LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling

 , , , , ,
, , , , , 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Acute Induction of PMT in Endotoxemia-Induced Oliguric Kidney Injury
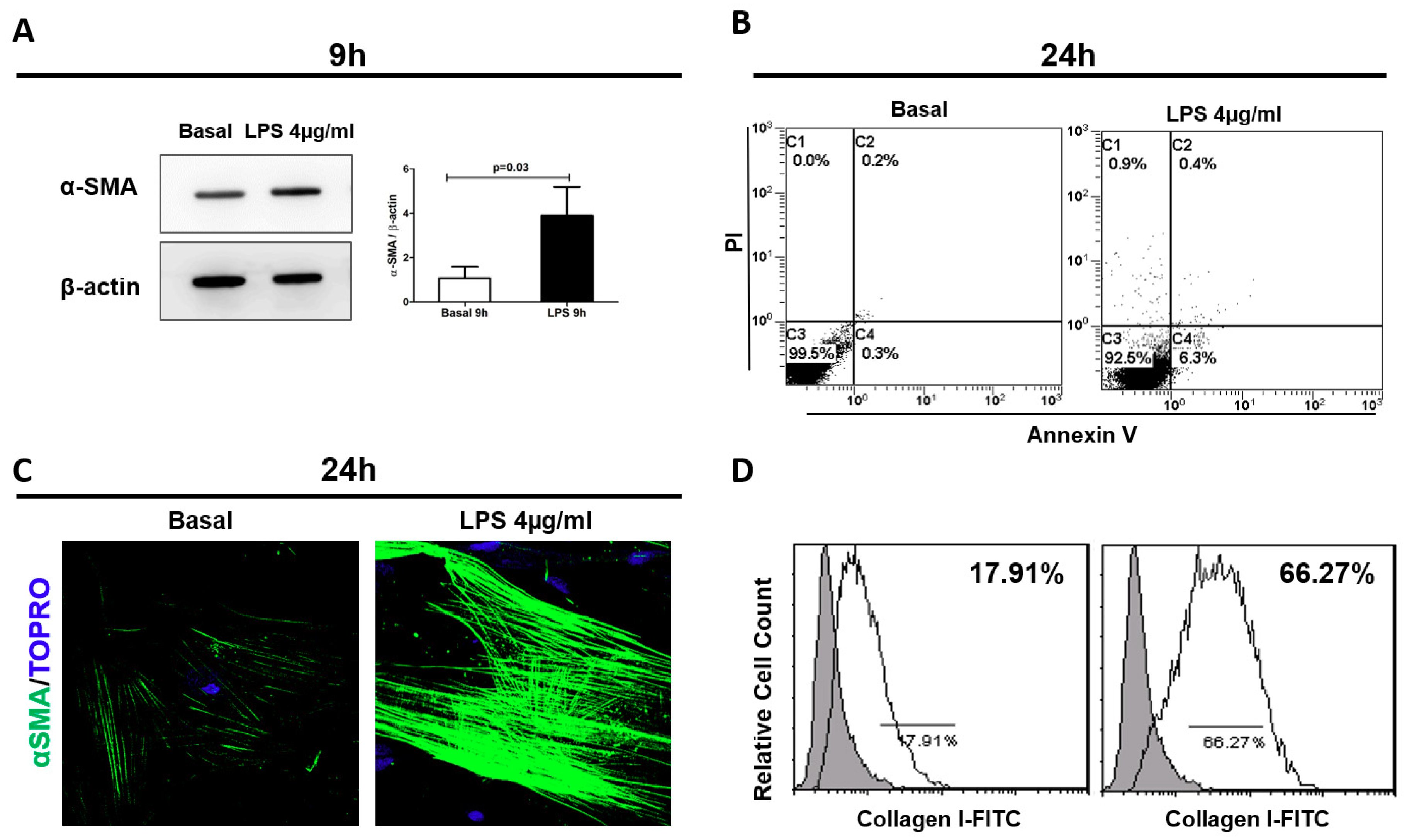
2.2. LPS-Mediated Early Pericyte-to-Myofibroblast Trans-Differentiation (PMT)
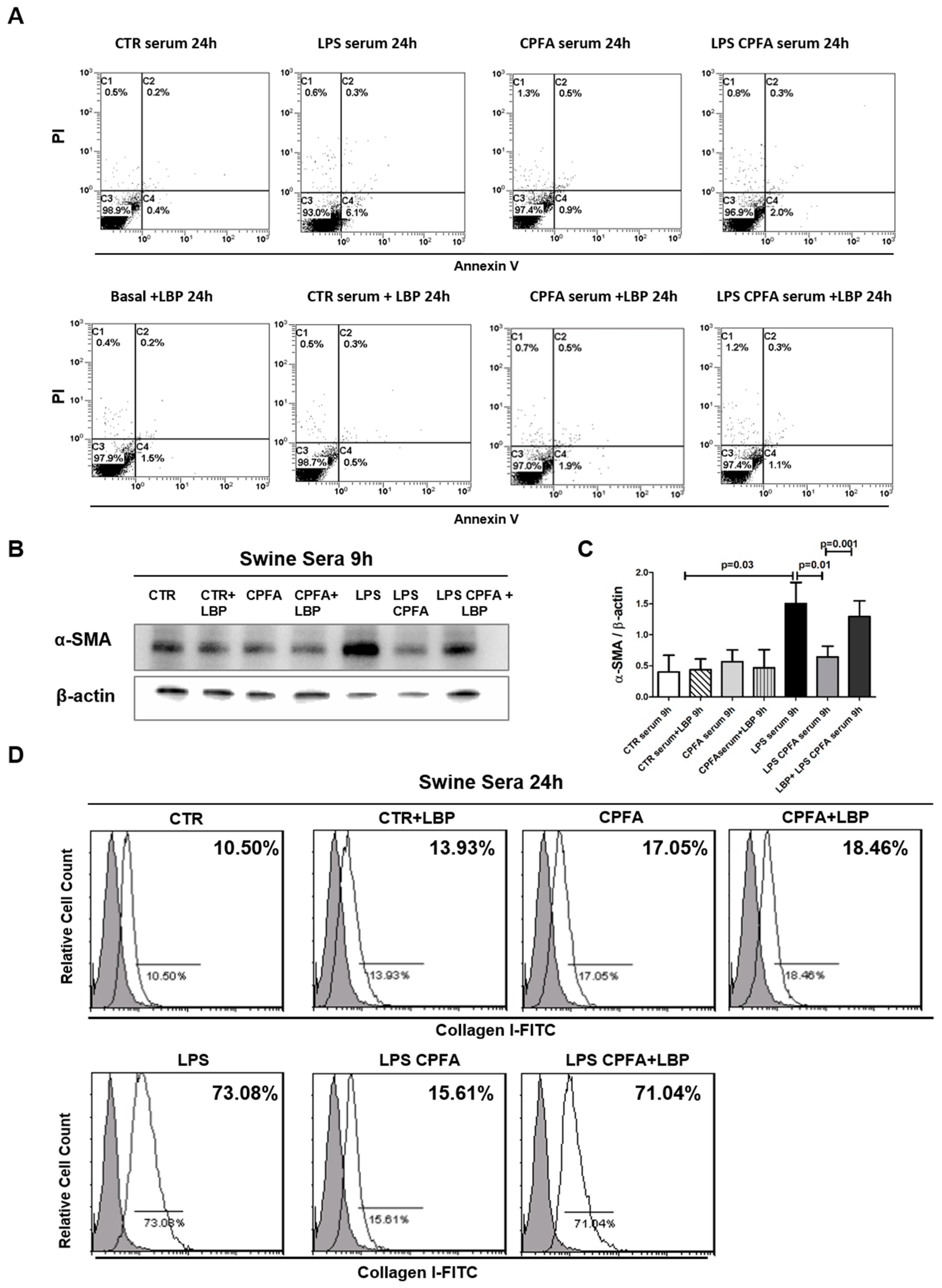
2.3. LPS Binding Protein (LBP) Was Critical in LPS-Mediated PMT
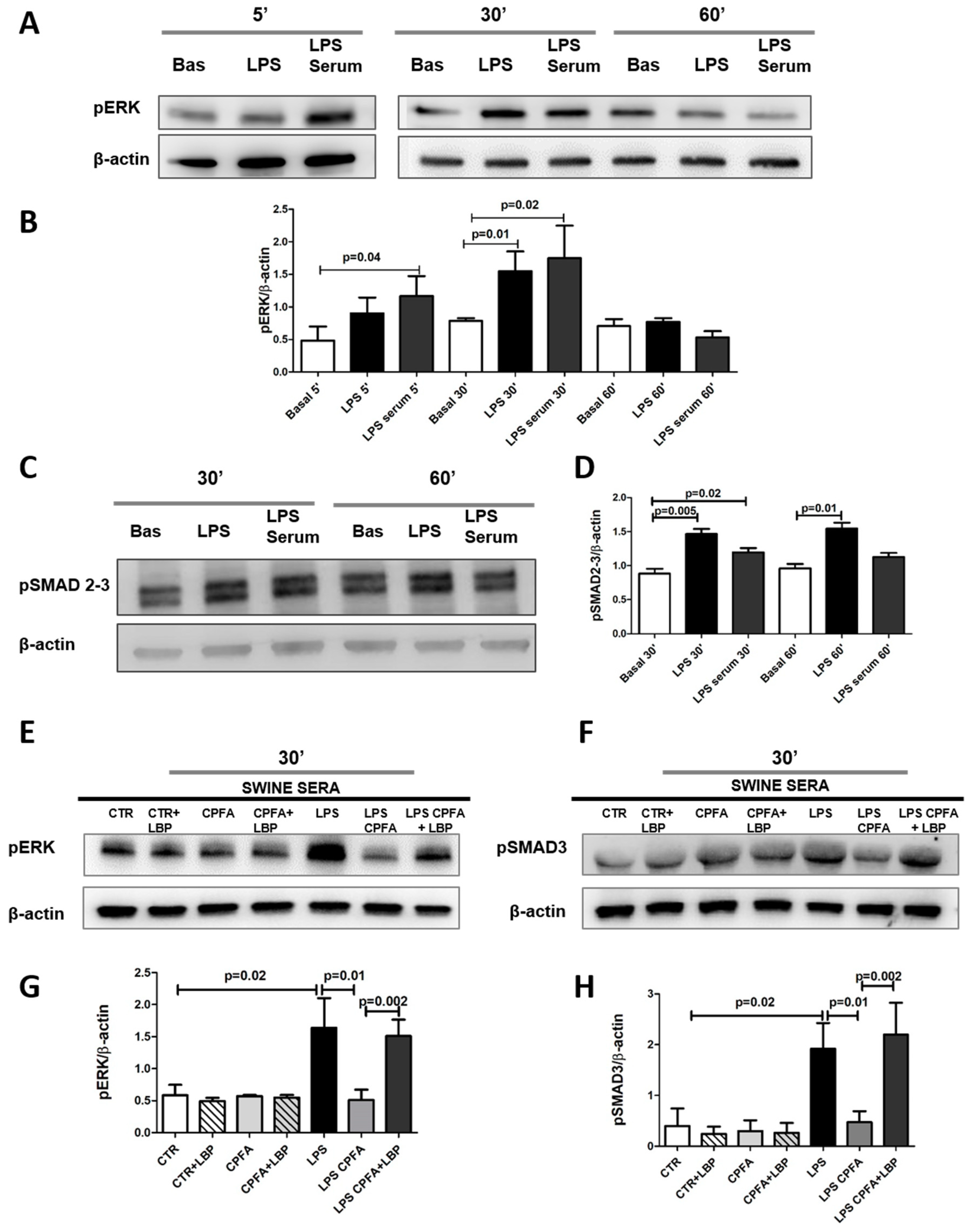
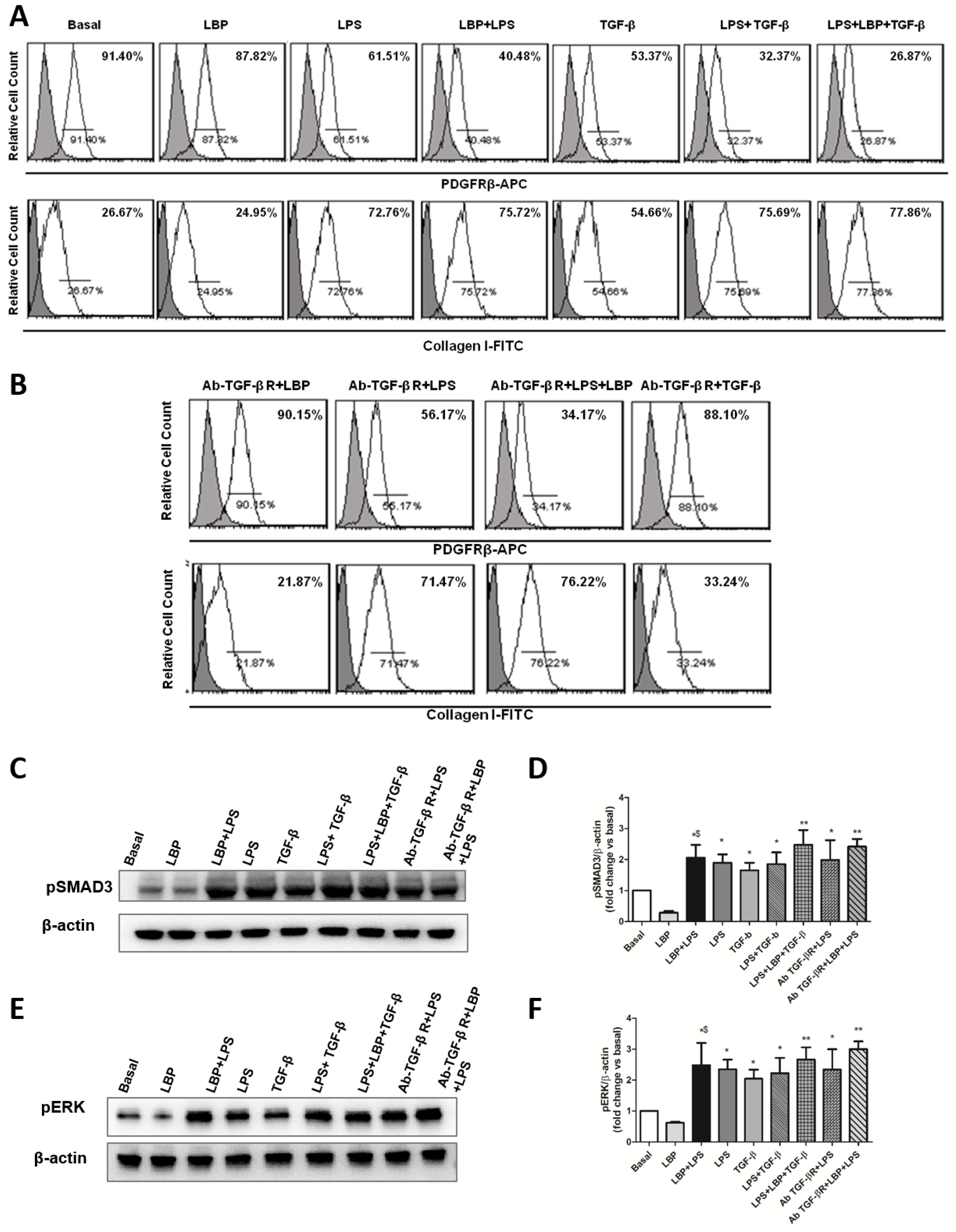
2.4. TLR4 Signaling Mediates PMT via Enhanced Canonical and Non-Canonical TGF-β Signaling
2.5. CPFA Treatment Significantly Decreased Pro-Fibrotic Factors in Endotoxemic Pigs
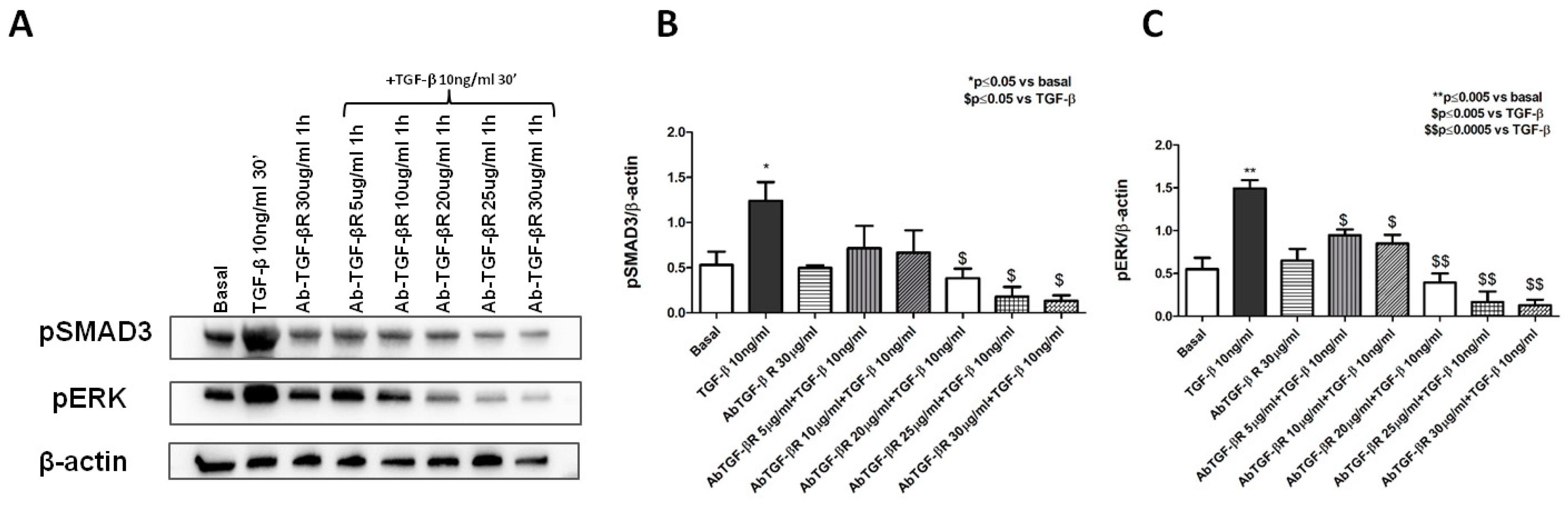
2.6. LBP-LPS Axis-Induced PMT was Characterized by Canonical and Non-Canonical TGF-β Signaling
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Collection of Samples
4.3. Cell Culture
4.4. Immunohistochemistry (IHC)
4.5. Confocal Laser Scanning Microscopy
4.6. Detection of Viable and Apoptotic Pericytes by Flow Cytometry Analysis (FACS)
4.7. Immunophenotypic Analysis
4.8. ELISA for TGF-β, ET-1, and LBP
4.9. Protein Extraction and Western Blotting
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PMT | Pericyte-to-Myofibroblast Trans-differentiation |
| LBP | LPS Binding Protein |
| AKI | Acute Kidney Injury |
| HepG2 | Human Hepato Cancer Cells |
| CKD | Chronic Kidney Disease |
| CPFA | Citrate-Based Coupled Plasma Filtration Adsorption |
| EC | Endothelial Cell |
| WB | Western Blot |
