossRef]
- Nichols, N.M.; Matthews, K.S. Human p53 phosphorylation mimic, s392e, increases nonspecific DNA affinity and thermal stability. Biochemistry 2002, 41, 170–178. [Google Scholar] [CrossRef]
- Friedler, A.; Veprintsev, D.B.; Freund, S.M.; von Glos, K.I.; Fersht, A.R. Modulation of binding of DNA to the c-terminal domain of p53 by acetylation. Structure 2005, 13, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Craik, D.J.; Pierens, G.; Bolger, R.E.; Otvos, L. Phosphorylation of the c-terminal sites of human p53 reduces non-sequence-specific DNA binding as modeled with synthetic peptides. Biochemistry 1998, 37, 13755–13764. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Murata, A.; Sakamoto, S.; Nanatani, K.; Wada, T.; Takahashi, S.; Kamagata, K. Activation of p53 facilitates the target search in DNA by enhancing the target recognition probability. J. Mol. Biol. 2016, 428, 2916–2930. [Google Scholar] [CrossRef] [PubMed]
- Khazanov, N.; Levy, Y. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011, 408, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Tafvizi, A.; Huang, F.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, T.; Kenzaki, H.; Takada, S. P53 searches on DNA by rotation-uncoupled sliding at c-terminal tails and restricted hopping of core domains. J. Am. Chem. Soc. 2012, 134, 14555–14562. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. Jaspar 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic. Acids Res. 2018, 46, D1284. [Google Scholar] [CrossRef]
- Cai, B.H.; Wu, P.H.; Chou, C.K.; Huang, H.C.; Chao, C.C.; Chung, H.Y.; Lee, H.Y.; Chen, J.Y.; Kannagi, R. Synergistic activation of the neu4 promoter by p73 and ap2 in colon cancer cells. Sci. Rep. 2019, 9, 950. [Google Scholar] [CrossRef]
- Ezawa, I.; Sawai, Y.; Kawase, T.; Okabe, A.; Tsutsumi, S.; Ichikawa, H.; Kobayashi, Y.; Tashiro, F.; Namiki, H.; Kondo, T.; et al. Novel p53 target gene fuca1 encodes a fucosidase and regulates growth and survival of cancer cells. Cancer Sci. 2016, 107, 734–745. [Google Scholar] [CrossRef]
- Baudot, A.D.; Crighton, D.; O’Prey, J.; Somers, J.; Sierra Gonzalez, P.; Ryan, K.M. P53 directly regulates the glycosidase fuca1 to promote chemotherapy-induced cell death. Cell Cycle 2016, 15, 2299–2308. [Google Scholar] [CrossRef]
- Balint, E.; Phillips, A.C.; Kozlov, S.; Stewart, C.L.; Vousden, K.H. Induction of p57(kip2) expression by p73beta. Proc. Natl. Acad. Sci. USA 2002, 99, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Beretta, C.; Chiarelli, A.; Testoni, B.; Mantovani, R.; Guerrini, L. Regulation of the cyclin-dependent kinase inhibitor p57kip2 expression by p63. Cell Cycle 2005, 4, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Apostolopoulou, K.; Niforou, K.; Kotsinas, A.; Gorgoulis, V.G. P57kip2: “Kip”ing the cell under control. Mol Cancer Res. 2009, 7, 1902–1919. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Caldarelli, I.; Bencivenga, D.; Criscuolo, M.; Cucciolla, V.; Tramontano, A.; Oliva, A.; Perrotta, S.; Della Ragione, F. P57(kip2) and cancer: Time for a critical appraisal. Mol. Cancer Res. 2011, 9, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xiao, Z.; Ren, E.C. Redefining the p53 response element. Proc. Natl. Acad. Sci. USA 2009, 106, 14373–14378. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in sv40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Linzer, D.I.; Levine, A.J. Characterization of a 54k dalton cellular sv40 tumor antigen present in sv40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. P63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Killick, R.; Niklison-Chirou, M.; Tomasini, R.; Bano, D.; Rufini, A.; Grespi, F.; Velletri, T.; Tucci, P.; Sayan, B.S.; Conforti, F.; et al. P73: A multifunctional protein in neurobiology. Mol. Neurobiol. 2011, 43, 139–146. [Google Scholar] [CrossRef]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Knights, C.D.; Catania, J.; Di Giovanni, S.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544. [Google Scholar] [CrossRef]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef]
- Nakamura, Y.; Futamura, M.; Kamino, H.; Yoshida, K.; Arakawa, H. Identification of p53-46f as a super p53 with an enhanced ability to induce p53-dependent apoptosis. Cancer Sci. 2006, 97, 633–641. [Google Scholar] [CrossRef]
