Effective Food Ingredients for Fatty Liver: Soy Protein β-Conglycinin and Fish Oil


Abstract
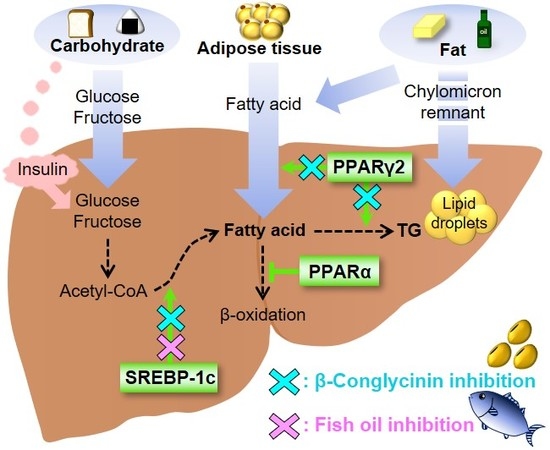
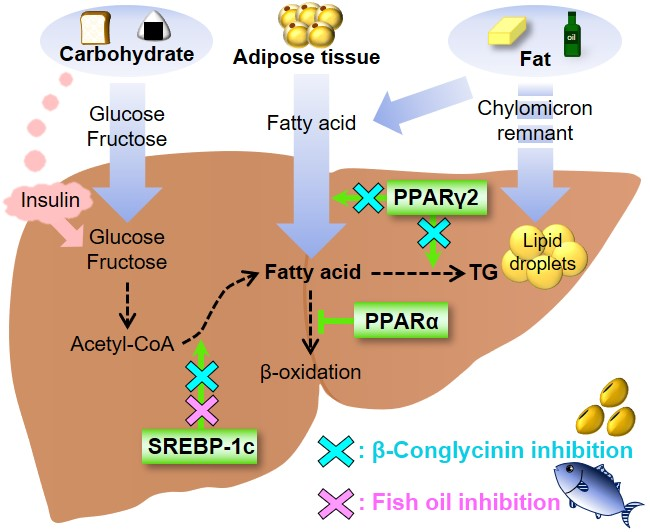{kind=link}
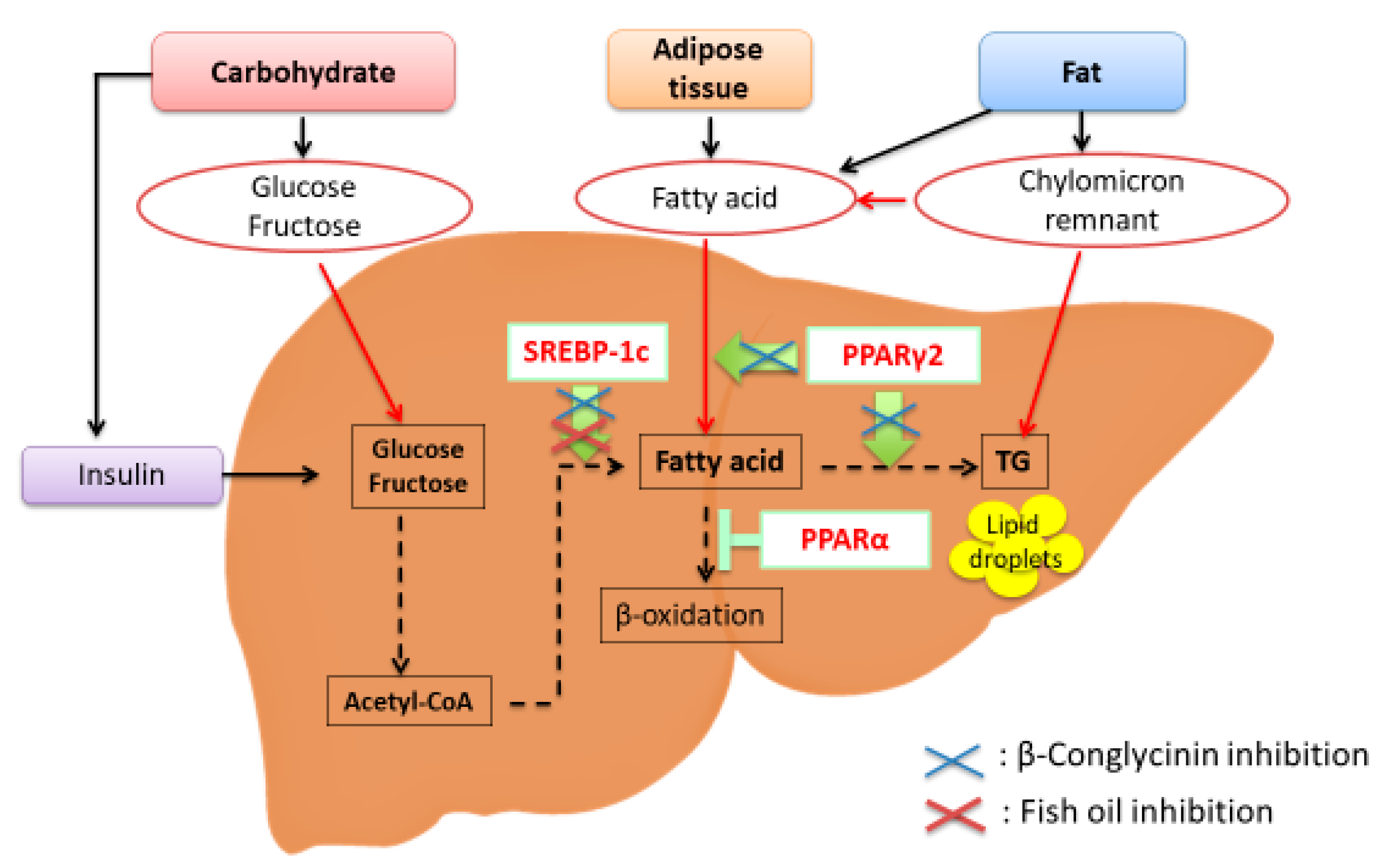{kind=link}
1. Introduction
2. Effects of β-Conglycinin
2.1. Preventive Effects of β-Conglycinin on Nonalcoholic Fatty Liver Disease (NAFLD)
2.2. Improvement Effects of β-Conglycinin on NAFLD in Diet-induced Obese Mice
2.3. Preventive Effects of β-Conglycinin on Alcohol-Induced Fatty Liver
2.4. How Does β-Conglycinin Work?
3. Effects of Fish Oil
3.1. Preventive Effects of Fish Oil on NAFLD Induced by a High-Fat Diet
3.2. Preventive Effects of Fish Oil on NAFLD Induced by High-Sucrose Diet
3.3. Fish Oil in Human NAFLD
3.4. Preventive Effects of Fish Oil on Ethanol-Induced Fatty Liver
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bes-Rastrollo, M.; Sanchez-Villegas, A.; Gomez-Gracia, E.; Martinez, J.A.; Pajares, R.M.; Martinez-Gonzalez, M.A. Predictors of weight gain in a Mediterranean cohort: The Seguimiento Universidad de Navarra Study 1. Am. J. Clin. Nutr. 2006, 83, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Swinburn, B.A.; Metcalf, P.A.; Ley, S.J. Long-term (5-year) effects of a reduced-fat diet intervention in individuals with glucose intolerance. Diabetes Care 2001, 24, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Vadiveloo, M.; Scott, M.; Quatromoni, P.; Jacques, P.; Parekh, N. Trends in dietary fat and high-fat food intakes from 1991 to 2008 in the Framingham Heart Study participants. Br. J. Nutr. 2014, 111, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.; Marques-Vidal, P.; Cortez-Pinto, H. Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol. 2006, 45, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Ni Mhurchu, C.; Parag, V.; Nakamura, M.; Patel, A.; Rodgers, A.; Lam, T.H. Body mass index and risk of diabetes mellitus in the Asia-Pacific region. Asia Pac. J. Clin. Nutr. 2006, 15, 127–133. [Google Scholar] [PubMed]
- Hartemink, N.; Boshuizen, H.C.; Nagelkerke, N.J.; Jacobs, M.A.; van Houwelingen, H.C. Combining risk estimates from observational studies with different exposure cutpoints: A meta-analysis on body mass index and diabetes type 2. Am. J. Epidemiol. 2006, 163, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Peeters, A.; de Courten, M.; Stoelwinder, J. The magnitude of association between overweight and obesity and the risk of diabetes: A meta-analysis of prospective cohort studies. Diabetes Res. Clin. Pract. 2010, 89, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hajifathalian, K.; Ezzati, M.; Woodward, M.; Rimm, E.B.; Danaei, G. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: A pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet 2014, 383, 970–983. [Google Scholar] [PubMed]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; et al. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Bo, S.; Uberti, B.; Biroli, G.; Pagano, G.; Cassader, M. Should nonalcoholic fatty liver disease be included in the definition of metabolic syndrome? A cross-sectional comparison with Adult Treatment Panel III criteria in nonobese nondiabetic subjects. Diabetes Care 2008, 31, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G. Nonalcoholic steatohepatitis: A study of 49 patients. Hum. Pathol. 