| TLR-4 | Toll Like Receptor-4 |
| ET-1 | Endothelin-1 |
| LPS | Lipopolysaccharide |
| PRR | Pattern Recognition Receptors |
| Ab | Antibody |
| TGF-βR | TGF-βReceptor |
| Ab-TGF-βR | Antibody Anti-TGF-βReceptor |
Appendix A


References
- Gómez, H.; Kellum, J.A. Sepsis-induced acute kidney injury. Curr. Opin. Crit. Care 2016, 22, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Gomez, H.; Kellum, J.A. Sepsis-induced acute kidney injury revisited: Pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care 2014, 20, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Fenhammar, J.; Rundgren, M.; Hultenby, K.; Forestier, J.; Taavo, M.; Kenne, E.; Weitzberg, E.; Eriksson, S.; Ozenci, V.; Wernerson, A.; et al. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit. Care 2014, 18, 488. [Google Scholar] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Schouten, M.; Wiersinga, W.J.; Levi, M.; van der Poll, T. Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol. 2008, 83, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Leaf, I.A.; Nakagawa, S.; Johnson, B.G.; Cha, J.J.; Mittelsteadt, K.; Guckian, K.M.; Gomez, I.G.; Altemeier, W.A.; Duffield, J.S. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J. Clin. Investig. 2017, 127, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Piazza, M.; Calabrese, V.; Damore, G.; Cighetti, R. Exploring the LPS/TLR4 signal pathway with small molecules. Biochem. Soc. Trans. 2010, 38, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Scannon, P.J.; Vincent, J.L.; White, M.; Carroll, S.F.; Palardy, J.E.; Parejo, N.A.; Pribble, J.P.; Lemke, J.H. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J. Infect. Dis. 1999, 180, 1584–1589. [Google Scholar] [CrossRef]