- Anazawa, Y.; Arakawa, H.; Nakagawa, H.; Nakamura, Y. Identification of stag1 as a key mediator of a p53-dependent apoptotic pathway. Oncogene 2004, 23, 7621–7627. [Google Scholar] [CrossRef]
- Ciribilli, Y.; Monti, P.; Bisio, A.; Nguyen, H.T.; Ethayathulla, A.S.; Ramos, A.; Foggetti, G.; Menichini, P.; Menendez, D.; Resnick, M.A.; et al. Transactivation specificity is conserved among p53 family proteins and depends on a response element sequence code. Nucleic Acids Res. 2013, 41, 8637–8653. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. Hocomoco: Towards a complete collection of transcription factor binding models for human and mouse via large-scale chip-seq analysis. Nucleic. Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. Promo: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the alggen server: Promo and malgen. Nucleic. Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [PubMed]
- Radoja, N.; Guerrini, L.; Lo Iacono, N.; Merlo, G.R.; Costanzo, A.; Weinberg, W.C.; La Mantia, G.; Calabrò, V.; Morasso, M.I. Homeobox gene dlx3 is regulated by p63 during ectoderm development: Relevance in the pathogenesis of ectodermal dysplasias. Development 2007, 134, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Takahashi, M.; Ozaki, T.; Watanabe Ki, K.; Todo, S.; Mizuguchi, H.; Hayakawa, T.; Nakagawara, A. Autoinhibitory regulation of p73 by delta np73 to modulate cell survival and death through a p73-specific target element within the delta np73 promoter. Mol. Cell. Biol. 2002, 22, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Huang, V.; Lu, X.; Jiang, Y.; Wang, J.Y. Effect of hydroxyurea on the promoter occupancy profiles of tumor suppressor p53 and p73. BMC Biol. 2009, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. Tap73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Yang, X. A critical role of glucose-6-phosphate dehydrogenase in tap73-mediated cell proliferation. Cell Cycle 2013, 12, 3720–3726. [Google Scholar] [CrossRef]
- Xie, N.; Vikhreva, P.; Annicchiarico-Petruzzelli, M.; Amelio, I.; Barlev, N.; Knight, R.A.; Melino, G. Integrin-β4 is a novel transcriptional target of tap73. Cell Cycle 2018, 17, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Bon, G.; Di Carlo, S.E.; Folgiero, V.; Avetrani, P.; Lazzari, C.; D’Orazi, G.; Brizzi, M.F.; Sacchi, A.; Soddu, S.; Blandino, G.; et al. Negative regulation of beta4 integrin transcription by homeodomain-interacting protein kinase 2 and p53 impairs tumor progression. Cancer Res. 2009, 69, 5978–5986. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Park, H.L.; Nagakawa, Y.; Begum, S.; Yamashita, K.; Wu, G.; Kim, M.S.; Trink, B.; Sidransky, D. A novel response element confers p63- and p73-specific activation of the wnt4 promoter. Biochem. Biophys. Res. Commun. 2006, 339, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Negishi, H.; Koyama, R.; Anbo, N.; Ohori, K.; Idogawa, M.; Mita, H.; Toyota, M.; Imai, K.; Shinomura, Y.; et al. P53 family members regulate the expression of the apolipoprotein d gene. J. Biol. Chem. 2009, 284, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Oshima, Y.; Koyama, R.; Maruyama, R.; Akashi, H.; Mita, H.; Toyota, M.; Shinomura, Y.; Imai, K.; Tokino, T. Identification of flotillin-2, a major protein on lipid rafts, as a novel target of p53 family members. Mol. Cancer Res. 2008, 6, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. Iapp-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Nic An tSaoir, C.B.; Blayney, J.K.; Oram, L.C.; Crawford, N.T.; D’Costa, Z.C.; Quinn, J.E.; Kennedy, R.D.; Harkin, D.P.; Mullan, P.B. Brca1 is a key regulator of breast differentiation through activation of notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic. Acids Res. 2013, 41, 8601–8614. [Google Scholar] [CrossRef]
- Sasaki, Y.; Naishiro, Y.; Oshima, Y.; Imai, K.; Nakamura, Y.; Tokino, T. Identification of pigment epithelium-derived factor as a direct target of the p53 family member genes. Oncogene 2005, 24, 5131–5136. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Software | Website | Reference | |
|---|---|---|---|
| JASAPR | http://jaspar.genereg.net/ | [87] | |
| Members | Motif | ||
| p53 model 1 (Motif ID: MA0106.1) |  | ||
| p53 model 2 (Motif ID: MA0106.2) |  | ||