1989, 20, 594–598. [Google Scholar] [CrossRef]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar] [CrossRef]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Tiikkainen, M.; Bergholm, R.; Vehkavaara, S.; Rissanen, A.; Hakkinen, A.M.; Tamminen, M.; Teramo, K.; Yki-Jarvinen, H. Effects of identical weight loss on body composition and features of insulin resistance in obese women with high and low liver fat content. Diabetes 2003, 52, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Krauss, R.M.; Christiansen, M.P.; Neese, R.A.; Hellerstein, M.K. Effects of a low-fat, high-carbohydrate diet on VLDL-triglyceride assembly, production, and clearance. J. Clin. Investig. 1999, 104, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Pacot, C.; Dugail, I.; Lemarchand, P.; Guichard, C.; Le Liepvre, X.; Berthelier-Lubrano, C.; Spiegelman, B.; Kim, J.B.; Ferre, P.; et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell. Biol. 1999, 19, 3760–3768. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.L.; Zhu, C.H.; Min, M.; Wang, J.; Lan, C.H.; Fan, L.L.; Sun, W.J.; Chen, D.F. The role of hepatic liver X receptor alpha- and sterol regulatory element binding protein-1c-mediated lipid disorder in the pathogenesis of non-alcoholic steatohepatitis in rats. J. Int. Med. Res. 2011, 39, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Jimenez-Linan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARgamma2 is an initial event to induce fatty liver in response to a diet high in butter: PPARgamma2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; Videla, L.A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: An additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. MeTable 2011, 96, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Yamazaki, T.; Kawano, Y.; Miura, S.; Ezaki, O. Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J. Hepatol. 2008, 49, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Horning, M.G.; Williams, E.A.; Maling, H.M.; Brodie, B.B. Depot fat as source of increased liver triglycerides after ethanol. Biochem. Biophys. Res. Commun. 1960, 3, 635–640. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chan, C.; Kaplowitz, N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J. Hepatol. 2006, 45, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H. Japan’s longevity challenge. Science 2015, 350, 1135. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Sawada, N. The JPHC study: Design and some findings on the typical Japanese diet. Jpn. J. Clin. Oncol. 2014, 44, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Grieshop, C.M.; Fahey, G.C., Jr. Comparison of quality characteristics of soybeans from Brazil, China, and the United States. J. Agric. Food Chem. 2001, 49, 2669–2673. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.C.; Torre, M.; Marina, M.L.; Laborda, F. Composition and characterization of soyabean and related products. Crit. Rev. Food Sci. Nutr. 1997, 37, 361–391. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, S.; Yamauchi, F. Determination of glycinin and β-conglycinin in soybean proteins by immunological methods. J. Agric. Food Chem. 1987, 35, 200–205. [Google Scholar] [CrossRef]