- Villar, J.; Pérez-Méndez, L.; Espinosa, E.; Flores, C.; Blanco, J.; Muriel, A.; Basaldúa, S.; Muros, M.; Blanch, L.; Artigas, A.; et al. Serum lipopolysaccharide binding protein levels predict severity of lung injury and mortality in patients with severe sepsis. PLoS ONE 2009, 4, e6818. [Google Scholar] [CrossRef]
- Castellano, G.; Stasi, A.; Intini, A.; Gigante, M.; Di Palma, A.M.; Divella, C.; Netti, G.S.; Prattichizzo, C.; Pontrelli, P.; Crovace, A.; et al. Endothelial dysfunction and renal fibrosis in endotoxemia-induced oliguric kidney injury: Possible role of LPS-binding protein. Crit. Care 2014, 18, 520. [Google Scholar] [CrossRef] [PubMed]
- Stasi, A.; Intini, A.; Divella, C.; Franzin, R.; Montemurno, E.; Grandaliano, G.; Ronco, C.; Fiaccadori, E.; Pertosa, G.B.; Gesualdo, L.; et al. Emerging role of Lipopolysaccharide binding protein in sepsis-induced acute kidney injury. Nephrol. Dial. Transplant. 2016, 32, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken barriers: A new take on sepsis pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef]
- Page, A.V.; Liles, W.C. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence 2013, 4, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Chang, F.-C.; Wu, C.-F.; Chou, Y.-H.; Hsu, H.-L.; Chiang, W.-C.; Shen, J.; Chen, Y.-M.; Wu, K.-D.; Tsai, T.