Efficient Production of the Flavoring Agent Zingerone and of both (R)- and (S)-Zingerols via Green Fungal Biocatalysis. Comparative Antifungal Activities between Enantiomers

Abstract
:
1. Introduction
2. Results
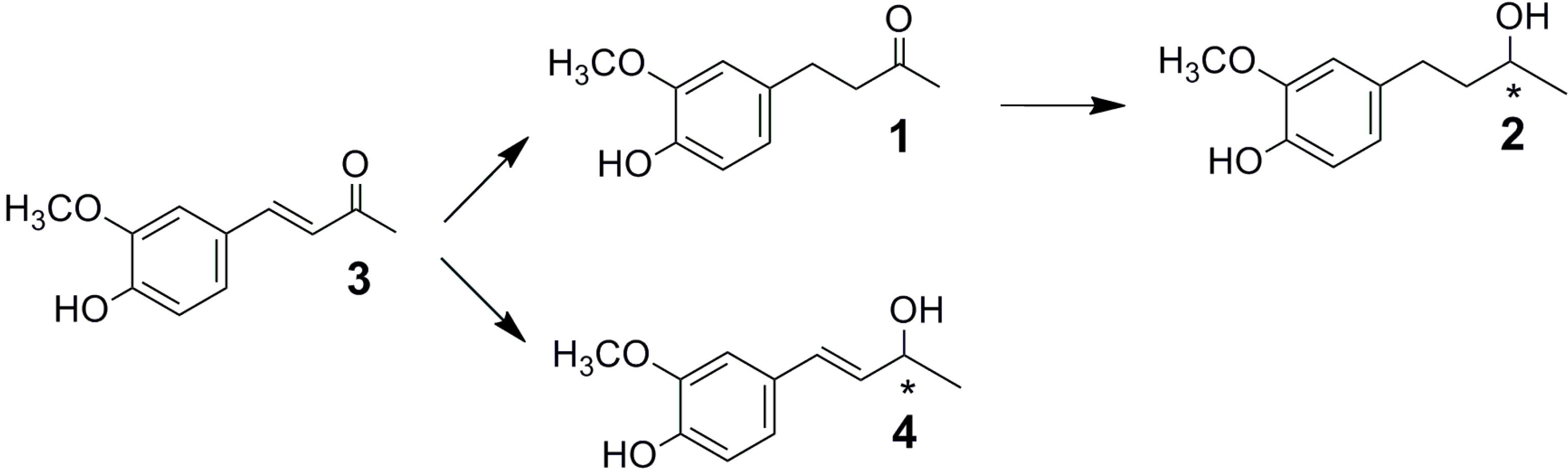
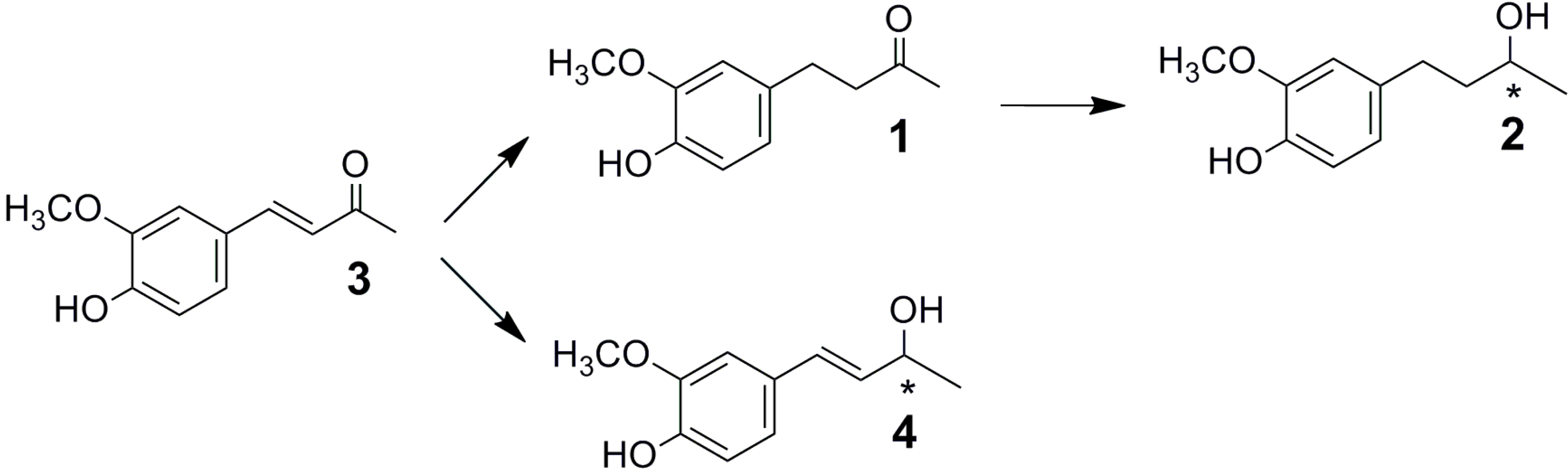
2.1. Fungal Biotransformation of 3
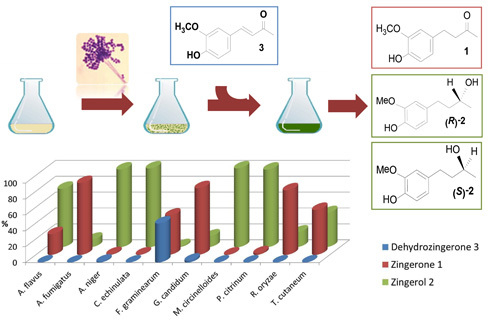
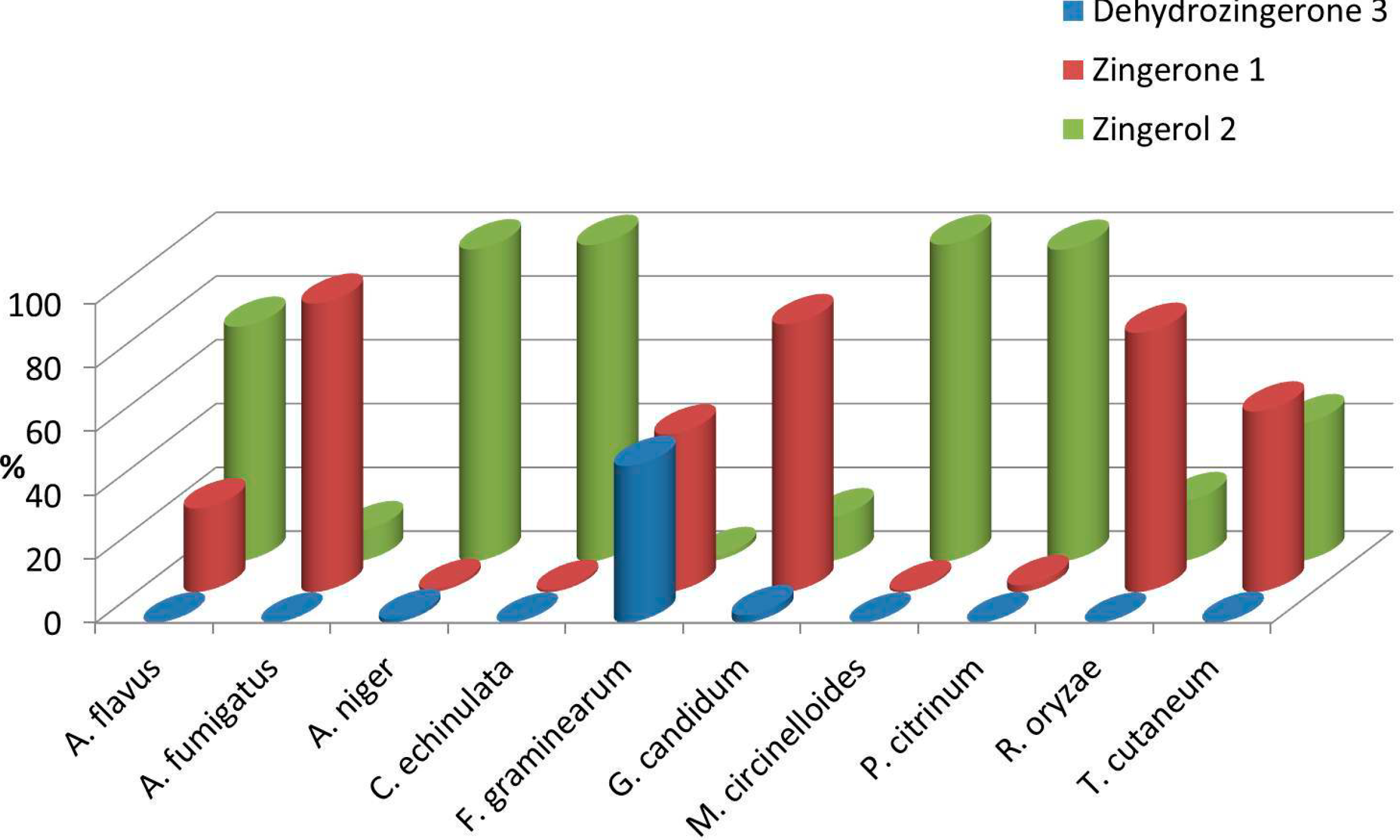
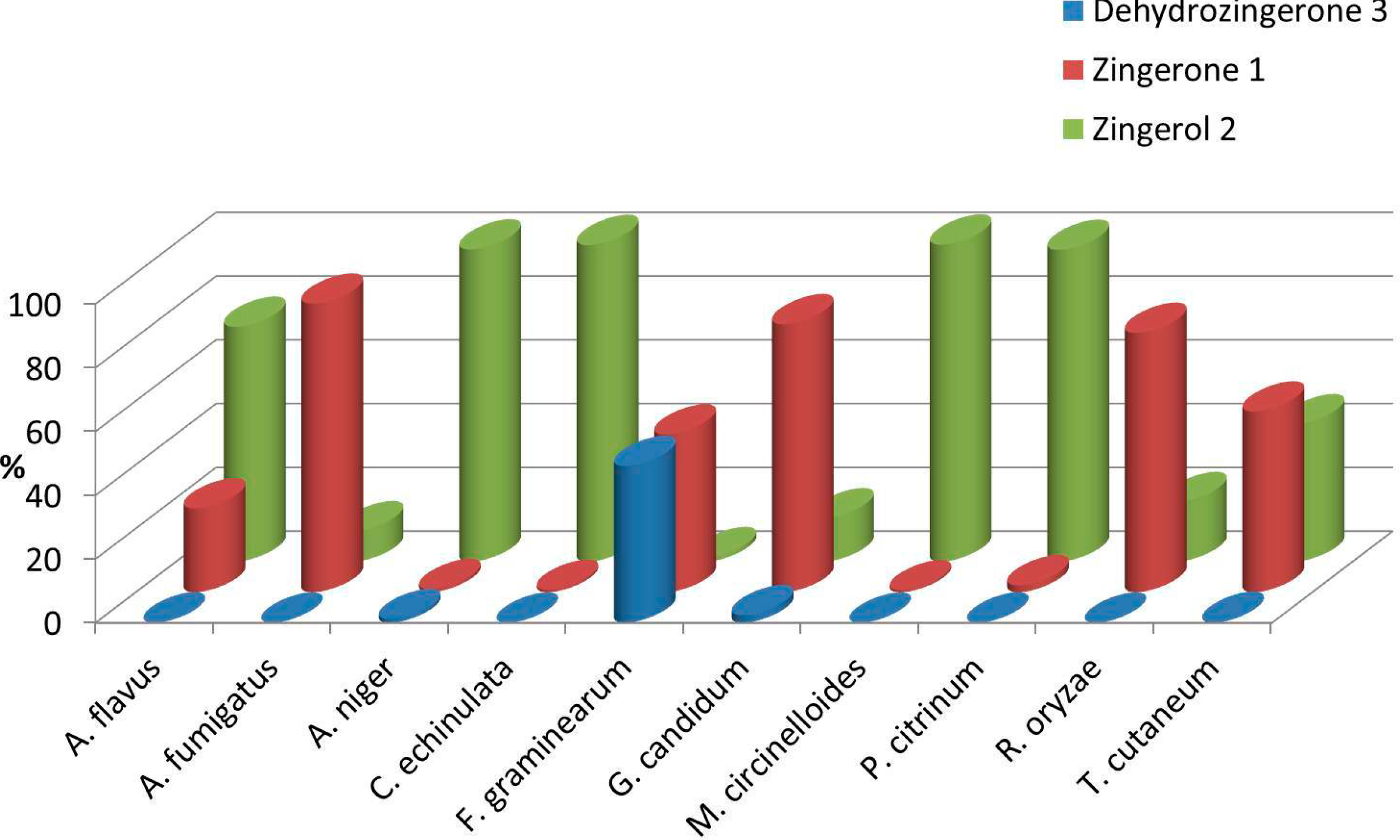
