Abstract
Following the epidemics of obesity due to the consumption of high-calorie diet and sedentary lifestyle, nonalcoholic fatty liver disease (NAFLD) is now the leading cause of liver disease in Western countries. NAFLD is epidemiologically associated with metabolic syndrome and insulin resistance, and in susceptible individuals it may progress to cirrhosis and hepatocellular carcinoma. Genetic factors play a key role in NAFLD predisposition by interacting with nutritional and other environmental factors. To date, there is no drug therapy for the treatment of NAFLD, and the main clinical recommendation is lifestyle modification. In the last years, nutrigenomics is promoting an increased understanding of how nutrition affects the switch from health to disease by altering the expression of an individual’s genetic makeup. The present review tries to summarize the most recent data evidencing how the interactions between nutrients and genetic factors can influence NAFLD development. The final goal should be to develop tools to quantify these complex interactions. The definition of a “nutrigenomic risk score” for each individual may represent a novel therapeutic approach for the management of NAFLD patients.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined by fat accumulation exceeding 5% of liver weight not explained by at-risk alcohol intake [1]. NAFLD is epidemiologically associated with metabolic syndrome and insulin resistance (IR) [2,3]. Following the epidemic of obesity due to the consumption of high-calorie diet and sedentary lifestyle, NAFLD is becoming a serious global health problem, representing the leading cause of liver disease [4,5,6]. NAFLD defines a wide pathological spectrum, ranging from indolent liver fat storage, which may remain uncomplicated to nonalcoholic steatohepatitis (NASH) characterized by hepatocellular damage, lobular necroinflammation, and the activation of fibrogenesis [7,8]. NASH potentially predisposes to liver cirrhosis and hepatocellular carcinoma (HCC), and is projected to become the first cause of liver-related morbidity and mortality in Western countries within ten years [9].
A growing body of evidence indicated that NAFLD develops as a result of a complex interaction between genetic susceptibility and other environmental factors [10], typically hypercaloric diet and physical inactivity. Furthermore, NAFLD development and progression is modulated by epigenetic factors such as liver-specific DNA-methylation and microRNAs, which regulate the liver transcriptome. There is currently no drug therapy for the treatment of NAFLD, and the mainstay management of the disease is lifestyle modification. The interface between the nutritional environment and cellular/genetic processes is being referred to as nutritional genomics or “nutrigenomics” [11]. In the last years, nutrigenomics has contributed to the understanding of how nutrition affects the switch from health to disease by modulating the expression of an individual’s genetic makeup.
In this review, we focus upon the interaction of genetic background and diet with regard to the development and progression of NAFLD, which can be considered a good candidate for nutrigenomic studies. Indeed, individuals vary in their nutrients requirement and response to diet according to their genetic features, suggesting that personalized nutrition may represent an individualistic therapeutic approach to the disease, with risk being modulated by nutritional, lifestyle, and genetic influences.
2. Pathophysiology of NAFLD
Hepatic fat accumulation results from an imbalance between the uptake and synthesis of fatty acids on one hand, and their utilization and secretion on the other hand. Esterification in the form of triglycerides (TGs) in hepatocellular lipid droplets represents the safest way to store fatty acids in the liver [2]. Excess hepatocellular TG derives from several sources, including dietary fatty acids, increased peripheral lipolysis due to adipose tissue insulin resistance, and elevated hepatic de novo lipogenesis (DNL) due to hyperinsulinemia [10,12,13]. A relative reduction of lipid secretion through very-low-density lipoproteins (VLDL) and of mitochondrial oxidation with the advent of oxidative damage are also involved in hepatic fat accumulation [13].
The multiple hit hypothesis proposed that TG accumulation sensitizes the liver to second insults, which are represented by (a) direct lipotoxicity and endoplasmic reticulum stress, (b) hepatocellular oxidative stress secondary to free radicals produced during β- and ω-oxidation of free fatty acids (FFAs), (c) inflammation triggered by endotoxin engaging Toll-like receptor-4 in Kupffer cells and hepatocytes due to increased intestinal permeability and to qualitative and quantitative changes in gut microbiota [14,15], (d) insulin resistance and altered profile of adipokines [16], (e) activation and senescence of hepatic stellate cells [17]. All these conditions lead to hepatic inflammation, cellular damage, and the activation of fibrogenesis. However, other organs (e.g., the adipose tissue, muscle, and intestine) are involved in the pathogenesis of NAFLD, which can thus be defined as a systemic metabolic disorder [18].
3. Genetics of NAFLD
Although many individuals share environmental risk factors such as obesity, not all at risk subjects develop fatty liver, suggesting that the genetic background plays a significant role in determining this condition. Epidemiological, familial, and twin studies [19,20] support a strong heritability component in NAFLD and NASH [21]. Indeed, there is a huge inter-ethnic variability in the predisposition towards NAFLD [4].