-J.; et al. Platelet-derived growth factor receptor signaling activates pericyte–myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Deng, Y.; Liu, A.; Shen, N.; Wang, W.; Du, X.; Tang, Q.; Li, S.; Odeh, Z.; Wu, T.; et al. Novel Mechanism of the Pericyte-Myofibroblast Transition in Renal Interstitial Fibrosis: Core Fucosylation Regulation. Sci. Rep. 2017, 7, 16914. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and Perivascular Fibroblasts Are the Primary Source of Collagen-Producing Cells in Obstructive Fibrosis of the Kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef]
- Wu, C.-F.; Chiang, W.-C.; Lai, C.-F.; Chang, F.-C.; Chen, Y.-T.; Chou, Y.-H.; Wu, T.-H.; Linn, G.R.; Ling, H.; Wu, K.-D.; et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef]
- Kuppe, C.; Kramann, R. Role of mesenchymal stem cells in kidney injury and fibrosis. Curr. Opin. Nephrol. Hypertens. 2016, 25, 372–377. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement activation during ischemia/reperfusion injury induces pericyte-to-myofibroblast transdifferentiation regulating peritubular capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; D’Intini, V.; Bellomo, R.; Ricci, Z.; Bonello, M.; Ratanarat, R.; Salvatori, G.; Bordoni, V.; Andricos, E.; Brendolan, A. Rationale for the use of extracorporeal treatments for sepsis. Anesteziol. Reanimatol. 2005, 2, 87–91. [Google Scholar]