- Maruyama, N.; Katsube, T.; Wada, Y.; Oh, M.H.; Barba De La Rosa, A.P.; Okuda, E.; Nakagawa, S.; Utsumi, S. The roles of the N-linked glycans and extension regions of soybean β-conglycinin in folding, assembly and structural features. Eur. J. Biochem. 1998, 258, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kishimoto, K.; Miura, S.; Ezaki, O. Dietary β-conglycinin prevents fatty liver induced by a high-fat diet by a decrease in peroxisome proliferator-activated receptor gamma2 protein. J. Nutr. Biochem. 2012, 23, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, N.; Iwaoka, Y.; Hirotsuka, M.; Horio, F.; Kohno, M. β-conglycinin lowers very-low-density lipoprotein-triglyceride levels by increasing adiponectin and insulin sensitivity in rats. Biosci. Biotechnol. Biochem. 2010, 74, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Fujiwara, Y.; Kato, M.; Funayama, A.; Ogawa, N.; Tachibana, N.; Kohno, M.; Ikeda, I. Soybean β-conglycinin improves carbohydrate and lipid metabolism in Wistar rats. Biosci. Biotechnol. Biochem. 2015, 79, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Hashidume, T.; Kato, A.; Tanaka, T.; Miyoshi, S.; Itoh, N.; Nakata, R.; Inoue, H.; Oikawa, A.; Nakai, Y.; Shimizu, M.; et al. Single ingestion of soy β-conglycinin induces increased postprandial circulating FGF21 levels exerting beneficial health effects. Sci. Rep. 2016, 6, 28183. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Yamashita, H.; Kawaguchi, T. Carbohydrate responsive element-binding protein (ChREBP): A key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 2002, 63, 2075–2080. [Google Scholar] [CrossRef]
- Li, D.; Ikaga, R.; Yamazaki, T. Soy protein β-conglycinin ameliorates fatty liver and obesity in diet-induced obese mice through the downregulation of PPARγ. Br. J. Nutr. 2018, 119, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Watanabe, K.; Inoue, N.; Nakagawa, Y.; Ishigaki, N.; Matsuzaka, T.; Takeuchi, Y.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; et al. Protein kinase Cbeta mediates hepatic induction of sterol-regulatory element binding protein-1c by insulin. J. Lipid Res. 2010, 51, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Goldstein, J.L.; Brown, M.S. Insulin induction of SREBP-1c in rodent liver requires LXRalpha-C/EBPbeta complex. Proc. Natl. Acad. Sci. USA 2016, 113, 8182–8187. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wu, J.; Chen, X.; Guo, T.; Chen, Z.J.; Goldstein, J.L.; Brown, M.S. BHLHE40, a third transcription factor required for insulin induction of SREBP-1c mRNA in rodent liver. eLife 2018, 7, e36826. [Google Scholar] [CrossRef] [PubMed]
- Ikaga, R.; Li, D.; Yamazaki, T. Dietary β-conglycinin prevents acute ethanol-induced fatty liver in mice. Biochem. Biophys. Res. Commun. 2017, 493, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Laws and Regulations, Ministry of Health. The National Health and Nutrition survey in Japan, 2015; Daiich Shuppan Publishing: Tokyo, Japan, 2017. [Google Scholar]