The rs738409 C > G variant in patatin-like phospholipase domain-containing 3 (PNPLA3)—encoding for the amino acid substitution I148M—has been identified as a major determinant of hepatic fat content [22,23]. The size effect of the I148M variant on the risk of NAFLD is the strongest ever reported for a common variant, modifying the genetic susceptibility of NAFLD [24,25] and also determining liver disease severity and the progression of NAFLD [23,24,26]. The polymorphism exerts its detrimental activity by leading to PNPLA3 accumulation at the surface of hepatocellular lipid droplets, where it likely inhibits the activity of other lipases, determining reduced TG turnover and dismissal [27]. Accumulation of the mutated I148M protein is triggered by acquired factors. Indeed, PNPLA3 is nutritionally regulated at the transcriptional level in response to increased insulin levels by sterol regulatory element-binding protein (SREBP)-1c through carbohydrate-mediated activation of liver X receptor/retinoid X receptor (LXR/RXR). Furthermore, fatty acids can inhibit PNPLA3 protein degradation [28,29,30]. As a consequence, a significant interaction exists between the effect of adiposity on the phenotypic expression of the PNPLA3 mutation and the risk of NAFLD and cirrhosis [31]. This represents a clear demonstration of a gene ↔* environment interaction in the pathogenesis of a complex disease.
The rs58542926 C > T genetic variant of the transmembrane 6 superfamily member 2 gene (TM6SF2), encoding the E167K variant, is another determinant of hepatic triglyceride content [32] and the full spectrum of liver damage linked to hepatic fat accumulation, including NASH, necroinflammation, and fibrosis. The protein is involved in VLDL lipidation and secretion, and the mechanism of the association is related to the retention of lipids within intracellular lipid droplets [32,33].
Variation in the GCKR (glucokinase regulatory gene) locus has been associated with fasting triglycerides levels [34,35,36,37] and NAFLD [38,39,40,41]. GCKR regulates DNL by controlling the influx of glucose in hepatocytes. A common missense loss-of-function GCKR mutation (rs1260326), encoding for the P446L protein variant, may represent the causal variant underlying the association with hepatic fat accumulation [37,42,43]. The P446L variant indeed affects GCKR’s ability to negatively regulate glucokinase in response to fructose-6-phosphate, thereby determining constitutive activation of hepatic glucose uptake [39]. This leads to decreased circulating fasting glucose and insulin levels, but on the other hand it would lead to increased glycolysis and the production of malonyl-CoA which favors hepatic fat accumulation by serving as a substrate for lipogenesis and by blocking fatty acid oxidation.
The rs641738 C > T variant linked to the 3’ untranslated region of the MBOAT7 (membrane bound O-acyltransferase domain-containing 7 gene) was recently associated with the risk of NAFLD, inflammation, and fibrosis by reducing protein expression and phosphatidyl-inositol desaturation in hepatocytes [44].
Finally, case–control studies demonstrated a role of other genetic variants implicated in inflammation, insulin signaling, oxidative stress, and fibrogenesis in NAFLD progression (reviewed in [45]).
4. Nutrients and NAFLD
Diet is an important contributor to the pathogenesis of NAFLD. Excessive energy intake results in excess adiposity, insulin resistance, and the consequent inappropriate release of fatty acids into the circulation. Dietary fat represents the primary source of adipose tissue fatty acids, which are subsequently released and re-esterified in the liver [46]. The composition of diet and the nature of fatty acids may also impact hepatic DNL by directly regulating the expression of genes involved in fatty acid synthesis. It has been established that—at least in animal models—polyunsaturated fatty acids (PUFAs) down-regulate sterol regulatory element binding protein-1c (SREBP-1c) [47], whereas saturated fatty acids (SFAs) stimulate its expression.
NAFLD patients tend to have a dietary pattern characterized by a higher consumption of SFAs, cholesterol, and fructose, and lower ingestion of PUFAs and antioxidants (vitamin C and E) [48]. It is possible that altering dietary macronutrient composition, which addresses specific molecular pathways and modifies gene and protein expression, modulates the clinical outcome.
4.1. Saturated and Unsaturated Fatty Acids
The consumption of diets rich in saturated fatty acids triggers DNL. The action of SFAs on hepatic lipid metabolism is influenced by the peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1beta (PGC-1β). A high-fat diet enriched with SFAs stimulates liver PGC-1β, which coactivates SREBP-1c and enhances its transcriptional activity in lipogenic genes, such as stearoyl-CoA desaturase-1 (SCD-1), fatty acid synthase (FAS), and diacylglycerol acyltransferase (DGAT) [49]. Dietary patterns enriched in SFAs have also been associated with a higher expression of the unfolded protein response (UPR)-related components, all elicited by endoplasmic reticulum stress. Moreover, an increase in caspase 3 activity was also observed [50].