- Ronco, C.; Brendolan, A.; Dan, M.; Piccinni, P.; Bellomo, R.; De Nitti, C.; Inguaggiato, P.; Tetta, C. Adsorption in sepsis. Kidney Int. Suppl. 2000, 76, S148–S155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Pablo, R.; Monserrat, J.; Reyes, E.; Díaz, D.; Rodríguez-Zapata, M.; de la Hera, A.; Prieto, A.; Alvarez-Mon, M. Sepsis-induced acute respiratory distress syndrome with fatal outcome is associated to increased serum transforming growth factor beta-1 levels. Eur. J. Intern. Med. 2012, 23, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.D.; Machado, F.S.; Tanowitz, H.B.; Desruisseaux, M.S. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. 2014, 118, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Kelley, K.; Melichian, D.S.; Tamaki, Z.; Fang, F.; Su, Y.; Feng, G.; Pope, R.M.; Budinger, G.R.S.; Mutlu, G.M.; et al. Toll-Like Receptor 4 Signaling Augments Transforming Growth Factor-β Responses. Am. J. Pathol. 2013, 182, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascual, F.; Busnadiego, O.; González-Santamaría, J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014, 118, 156–164. [Google Scholar] [CrossRef]
- Dunzendorfer, S.; Lee, H.-K.; Soldau, K.; Tobias, P.S. Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: Roles of LBP and sCD14 in mediating LPS responses. FASEB J. 2004, 18, 1117–1119. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Kennedy-Lydon, T.M.; Crawford, C.; Wildman, S.S.P.; Peppiatt-Wildman, C.M. Renal pericytes: Regulators of medullary blood flow. Acta Physiol. 