- Kohno, M.; Hirotsuka, M.; Kito, M.; Matsuzawa, Y. Decreases in serum triacylglycerol and visceral fat mediated by dietary soybean β-conglycinin. J. Atheroscler. Thromb. 2006, 13, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Qin, G.X.; Sun, Z.W.; Zhao, Y. Advances of research on glycinin and β-conglycinin: A review of two major soybean allergenic proteins. Crit. Rev. Food Sci. Nutr. 2014, 54, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Lovati, M.R.; Manzoni, C.; Gianazza, E.; Arnoldi, A.; Kurowska, E.; Carroll, K.K.; Sirtori, C.R. Soy protein peptides regulate cholesterol homeostasis in Hep G2 cells. J. Nutr. 2000, 130, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, Y.; Maebuchi, M.; Kohno, M.; Hirotsuka, M.; Wadahama, H.; Moriyama, T.; Kawada, T.; Urade, R. Changes in lipid metabolism by soy β-conglycinin-derived peptides in HepG2 cells. J. Agric. Food Chem. 2009, 57, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Villaluenga, C.; Rupasinghe, S.G.; Schuler, M.A.; Gonzalez de Mejia, E. Peptides from purified soybean β-conglycinin inhibit fatty acid synthase by interaction with the thioesterase catalytic domain. FEBS J. 2010, 277, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Ede, S.; Silva, M.A.; Demonte, A.; Neves, V.A. Soy β-conglycinin (7S globulin) reduces plasma and liver cholesterol in rats fed hypercholesterolemic diet. J. Med. Food 2011, 14, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.d.S.; Silva, M.A.; Demonte, A.; Neves, V.A. β-Conglycinin (7S) and glycinin (11S) exert a hypocholesterolemic effect comparable to that of fenofibrate in rats fed a high-cholesterol diet. J. Funct. Foods 2010, 2, 275–283. [Google Scholar] [CrossRef]
- Wall, R.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Capanni, M.; Calella, F.; Biagini, M.R.; Genise, S.; Raimondi, L.; Bedogni, G.; Svegliati-Baroni, G.; Sofi, F.; Milani, S.; Abbate, R.; et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: A pilot study. Aliment. Pharmacol. Ther. 2006, 23, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Takahashi, M.; Ezaki, O. Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for down-regulation of lipogenic enzyme mRNAs. J. Biol. Chem. 1999, 274, 25892–25898. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Kim, H.J.; Kaburagi, Y.; Yasuda, K.; Ezaki, O. A low fish oil inhibits SREBP-1 proteolytic cascade, while a high-fish-oil feeding decreases SREBP-1 mRNA in mice liver: Relationship to anti-obesity. J. Lipid Res. 2003, 44, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Harris, W.S. Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J. Lipid Res. 2003, 44, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Kemuriyama, N.; Matsuzaka, T.; Kuba, M.; Ohno, H.; Han, S.I.; Takeuchi, Y.; Isaka, M.; Kobayashi, K.; Iwasaki, H.; Yatoh, S.; et al. Different Effects of Eicosapentaenoic and Docosahexaenoic Acids on Atherogenic High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. PLoS ONE 2016, 11, e0157580. [Google Scholar] [CrossRef] [PubMed]
- Depner, C.M.; Philbrick, K.A.; Jump, D.B. Docosahexaenoic acid attenuates hepatic inflammation, oxidative stress, and fibrosis without decreasing hepatosteatosis in a Ldlr(−/−) mouse model of western diet-induced nonalcoholic steatohepatitis. J. Nutr. 2013, 143, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lytle, K.A.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid blocks progression of western diet-induced nonalcoholic steatohepatitis in obese Ldlr−/− mice. PLoS ONE 2017, 12, e0173376. [Google Scholar] [CrossRef] [PubMed]