The ingestion of monounsaturated fatty acids (MUFAs) lowers cardiovascular risk and improves lipid profile. Replacement of SFAs with MUFAs ameliorates serum glucose levels and blood pressure [51]. MUFAs inhibit the oxidation of LDL-cholesterol and decrease the serum level of triglycerides by activation of peroxisome proliferator-activated receptor α (PPARα). PUFAs—in particular docosahexaenoic acid (DHA)—are long-chain n-3 fatty acids that reduce TG accumulation and improve hepatic steatosis [52]. PUFAs might prevent NAFLD by activation of peroxisome proliferator-activated receptors (PPARs) and inhibition of SREBP-1c. PPARs are the most extensively characterized nuclear receptors that are regulated by fatty acids [53]. There are three major isoforms of PPARs: PPARα, PPARβ, and PPARγ, with PPARα being the predominant isoform in liver. Ligand activation of PPARα is associated with transcriptional up-regulation of a wide range of genes for proteins associated with fatty acid oxidation and lipoprotein metabolism [52].
4.2. Carbohydrates
Carbohydrates are the main substrate that drive DNL in the liver and the adipose tissue. In particular, the intake of sucrose-sweetened beverages has been associated with a higher amount of visceral adipose tissue (VAT) and liver fat accumulation compared with milk, diet cola, and water [54]. A study which compared the consumption of fructose-sweetened with glucose-sweetened beverages revealed that mainly fructose-containing drinks increase visceral adipose volume, promote DNL, and decrease insulin sensitivity in overweight/obese subjects [55]. Fructose consumption induces hepatic lipid accumulation by (a) activation of lipogenic gene expression and (b) direct flow of fructose carbon into the glycolytic pathway [56]. Fructose but not glucose ingestion activates hepatic carbohydrate-responsive element-binding protein (ChREBP), which regulates glycolytic and lipogenic gene expression programs [57]. Moreover, dietary fructose impairs the insulin signaling pathway by increasing c-Jun N-terminal kinase (JNK) activity and insulin receptor substrate 1 (IRS-1) phosphorylation [58]. Finally, elevated dietary fructose intake facilitates bacterial overgrowth in the small intestine, accompanied by heightened intestinal permeability. It increases endotoxin levels in the portal vein and contributes to the mechanism of NAFLD [59].
4.3. Proteins
Although the effects of dietary protein on hepatic lipids is not clear, clinical trials show that protein intake has a beneficial impact on the course of NAFLD [60]. Experiments in rodents demonstrated that a high-protein and low-carbohydrate diet reduces adipose tissue deposition, improves glucose homeostasis, and decreases steatosis by inhibition of DNL [61]. Dietary protein has a beneficial effect on glucose metabolism and decreases insulin output. Protein intake is essential for the regeneration of hepatocytes and supplies crucial amino acids for the inclusion of fat into lipoproteins for liver export, thus preventing steatosis [62].
Moreover, as amino acid catabolism is an energy-requiring process, a higher protein consumption might trigger hepatic lipid oxidation through an increase in hepatic energy expenditure [63].
4.4. Metals and Other Dietary Components
NAFLD is also characterized by a derangement in the homeostasis of metals, possibly reflecting an increased oxidative stress and an inflammation condition. In particular, it has been demonstrated that iron and copper dys-homeostasis can contribute to NAFLD pathogenesis.
Iron (Fe) is an essential nutrient required for erythropoiesis and multiple cellular metabolic functions. However, an excess of iron may be detrimental, mostly via the formation of reactive oxygen species, which may lead to severe organ damage. Iron perturbations are frequently observed in patients with NAFLD. Hepatic iron overload in conjunction with metabolic syndrome is commonly observed, and has been termed dysmetabolic iron overload syndrome (DIOS). Increased ferritin levels are usually associated with NASH and the severity of liver damage, whereas in patients with mild iron overload, iron depletion may decrease insulin resistance and liver damage [64,65]. Moreover, iron depletion up-regulated glucose uptake and increased insulin receptor expression and signaling in hepatocytes in vitro and in vivo [66], whereas dietary iron supplementation induced insulin resistance and dyslipidemia [67]. To investigate the mechanisms underlying iron accumulation in NAFLD patients, we recently examined the effect of fatty acids on hepatic iron metabolism. The exposure of hepatocytes to FFAs, leading to steatosis, was associated with a subversion of iron metabolism characterized by increased expression of transferrin receptor and the facilitation of iron storage [68].
Copper is known to be essential for many physiologic processes, such as antioxidant defense, lipid peroxidation, and mitochondrial function. A copper (Cu)-restricted diet induced NAFLD in animal models [69]. In addition, hepatic Cu deficiency is observed in human NAFLD and is associated with steatosis, NASH progression, and insulin resistance [70,71]. Additionally, NAFLD patients with low copper levels have increased iron stores [70]. In experimental models, dietary copper deficiency determined increased hepatic iron content as well as steatosis and insulin resistance [71]. Intestine-specific genetic inactivation of high-affinity copper import in mice also resulted in hepatic iron overload [72]. Interestingly, fructose feeding exacerbates complications of copper deficiency. In rats, fructose consumption impaired copper status and precipitated copper deficiency, possibly inhibiting its absorption through the intestinal epithelium.