| p53 model 3 (Motif ID: MA0106.3) |  | ||
| p63 model 1 (Motif ID: MA0525.1) |  | ||
| p63 model 2 (Motif ID: MA0525.2) |  | ||
| p73 model (Motif ID: MA0861.1) |  | ||
| Software | Website | Reference | |
| CIS-BP | http://cisbp.ccbr.utoronto.ca/index.php | [111] | |
| Members | Motif | ||
| p53 model 1 (Motif ID: M09337_2.00) |  | ||
| p53 model 2 (Motif ID: M09621_2.00) |  | ||
| p53 model 3 (Motif ID: M11197_2.00) |  | ||
| p53 model 4 (Motif ID: M11198_2.00) * half-site only |  | ||
| p63 model 1 (Motif ID: M03437_2.00) |  | ||
| p63 model 2 (Motif ID: M09335_2.00) |  | ||
| p73 model 1 (Motif ID: M08035_2.00) |  | ||
| p73 model 2 (Motif ID:M08036_2.00) |  | ||
| p73 model 3 (Motif ID:M08037_2.00) |  | ||
| p73 model 4 (Motif ID:M09336_2.00) |  | ||
| Software | Website | Reference | |
| HOCOMOCO | http://hocomoco11.autosome.ru/ | [112] | |
| Members | Motif | ||
| p53 model 1 (Motif ID: P53_HUMAN.H11MO.0.A) |  | ||
| p53 model 2 (Motif ID: P53_HUMAN.H11MO.1.A) * half-site only |  | ||
| p63 model 1 (Motif ID: P63_ HUMAN.H11MO.0.A) |  | ||
| p63 model 2 (Motif ID: P63_HUMAN.H11MO.1.A) * half-site only |  | ||
| p73 model 1 (Motif ID: P73_HUMAN.H11MO.0.A) |  | ||
| p73 model 2 (Motif ID: P73_HUMAN.H11MO.1.A) * half-site only |  | ||
| Software | Website | Reference | |
| PROMO | http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3 | [113,114] | |
| Members | Motif | ||
| p53 model * half-site only |  | ||
| Gene | Response Element Sequences | Noncanonical | p53 | p63 | p73 | Function | Reference |
|---|---|---|---|---|---|---|---|
| FUCA1 | +2609 GGGCAAGTTCATGCAAGTTC +2628 | O | ND | X | 3 | [89,90] | |
| Dlx3 | −89 AAGCAAGACTTGCAG −75 | 1.5× half-site | X | O | ND | 8 | [115] |
| SMARCD3 | −226 GGGCGTGCAGATGCAAGCAC −207 | CGTG core sequence | X | O | ND | 8 | [18] |
| KRT14 | −140 AGACATGATG −131 | half-site | X | O | ND | 8 | [16] |
| ΔNp73 | −76 GGGCAAGCTGAGGCCTGCCC −57 | CCTG core sequence | X | X | O | 7 | [116] |
| NEU4 | −504 GGTCCTGGTCTCGTCATGCTT −484 | CCTG core sequence | X | ND | O | 3 | [88] |
| MLH3 | + 56 GCGCATGCTC +65 | half-site | X | ND | O | 5 | [117] |
| G6PD | +6085 GGTCATGAGCAAACATGACC +6104 | X | X | O | 3 | [118,119,120] | |
| ITGB4 | +17 GCTCCTGCCCCGACAGGTGC +36 | CCTG and CAGG core sequence | X | O | O | 8 | [121,122] |
| WNT4 | −141 GGGCAGGCTGCCGGCAGGCAC −121 | two CAGG core sequence | X | O | O | 8 | [123] |
| MDR1 | −210 CTACTTGCCCTTTCTAGAGA −191 | Second half-site with 5′ PyPyPy and 3′ PuPuPu flanking sequence | X | O | O | 4 | [72] |
| apoD | −419 ATACCAGATGTTTGAAAACATG TTGCAACACGTCCTGCTG −380 | CCAG and CCTG core sequence | X | O | O | 4 | [124] |
| FLOT2 | −4029 GGACTTGGCCAGTCTGGCCT −4010 | CTGG core sequence | X | O | O | 8 | [125] |
| IAPP | −1756 CAACATGAGGCTGCATGTCA −1763 (site 1) mouse genome +706 GAACATGTTTTAGGACATACAGG GGC +732 (site 2) mouse genome | First half-site with a 3′ PuPuPu flanking sequence (site 1) CAGG core sequence (site 2) | X | O | O | 3 | [126] |
| JAG1 | +5597 AGGCTTCTTGTTCAGGCTTGCTCT GTGTGAACCAGACCGTTGTGCTTGGCT +5647 | A 1.5× half-site plus a whole-site with a CCAG core sequence | X | O | O | 8 | [10,127] |
| PEDF | −1366 AAACTTGTTTTAAAACAAGCTTG TGTAACCCATGACC −1330 | AT-rich of flanking sequence of first and second core sequence | X | O | O | 1 | [128] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, B.-H.; Chao, C.-F.; Huang, H.-C.; Lee, H.-Y.; Kannagi, R.; Chen, J.-Y. Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. Int. J. Mol. Sci. 2019, 20, 3681. https://doi.org/10.3390/ijms20153681
Cai B-H, Chao C-F, Huang H-C, Lee H-Y, Kannagi R, Chen J-Y. Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. International Journal of Molecular Sciences. 2019; 20(15):3681. https://doi.org/10.3390/ijms20153681
Chicago/Turabian StyleCai, Bi-He, Chung-Faye Chao, Hsiang-Chi Huang, Hsueh-Yi Lee, Reiji Kannagi, and Jang-Yi Chen. 2019. "Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation" International Journal of Molecular Sciences 20, no. 15: 3681. https://doi.org/10.3390/ijms20153681
APA StyleCai, B.-H., Chao, C.-F., Huang, H.-C., Lee, H.-Y., Kannagi, R., & Chen, J.-Y. (2019). Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. International Journal of Molecular Sciences, 20(15), 3681. https://doi.org/10.3390/ijms20153681