- Froyland, L.; Madsen, L.; Vaagenes, H.; Totland, G.K.; Auwerx, J.; Kryvi, H.; Staels, B.; Berge, R.K. Mitochondrion is the principal target for nutritional and pharmacological control of triglyceride metabolism. J. Lipid Res. 1997, 38, 1851–1858. [Google Scholar] [PubMed]
- Kimura, R.; Takahashi, N.; Lin, S.; Goto, T.; Murota, K.; Nakata, R.; Inoue, H.; Kawada, T. DHA attenuates postprandial hyperlipidemia via activating PPARalpha in intestinal epithelial cells. J. Lipid Res. 2013, 54, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shao, Y.; Yuan, F.; Feng, H.; Li, N.; Zhang, H.; Wu, C.; Liu, Z. Fish Oil Feeding Modulates the Expression of Hepatic MicroRNAs in a Western-Style Diet-Induced Nonalcoholic Fatty Liver Disease Rat Model. BioMed Res. Int. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Nakamori, A.; Sasaki, E.; Wada, S.; Ezaki, O. Fish oil prevents sucrose-induced fatty liver but exacerbates high-safflower oil-induced fatty liver in ddy mice. Hepatology 2007, 46, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kishimoto, K.; Ezaki, O. The ddY mouse: A model of postprandial hypertriglyceridemia in response to dietary fat. J. Lipid Res. 2012, 53, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Shionoiri, F.; Ohashi, K.; Osuga, J.; et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem. 1999, 274, 35840–35844. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Shimano, H.; Yahagi, N.; Ide, T.; Amemiya-Kudo, M.; Matsuzaka, T.; Nakakuki, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem. 2002, 277, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Xu, Z.; Harvey, K.A.; Pavlina, T.M.; Becker, M.J.; Zaloga, G.P. Comparative study of the modulation of fructose/sucrose-induced hepatic steatosis by mixed lipid formulations varying in unsaturated fatty acid content. Nutr. Metab. 2015, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- De Castro, G.S.; Deminice, R.; Simoes-Ambrosio, L.M.; Calder, P.C.; Jordao, A.A.; Vannucchi, H. Dietary docosahexaenoic acid and eicosapentaenoic acid influence liver triacylglycerol and insulin resistance in rats fed a high-fructose diet. Mar. Drugs 2015, 13, 1864–1881. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Peng, C.; Ai, Y.; Wang, H.; Xiao, X.; Li, J. Docosahexaenoic Acid Ameliorates Fructose-Induced Hepatic Steatosis Involving ER Stress Response in Primary Mouse Hepatocytes. Nutrients 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Kris-Etherton, P.M.; Taylor, D.S.; Yu-Poth, S.; Huth, P.; Moriarty, K.; Fishell, V.; Hargrove, R.L.; Zhao, G.; Etherton, T.D. Polyunsaturated fatty acids in the food chain in the United States. Am. J. Clin. Nutr. 2000, 71, 179S–188S. [Google Scholar] [CrossRef] [PubMed]