Moreover, dietary copper deficiency and fructose feeding work together to exacerbate liver damage and accelerate hepatic fat accumulation [69,73]. Hepatic carnitine palmitoyltransferase 1 CPT-1 expression was significantly inhibited whereas FAS was markedly upregulated [69] in copper-deficient rats fed with fructose. Finally, rats fed either high-sucrose or low-copper diet had increased hepatic inflammation and fibrogenesis and hepatic stellate cell activation, while the combination of dietary factors induced fasting hepatic insulin resistance and liver damage [74].
5. Epigenetics in NAFLD
The emerging field of epigenetics, a heritable but reversible phenomenon that allows for fine-tuning gene expression without altering DNA sequence, provides a new perspective on the pathogenesis of NAFLD [75]. The epigenetic modulation of gene expression includes chromatin remodeling (histone modifications), DNA methylation, and regulation of RNA processing, stability, and translation through specific binding of small RNA molecules (microRNAs). Epigenetic modifications are known to be involved in hepatic lipid metabolism, insulin resistance, mitochondrial dysfunction, and oxidative stress—all of which contribute to NAFLD development and progression [76,77]
There has been considerable progress in understanding epigenetic mechanisms by which the fetal liver might be “primed” by gestational over-nutrition, and epigenetic modifications can be inherited from parents to their offspring. For example, increased methylation leading to down-regulation of PGC1α and mitochondrial biogenesis has been reported in patients with NAFLD [78]. In addition, fetal exposure to high fat diet (HFD) has been associated with histone modifications of the phosphoenol pyruvate carboxy kinase 1 gene (PCK1), the rate-limiting enzyme in the gluconeogenic pathway. These data suggest that epigenetic mechanisms prime offspring liver for increased hepatic glucose production [79]. Moreover, the livers of offspring mice exposed to maternal HFD were hypomethylated at the Cdkn1a gene, which codes for a cell cycle inhibitory protein, impairing the regenerative capacity and response to damage [80].
DNA methylation is considered a key factor in the process leading from simple steatosis to NASH, and it may be modulated by dietary deficiency of fundamental methyl donors such as betaine, choline, and folate [81]. Supplementation of betaine is associated with a reduced methylation of the promoter of microsomal triglyceride transfer protein (MTTP), involved in VLDL lipidation, consequently promoting TG export from the liver [82], whereas folate deficiency induces triglycerides accumulation in the liver by influencing the expression of genes involved in fatty acids synthesis [83].
Moreover, in a study that included obese patients with NAFLD and healthy controls, NAFLD-specific methylation differences were seen for nine genes coding for key enzymes catalyzing the initial steps in glucose, lipid, acetyl-CoA, and oligosaccharide synthesis and pathway members of insulin signaling. These NAFLD-specific methylation changes were partially reversible after bariatric surgery [84].
6. A Role for Microbiota?
Each individual contains about 1.5–2 Kg of micro-organisms (bacteria, fungi, viruses, and bacteriophages) within the gut. The intestinal microbiota contains >100-fold genes than the human genome, which are responsible for the production and modifications of a wide range of circulating metabolites, regulating the function of organs (reviewed in [85]). Human diseases are associated with modification of the human microbiota, and the diet is able to profoundly reshape the microbiotal composition within hours. It has already been demonstrated that the beneficial effect of some drugs (e.g., metformin) is partly mediated by their effect on the microbiota [86]. Therefore, it is likely that the effect of diet on the risk of NAFLD is partly mediated by alterations of the microbiota.
Intake of alcohol or a diet rich in saturated fatty acids, cholesterol, and fructose impairs the intestinal barrier by weakening the mucus-associated defense, impairing tight junction function and causing intestinal inflammation with subsequent translocation of bacterial pathogens into the bloodstream and to the liver. Several lines of evidence suggest that endotoxemia is involved in the pathogenesis of NAFLD/NASH. Endotoxin induces systemic inflammation and hepatic necroinflammation mainly through its action on toll-like receptor 4 (TLR4), and NAFLD/NASH patients have increased endotoxin levels in the blood. Indeed, patients with NAFLD have significantly increased gut permeability [15] and a higher prevalence of small intestine bacterial overgrowth compared to healthy controls, which in turn increase the absorption of endotoxin. Specifically, a dysbiotic microbiome is often observed among obese individuals and is considered to be one of the major risk factors for NAFLD [87]. Both obesity and NAFLD are associated with a higher proportion of Gram-negative bacterial species in the gut microbiome, and microbial populations of NASH patients have been suggested to have a higher ability to produce alcohol [88]. Given the association between specific microbial population and NAFLD, the development of a specific metabolic diagnostic profile could possibly represent a non-invasive therapeutic approach [89,90], and may be also useful for monitoring fibrosis stage.