2013, 207, 212–225. [Google Scholar] [CrossRef]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, C.; Wan, L.; Egi, M.; May, C.N.; Bellomo, R. Renal blood flow in experimental septic acute renal failure. Kidney Int. 2006, 69, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Banas, B.; Schlöndorff, D. Signaling danger: Toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. 2004, 15, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.P.; Tsuji, T.; Baranova, I.N.; Bocharov, A.V.; Wilkins, K.J.; Street, J.M.; Alvarez-Prats, A.; Hu, X.; Eggerman, T.; Yuen, P.S.T.; et al. TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol. Rep. 2015, 3, e12558. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, X.; Zou, F.; Xu, T.; Pan, P.; Hu, C. Toll-like receptor-4 deficiency alleviates chronic intermittent hypoxia-induced renal injury, inflammation, and fibrosis. Sleep Breath. 2019, 23, 503–513. [Google Scholar] [CrossRef]
- Jiang, H.; Qu, P.; Wang, J.-W.; Li, G.-H.; Wang, H.-Y. Effect of NF-κB inhibitor on Toll-like receptor 4 expression in left ventricular myocardium in two-kidney-one-clip hypertensive rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3224–3233. [Google Scholar]
- Campbell, M.T.; Hile, K.L.; Zhang, H.; Asanuma, H.; Vanderbrink, B.A.; Rink, R.R.; Meldrum, K.K. Toll-Like Receptor 4: A Novel Signaling Pathway During Renal Fibrogenesis. J. Surg. Res. 2011, 168, e61–e69. [Google Scholar] [CrossRef]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.-J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ishida, W.; Bhattacharyya, S.; Li, Y.; Platanias, L.C.; Varga, J. Selective inhibition of activin receptor-like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum. 2004, 50, 4008–4021. [Google Scholar] [CrossRef]