2.2. Biotransformation of 3 with the Whole Fungal Panel

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entries | Fungal spp. | Conversion Rate (%) | 1 (%) | 2 (%) | Absolute Configuration | (0.75, CHCl3) | ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | A. flavus | >99 | 26 | 74 | S | +3.1 ± 0.2 | 20 |
| 2 | A. fumigatus | >99 | 90 | 10 | S | +2.3 ± 0.4 | 14 |
| 3 | A. niger | 99 | 1 | 98 | R | −15.5 ± 0.7 | 94 |
| 4 | C. echinulata | >99 | 1 | 99 | S | +11.3 ± 0.7 | 70 |
| 5 | F. graminearum | 51 | 50 | 1 | R | −11.8 ± 0.4 | 72 |
| 6 | G. candidum | 98 | 84 | 14 | R | −16.1 ± 0.5 | 98 |
| 7 | M. circinelloides | >99 | 1 | 99 | R | −10.4 ± 0.7 | 64 |
| 8 | P. citrinum. | >99 | 2 | 98 | S | +11.7 ± 0.8 | 94 |
| 9 | R. oryzae | >99 | 81 | 19 | S | +14.2 ± 0.4 | 88 |
| 10 | T. cutaneum | >99 | 57 | 43 | R | −4.6 ± 0.3 | 28 |