7. Interaction between Genetic and Nutritional Factors
The PNPLA3 I148M variant represents a first and until now almost unique example of a genetic factor for which a clear interaction between the gene and environment has been robustly demonstrated. The PNPLA3 I148M variant interacts with environmental stressors such as obesity [31,91] and alcohol consumption [92], which induce fatty liver. Similarly, individuals who carry the I148M variant are more susceptible to increased fat content when dietary carbohydrate intake—specifically sugar—is high [93]. A nutrigenetic analysis with the I148M variant in Hispanic children revealed that hepatic TG accumulation was related to carbohydrate and sugar intake in the homozygous 148M/M group, whereas no effect was observed in I/I and I/M individuals. In Italian adolescents, the 148M variant interacted with dietary intake of fructose-sweetened beverages in determining NAFLD [94,95]. These results are consistent with the notion that the reduced capacity of subjects with the 148M/M genotype to hydrolyze TG in the liver would be exacerbated in the context of high dietary sugar, because carbohydrate-mediated up-regulation of PNPLA3 would favor the accumulation of the pathological protein on the surface of lipid droplets, resulting in the inhibition of other lipases, mainly PNPLA2 (Figure 1). Consistently, a short-term dietary intervention study provided evidence that a hypocaloric and low-carbohydrate diet reduces hepatic fat to a greater extent in PNPLA3 148M/M carriers as compared to wild-type individuals, despite similar weight loss in both genotype groups.
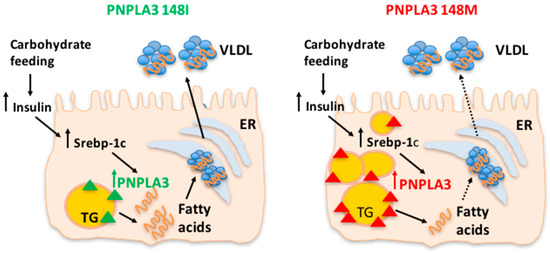
Figure 1.
The patatin-like phospholipase domain-containing 3 (PNPLA3) 148M variant favors triglyceride (TG) accumulation upon carbohydrate feeding. The reduced capacity of subjects with the PNPLA3 148M/M genotype to hydrolyze TG in the liver is exacerbated by high dietary sugar intake because carbohydrate-mediated up-regulation of PNPLA3 would favor the accumulation of the pathological protein on the surface of lipid droplets. Personalized dietary intervention based on reduction of carbohydrates intake in individuals with the PNPLA3 at risk genotype may lead to more effective clinical outcomes for NAFLD patients. ER: endoplasmic reticulum; VLDL: very low-density lipoprotein. Solid lines indicate unmodified pathways, dashed lines indicate pathways which are reduced.
In addition, hepatic fat accumulation could be modulated by the interaction between PNPLA3 I148M and dietary omega 6/omega 3 PUFA [96]. The I148M variant has decreased activity in hydrolyzing the n-9 fatty acids [97]. The n-9 represent the most common fatty acids in the diet, but they are also synthesized starting from n-6. It could be speculated that the newly-synthetized TGs tend to accumulate in the liver in patients carrying the 148M risk variant, leading to steatosis. In the meantime, the excess of n-6 not incorporated in TG will be utilized as substrate for the synthesis of proinflammatory species, triggering NASH [96]. Recently, data from the WELCOME trial revealed that in patients with NAFLD the PNPLA3 148M/M genotype was associated with higher liver fat percentage and lower DHA tissue enrichment and reduced amelioration of aminotransferases after DHA+EPA supplementation for 15–18 months [98]. Consistently, in a randomized trial of short-term DHA supplementation in children with NAFLD, carriage of the 148M risk variant reduced the beneficial effect on liver enzymes and circulating lipids [99]. These results suggest that PNPLA3 I148M is involved in omega-3 fatty acids mobilization in the liver, and subjects with PNPLA3 148M/M genotype have lower levels of DHA [96]. Additionally, omega-3 fatty acids decrease the expression of SREBP1c [100], and carriers of the PNPLA3 148M allele tend to have decreased DNL as a feed-back mechanism to avoid excessive lipid accumulation [101]. Thus, it is possible that the lack of response to omega-3 fatty acids supplementation in decreasing liver fat percentage in subjects with PNPLA3 I148M/M is explained by the fact that these subjects already have low levels of DNL.
GCKR
Initial evidence has been gathered on the interaction between GCKR variation and diet in the pathogenesis of dyslipidemia, although no data are yet available specifically for NAFLD. GCKR variation determines higher fasting TG levels and higher absolute plasma postprandial TG and VLDL-cholesterol levels following a fat-meal challenge [102,103]. Supporting the presence of gene–diet interaction, in metabolic syndrome individuals participating to the LIPGENE study it has been reported a statistically significant association between GCKR rs1260326 polymorphism and plasma omega-3 fatty acids modulating insulin resistance and inflammatory markers. Moreover, an interaction has been demonstrated between the P446L GCKR variant and Mediterranean diet on circulating TG concentrations. Indeed, adherence to this dietary pattern blunted the negative effect of the risk 446L on circulating TG, likely reflecting the activation of DNL [104]. Consistent with these latter findings, the influence of GCKR genotype on serum cholesterol was reported to depend on the MUFA: SFA ratio in the diet [105].