- Meng, X.-M.; Huang, X.R.; Xiao, J.; Chung, A.C.K.; Qin, W.; Chen, H.; Lan, H.Y. Disruption of Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef]
- Gu, H.; Mickler, E.A.; Cummings, O.W.; Sandusky, G.E.; Weber, D.J.; Gracon, A.; Woodruff, T.; Wilkes, D.S.; Vittal, R. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014, 28, 4223–4234. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Midwood, K.S.; Yin, H.; Varga, J. Toll-Like Receptor-4 Signaling Drives Persistent Fibroblast Activation and Prevents Fibrosis Resolution in Scleroderma. Adv. Wound Care 2017, 6, 356–369. [Google Scholar] [CrossRef]
- Jerala, R. Structural biology of the LPS recognition. Int. J. Med. Microbiol. 2007, 297, 353–363. [Google Scholar] [CrossRef]
- Wang, S.C.; Klein, R.D.; Wahl, W.L.; Alarcon, W.H.; Garg, R.J.; Remick, D.G.; Su, G.L. Tissue Coexpression of LBP and CD14 mRNA in a Mouse Model of Sepsis. J. Surg. Res. 1998, 76, 67–73. [Google Scholar] [CrossRef]
- Stromberg, L.R.; Mendez, H.M.; Kubicek-Sutherland, J.Z.; Graves, S.W.; Hengartner, N.W.; Mukundan, H. Presentation matters: Impact of association of amphiphilic LPS with serum carrier proteins on innate immune signaling. PLoS ONE 2018, 13, e0198531. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.R.; Galley, H.F. Immunomodulation in the critically ill. Br. J. Anaesth. 2009, 103, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Wan, L.; Langenberg, C.; May, C. Septic Acute Kidney Injury: New Concepts. Nephron Exp. Nephrol. 