- Feskens, E.J.; Kromhout, D. Epidemiologic studies on Eskimos and fish intake. Ann. N. Y. Acad. Sci. 1993, 683, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: Results from the Welcome* study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Argo, C.K.; Patrie, J.T.; Lackner, C.; Henry, T.D.; de Lange, E.E.; Weltman, A.L.; Shah, N.L.; Al-Osaimi, A.M.; Pramoonjago, P.; Jayakumar, S.; et al. Effects of n-3 fish oil on metabolic and histological parameters in NASH: A double-blind, randomized, placebo-controlled trial. J. Hepatol. 2015, 62, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Dasarathy, J.; Khiyami, A.; Yerian, L.; Hawkins, C.; Sargent, R.; McCullough, A.J. Double-blind randomized placebo-controlled clinical trial of omega 3 fatty acids for the treatment of diabetic patients with nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2015, 49, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Sano, K.; Horiuchi, A.; Tanaka, E.; Kiyosawa, K.; Aoyama, T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2008, 42, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 2014, 147, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Rise, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Della Corte, C.; Rise, P.; Galli, C.; Agostoni, C.; Bedogni, G. Docosahexaenoic acid for the treatment of fatty liver: Randomised controlled trial in children. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.M.; Johnson, N.A.; Burdon, C.A.; Cohn, J.S.; O’Connor, H.T.; George, J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 56, 944–951. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Wu, X.L.; Chen, R.P.; Chen, C.; Liu, X.G.; Wu, B.J.; Huang, Z.M. Effectiveness of Omega-3 Polyunsaturated Fatty Acids in Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0162368. [Google Scholar] [CrossRef] [PubMed]
- De Castro, G.S.; Calder, P.C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 2018, 37, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Musa-Veloso, K.; Venditti, C.; Lee, H.Y.; Darch, M.; Floyd, S.; West, S.; Simon, R. Systematic review and meta-analysis of controlled intervention studies on the effectiveness of long-chain omega-3 fatty acids in patients with nonalcoholic fatty liver disease. Nutr. Rev. 2018, 76, 581–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Wang, Y.F.; Xu, Q.H.; Chen, S.S. Omega-3 fatty acids as a treatment for non-alcoholic fatty liver disease in children: A systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2018, 37, 516–521. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, T.; Li, D.; Ikaga, R. Effective Food Ingredients for Fatty Liver: Soy Protein β-Conglycinin and Fish Oil. Int. J. Mol. Sci. 2018, 19, 4107. https://doi.org/10.3390/ijms19124107
Yamazaki T, Li D, Ikaga R. Effective Food Ingredients for Fatty Liver: Soy Protein β-Conglycinin and Fish Oil. International Journal of Molecular Sciences. 2018; 19(12):4107. https://doi.org/10.3390/ijms19124107
Chicago/Turabian StyleYamazaki, Tomomi, Dongyang Li, and Reina Ikaga. 2018. "Effective Food Ingredients for Fatty Liver: Soy Protein β-Conglycinin and Fish Oil" International Journal of Molecular Sciences 19, no. 12: 4107. https://doi.org/10.3390/ijms19124107
APA StyleYamazaki, T., Li, D., & Ikaga, R. (2018). Effective Food Ingredients for Fatty Liver: Soy Protein β-Conglycinin and Fish Oil. International Journal of Molecular Sciences, 19(12), 4107. https://doi.org/10.3390/ijms19124107