8. Future Perspectives
The field is clearly only in its early stages, and in order to develop a full nutrigenomic approach to the management of NAFLD, key information is still missing.
In particular, large well-characterized cohorts with contemporary determination of (a) nutritional (b) genomics (and possibly epigenetics, transcriptomics, and proteomics) (c) microbiota composition (d) hepatic fat and liver damage assessment will be needed together with long-term follow-up of patients. The integration of this information could be used to define biomarkers and to develop tools to calculate a NAFLD risk score by changing our ability to control or cure the disease.
To apply these findings to the treatment of patients with NAFLD, randomized controlled trials aimed at the study of the interaction between diet and genetic/epigenetic factors on health-related outcomes will have to be conducted, in order to optimize the individual’s response to the intervention.
9. Conclusions
NAFLD is becoming a serious global health problem, representing the leading cause of liver disease. To date there are no approved drugs for the treatment of NAFLD, and the main clinical recommendation as an initial step is lifestyle modification, that is reduced caloric intake and physical exercise. Supporting a key role of gene–diet interaction in NAFLD development, it is important to understand how dietary composition, which influences specific molecular pathways and modifies gene and protein expression, could modulate the clinical outcome. Personalized dietary intervention based on the reduction of carbohydrates and sugar intake in genetically predisposed individuals with the PNPLA3 at-risk genotype may lead to more effective clinical outcomes for NAFLD patients. Since an individual’s genetic makeup influences how nutrients are assimilated, stored and excreted, personalized nutrition which takes into account the genetic features of patients could have immediate potential for clinical translation, representing an individualized therapeutic approach to the disease (Figure 2).
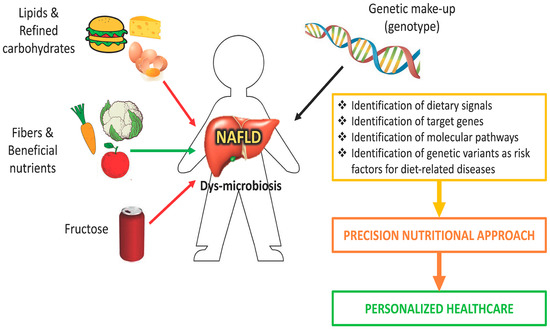
Figure 2.
A nutrigenomic approach for nonalcoholic fatty liver disease (NAFLD). Dietary intervention based on knowledge of nutritional requirement, nutritional status, and genetic makeup (personalized medicine) can be used in the management of NAFLD patients.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| TG | Triglycerides |
| DNL | De novo lipogenesis |
| VLDL | Very low-density lipoproteins |
| FFAs | Free fatty acids |
| PNPLA3 | Patatin-like phospholipase domain-containing 3 |
| SREBP | Sterol regulatory element-binding protein |
| LXR | Liver X receptor |
| RXR | Retinoid X receptor |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| GCKR | Glucokinase regulator |
| MBOAT7 | Membrane bound O-acyltransferase domain-containing 7 |
| PUFAs | Polyunsaturated fatty acids |
| SFAs | Saturated fatty acids |
| MUFAs | Monounsaturated fatty acids |
| PGC-1 | PPAR-γ coactivator-1beta |
| SCD1 | Stearoyl-CoA desaturase-1 |
| FAS | Fatty acid synthase |
| DGAT | Diacylglycerol acyltransferase DGAT |
| PPARs | Peroxisome proliferator-activated receptors |
| ChREBP | Carbohydrate-Responsive Element-Binding Protein |
| JNK | c-Jun N-terminal kinases |
| IRS-1 | Insulin receptor substrate 1 |
| Fe | Iron |
| Cu | Copper |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1a |
| PCK1 | Pyruvate carboxy kinase 1 gene |
| MTTP | Microsomal triglyceride transfer protein |
| SIRT1 | Sirtuin 1 |
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Rametta, R.; Meroni, M.; Valenti, L. The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development—a potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Blachier, M.; Leleu, H.; Peck-Radosavljevic, M.; Valla, D.C.; Roudot-Thoraval, F. The burden of liver disease in Europe: A review of available epidemiological data. J. Hepatol. 2012, 58, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Anstee, Q.M.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, J. Personalised nutrition: How far has nutrigenomics progressed? Eur. J. Clin. Nutr. 2013, 67, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Ludovica Fracanzani, A.; Fargion, S. The immunopathogenesis of alcoholic and nonalcoholic steatohepatitis: Two triggers for one disease? Semin. Immunopathol. 2009, 31, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 Variant Associated with Fatty Liver Disease (I148M) Accumulates on Lipid Droplets by Evading Ubiquitylation. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA. 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [PubMed]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.C.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Stender, S.; Kozlitina, J.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Hobbs, H.H.; Cohen, J.C. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [PubMed]