2008, 109, e95–e100. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellano, G.; Stasi, A.; Franzin, R.; Sallustio, F.; Divella, C.; Spinelli, A.; Netti, G.S.; Fiaccadori, E.; Cantaluppi, V.; Crovace, A.; et al. LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling. Int. J. Mol. Sci. 2019, 20, 3682. https://doi.org/10.3390/ijms20153682
Castellano G, Stasi A, Franzin R, Sallustio F, Divella C, Spinelli A, Netti GS, Fiaccadori E, Cantaluppi V, Crovace A, et al. LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling. International Journal of Molecular Sciences. 2019; 20(15):3682. https://doi.org/10.3390/ijms20153682
Chicago/Turabian StyleCastellano, Giuseppe, Alessandra Stasi, Rossana Franzin, Fabio Sallustio, Chiara Divella, Alessandra Spinelli, Giuseppe Stefano Netti, Enrico Fiaccadori, Vincenzo Cantaluppi, Antonio Crovace, and et al. 2019. "LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling" International Journal of Molecular Sciences 20, no. 15: 3682. https://doi.org/10.3390/ijms20153682
APA StyleCastellano, G., Stasi, A., Franzin, R., Sallustio, F., Divella, C., Spinelli, A., Netti, G. S., Fiaccadori, E., Cantaluppi, V., Crovace, A., Staffieri, F., Lacitignola, L., Grandaliano, G., Simone, S., Pertosa, G. B., & Gesualdo, L. (2019). LPS-Binding Protein Modulates Acute Renal Fibrosis by Inducing Pericyte-to-Myofibroblast Trans-Differentiation through TLR-4 Signaling. International Journal of Molecular Sciences, 20(15), 3682. https://doi.org/10.3390/ijms20153682