- Sparso, T.; Andersen, G.; Nielsen, T.; Burgdorf, K.S.; Gjesing, A.P.; Nielsen, A.L.; Albrechtsen, A.; Rasmussen, S.S.; Jorgensen, T.; Borch-Johnsen, K.; et al. The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT-related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia 2008, 51, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Miele, L.; Bugianesi, E.; Camma, C.; Rosso, C.; Boccia, S.; Cabibi, D.; Di Marco, V.; Grimaudo, S.; Grieco, A.; et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e87523. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Zain, S.M.; Mohamed, Z.; Mohamed, R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: A meta-analysis. J. Gastroenterol. Hepatol. 2015, 30, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Vaxillaire, M.; Veslot, J.; Dina, C.; Proenca, C.; Cauchi, S.; Charpentier, G.; Tichet, J.; Fumeron, F.; Marre, M.; Meyre, D.; et al. Impact of common type 2 diabetes risk polymorphisms in the DESIR prospective study. Diabetes 2008, 57, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Koster, B.; Fenger, M.; Poulsen, P.; Vaag, A.; Bentzen, J. Novel polymorphisms in the GCKR gene and their influence on glucose and insulin levels in a Danish twin population. Diabet Med. 2005, 22, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. Genetics of nonalcoholic fatty liver disease. Metabolism 2016, 65, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr. Opin. Lipidol. 2002, 13, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Lottenberg, A.M.; Afonso Mda, S.; Lavrador, M.S.; Machado, R.M.; Nakandakare, E.R. The role of dietary fatty acids in the pathology of metabolic syndrome. J. Nutr. Biochem. 2012, 23, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; German, J.B.; Sanyal, A.J. Comparative review of diets for the metabolic syndrome: Implications for nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2007, 86, 285–300. [Google Scholar] [PubMed]
- Masterton, G.S.; Plevris, J.N.; Hayes, P.C. Review article: Omega-3 fatty acids - a promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 31, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Plutzky, J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Maersk, M.; Belza, A.; Stodkilde-Jorgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2012, 95, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2016, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Pagliassotti, M.J. Fructose selectively modulates c-Jun N-terminal kinase activity and insulin signaling in rat primary hepatocytes. J. Nutr. 2005, 135, 1642–1646. [Google Scholar] [PubMed]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Konigsrainer, A.; Maier, K.P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [PubMed]
- Arciero, P.J.; Gentile, C.L.; Pressman, R.; Everett, M.; Ormsbee, M.J.; Martin, J.; Santamore, J.; Gorman, L.; Fehling, P.C.; Vukovich, M.D.; et al. Moderate protein intake improves total and regional body composition and insulin sensitivity in overweight adults. Metabolism 2008, 57, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Mariotti, F.; Azzout-Marniche, D.; Bos, C.; Mathe, V.; Tome, D.; Huneau, J.F. The reduced energy intake of rats fed a high-protein low-carbohydrate diet explains the lower fat deposition, but macronutrient substitution accounts for the improved glycemic control. J. Nutr. 2006, 136, 1849–1854. [Google Scholar] [PubMed]
- Leclercq, I.A.; Horsmans, Y. Nonalcoholic fatty liver disease: The potential role of nutritional management. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Chalvon-Demersay, T.; Even, P.C.; Tome, D.; Chaumontet, C.; Piedcoq, J.; Gaudichon, C.; Azzout-Marniche, D. Low-protein diet induces, whereas high-protein diet reduces hepatic FGF21 production in mice, but glucose and not amino acids up-regulate FGF21 in cultured hepatocytes. J. Nutr. Biochem. 2016, 36, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: Evidence from a case-control study. Am. J. Gastroenterol. 2007, 102, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Ludovica Fracanzani, A.; Gatti, S.; Cairo, G.; Fargion, S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Ruscica, M.; Rametta, R.; Recalcati, S.; Steffani, L.; Gatti, S.; Girelli, D.; Cairo, G.; Magni, P.; Fargion, S.; et al. Dietary iron overload induces visceral adipose tissue insulin resistance. Am. J. Pathol. 2013, 182, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Lanti, C.; Gatti, S.; Rametta, R.; Recalcati, S.; Maggioni, M.; Fracanzani, A.L.; Riso, P.; Cairo, G.; Fargion, S.; et al. High fat diet subverts hepatocellular iron uptake determining dysmetabolic iron overload. PLoS ONE 2015, 10, e0116855. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Chen, T.; Pierce, W.M., Jr.; Wang, R.; Johnson, W.T.; McClain, C.J. High fructose feeding induces copper deficiency in Sprague-Dawley rats: A novel mechanism for obesity related fatty liver. J. Hepatol. 2012, 56, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Haufe, H.; Seifert, M.; Hohla, F.; Scharinger, L.; Stickel, F.; Mourlane, F.; Weiss, G.; Datz, C. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology 2008, 135, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Strasser, M.; Haufe, H.; Sonnweber, T.; Hohla, F.; Stadlmayr, A.; Solioz, M.; Tilg, H.; Patsch, W.; Weiss, G.; et al. A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2010, 105, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Holbrook, J.; Scholfield, D.; Smith, J.C., Jr.; Reiser, S. Effect of fructose or starch on copper-67 absorption and excretion by the rat. J. Nutr. 1986, 116, 625–632. [Google Scholar] [PubMed]
- Tallino, S.; Duffy, M.; Ralle, M.; Cortes, M.P.; Latorre, M.; Burkhead, J.L. Nutrigenomics analysis reveals that copper deficiency and dietary sucrose up-regulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2015, 26, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, V.; Lammert, F. Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Borghesan, M.; Greco, A.; Pazienza, V.; Mazzoccoli, G.; Vinciguerra, M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr. Pharm. Des. 2013, 19, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueno, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castano, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Strakovsky, R.S.; Zhang, X.; Zhou, D.; Pan, Y.X. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J. Physiol. 2011, 589, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011, 6, e21662. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Pooya, S.; Blaise, S.; Moreno Garcia, M.; Giudicelli, J.; Alberto, J.M.; Gueant-Rodriguez, R.M.; Jeannesson, E.; Gueguen, N.; Bressenot, A.; Nicolas, B.; et al. Methyl donor deficiency impairs fatty acid oxidation through PGC-1α hypomethylation and decreased ER-α, ERR-α, and HNF-4α in the rat liver. J. Hepatol. 2012, 57, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. 2009, 34, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet. 2009, 42, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; Le, K.A.; Walker, R.W.; Vikman, S.; Spruijt-Metz, D.; Weigensberg, M.J.; Allayee, H.; Goran, M.I. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am. J. Clin. Nutr. 2010, 92, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Liccardo, D.; Bedogni, G.; Salvatori, G.; Gnani, D.; Bersani, I.; Alisi, A.; Valenti, L.; Raponi, M. Influence of dietary pattern, physical activity, and I148M PNPLA3 on steatosis severity in at-risk adolescents. Genes Nutr. 2014, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Sevastianova, K.; Kotronen, A.; Gastaldelli, A.; Perttila, J.; Hakkarainen, A.; Lundbom, J.; Suojanen, L.; Orho-Melander, M.; Lundbom, N.; Ferrannini, E.; et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am. J. Clin. Nutr. 2011, 94, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Savoye, M.; Kim, G.; Marotto, K.; Shaw, M.M.; Pierpont, B.; Caprio, S. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS ONE 2012, 7, e37827. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; West, A.L.; Bhatia, L.; Hoile, S.P.; McCormick, K.G.; Burdge, G.C.; Lillycrop, K.A.; Clough, G.F.; Calder, P.C.; Byrne, C.D. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: Results from the WELCOME trial. J. Hepatol. 2015, 63, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Bedogni, G.; Donati, B.; Alisi, A.; Valenti, L. The I148M variant of PNPLA3 reduces the response to docosahexaenoic acid in children with non-alcoholic fatty liver disease. J. Med. Food 2013, 16, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, C.; Lohe, K.; Kuang, C.; Xiao, Y.; Jouni, Z.; Poels, E. Modulation of adipocyte differentiation by omega-3 polyunsaturated fatty acids involves the ubiquitin-proteasome system. J. Cell. Mol. Med. 2014, 18, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Matikainen, N.; Maglio, C.; Soderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Pollin, T.I.; Damcott, C.M.; McLenithan, J.C.; Mitchell, B.D.; Shuldiner, A.R. Glucokinase regulatory protein gene polymorphism affects postprandial lipemic response in a dietary intervention study. Hum. Genet. 2009, 126, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Corella, D.; Shen, J.; Arnett, D.K.; Yiannakouris, N.; Tai, E.S.; Orho-Melander, M.; Tucker, K.L.; Tsai, M.; Straka, R.J.; et al. Association between glucokinase regulatory protein (GCKR) and apolipoprotein A5 (APOA5) gene polymorphisms and triacylglycerol concentrations in fasting, postprandial, and fenofibrate-treated states. Am. J. Clin. Nutr. 2009, 89, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Sotos-Prieto, M.; Guillen, M.; Vicente Sorli, J.; Portoles, O.; Guillem-Saiz, P.; Ignacio Gonzalez, J.; Qi, L.; Corella, D. Relevant associations of the glucokinase regulatory protein/glucokinase gene variation with TAG concentrations in a high-cardiovascular risk population: modulation by the Mediterranean diet. Br. J. Nutr. 2013, 109, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jang, H.B.; Kim, H.J.; Ahn, Y.; Hong, K.W.; Cho, S.B.; Kang, J.H.; Park, S.I. The dietary monounsaturated to saturated fatty acid ratio modulates the genetic effects of GCKR on serum lipid levels in children. Clin. Chim. Acta 2015, 450, 155–161. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).