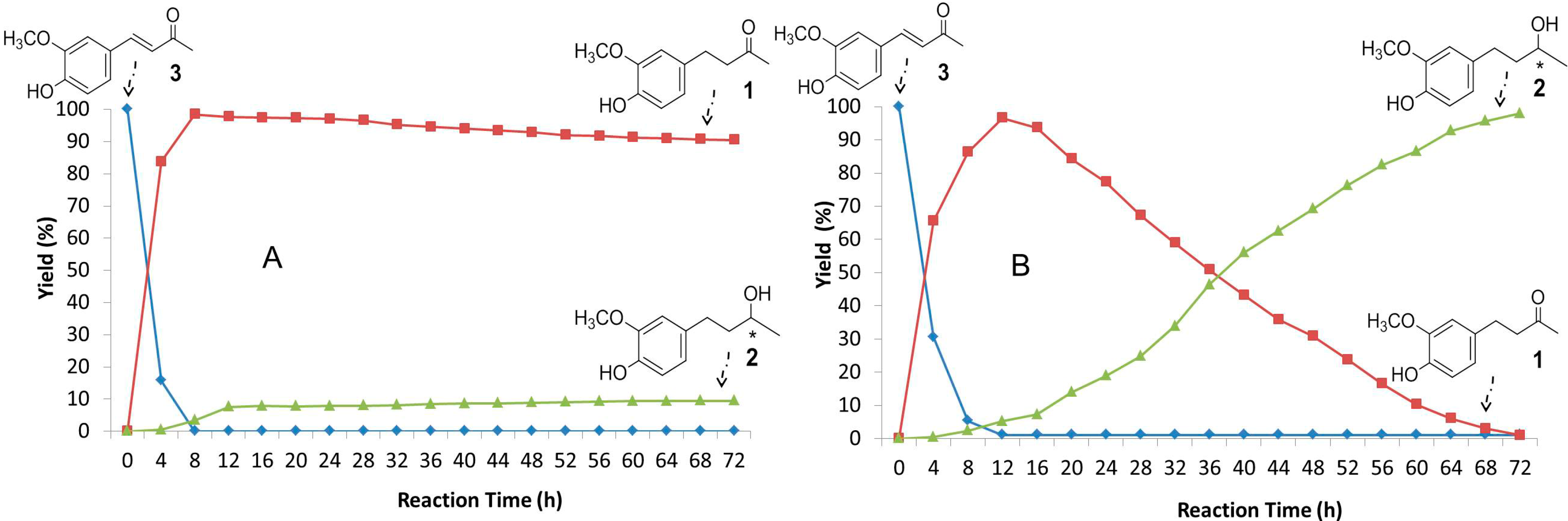
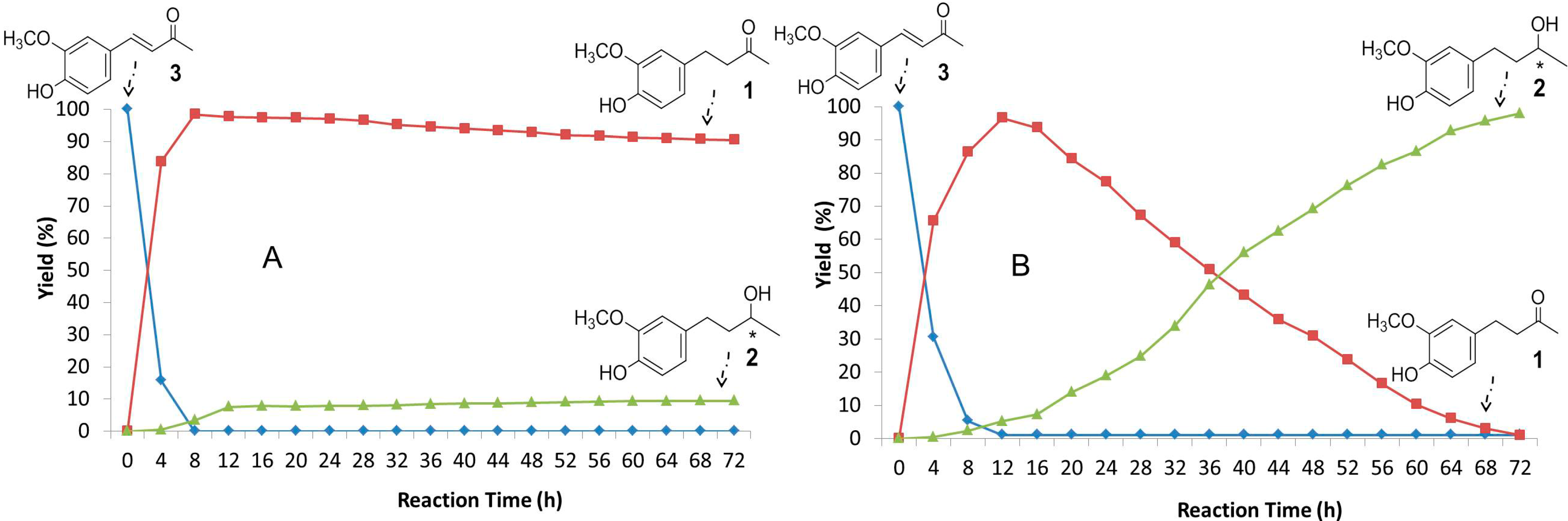
2.3. Time-Course of the Biotransformation of 3 with Selected Fungi
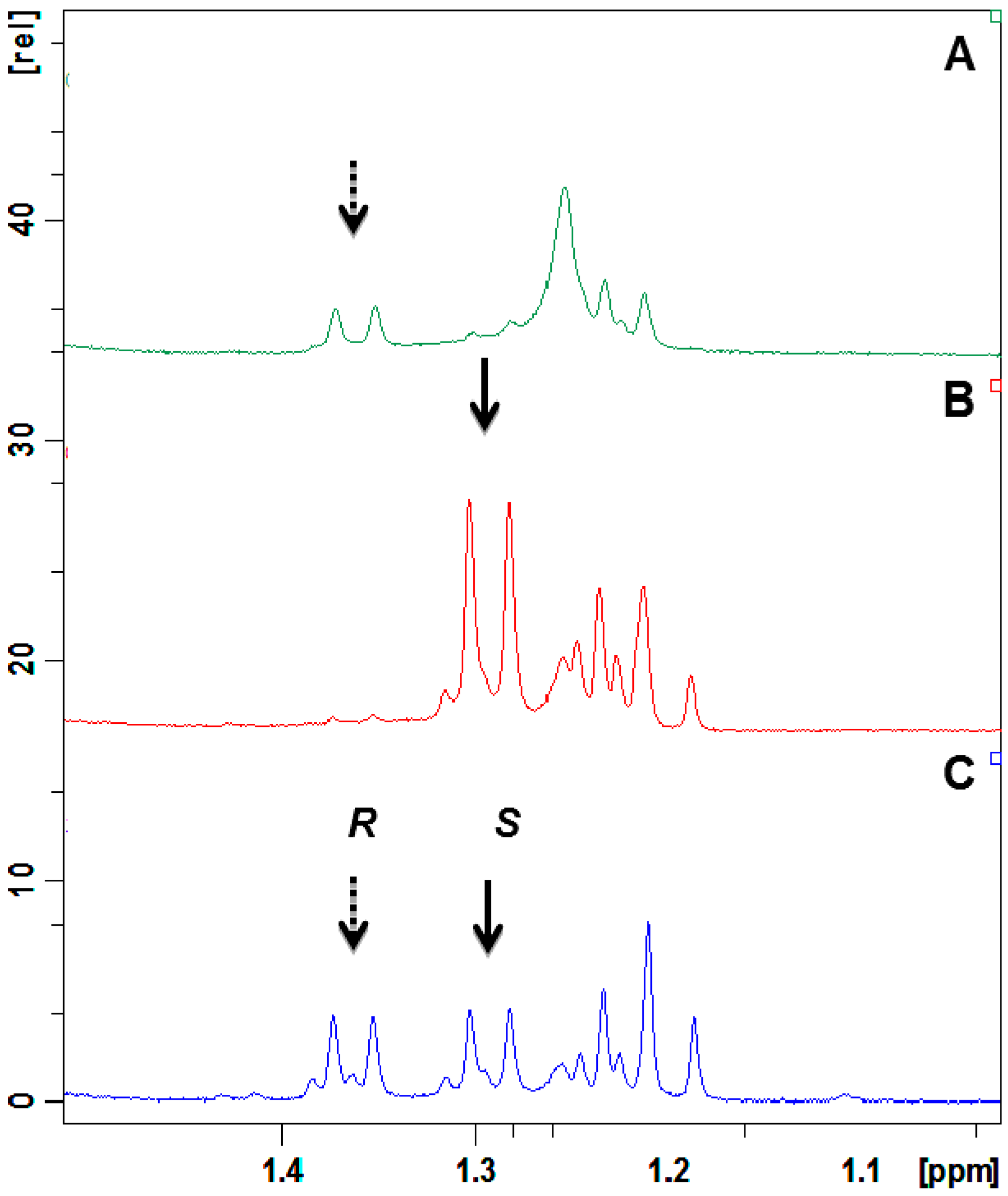
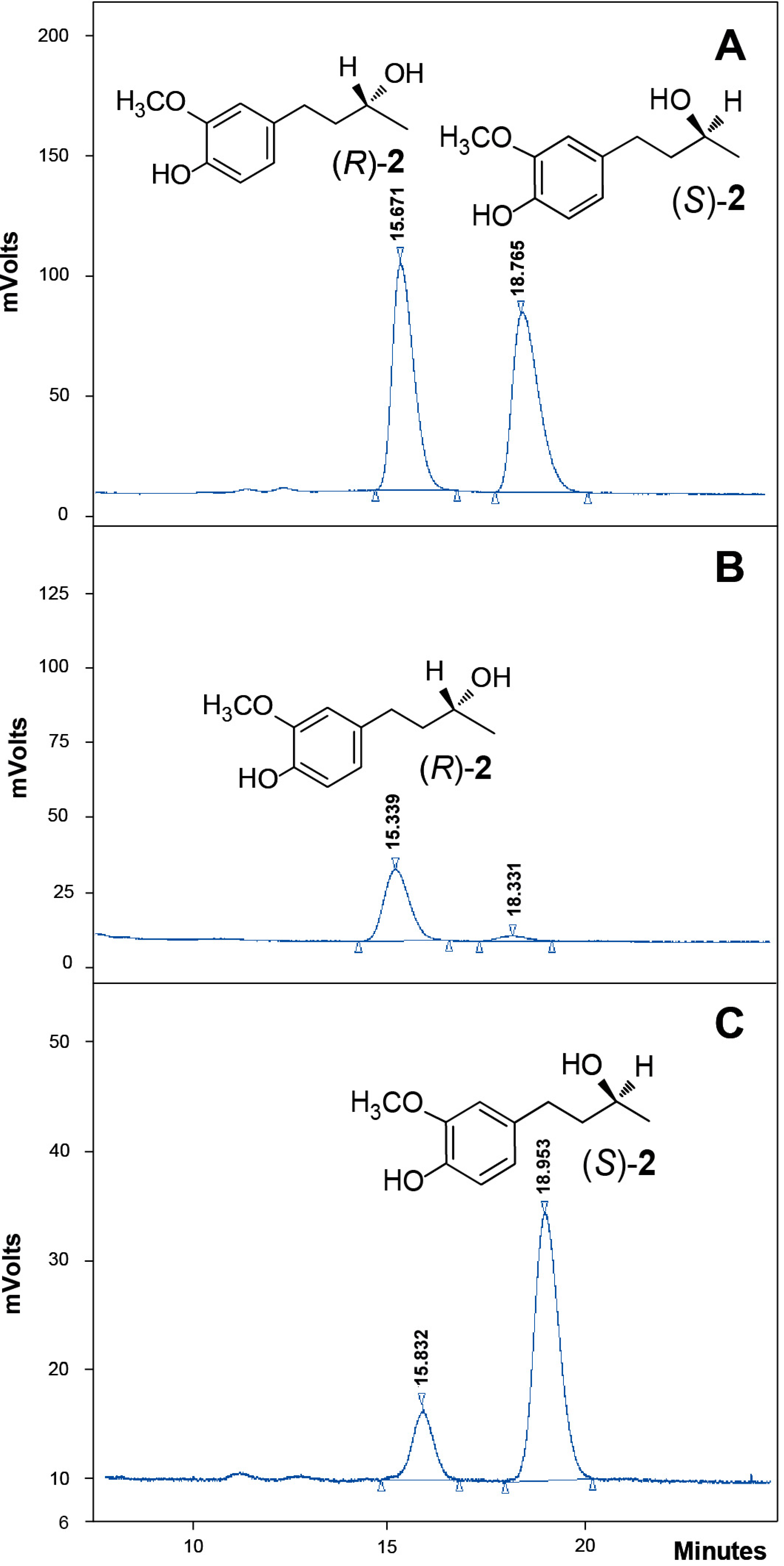
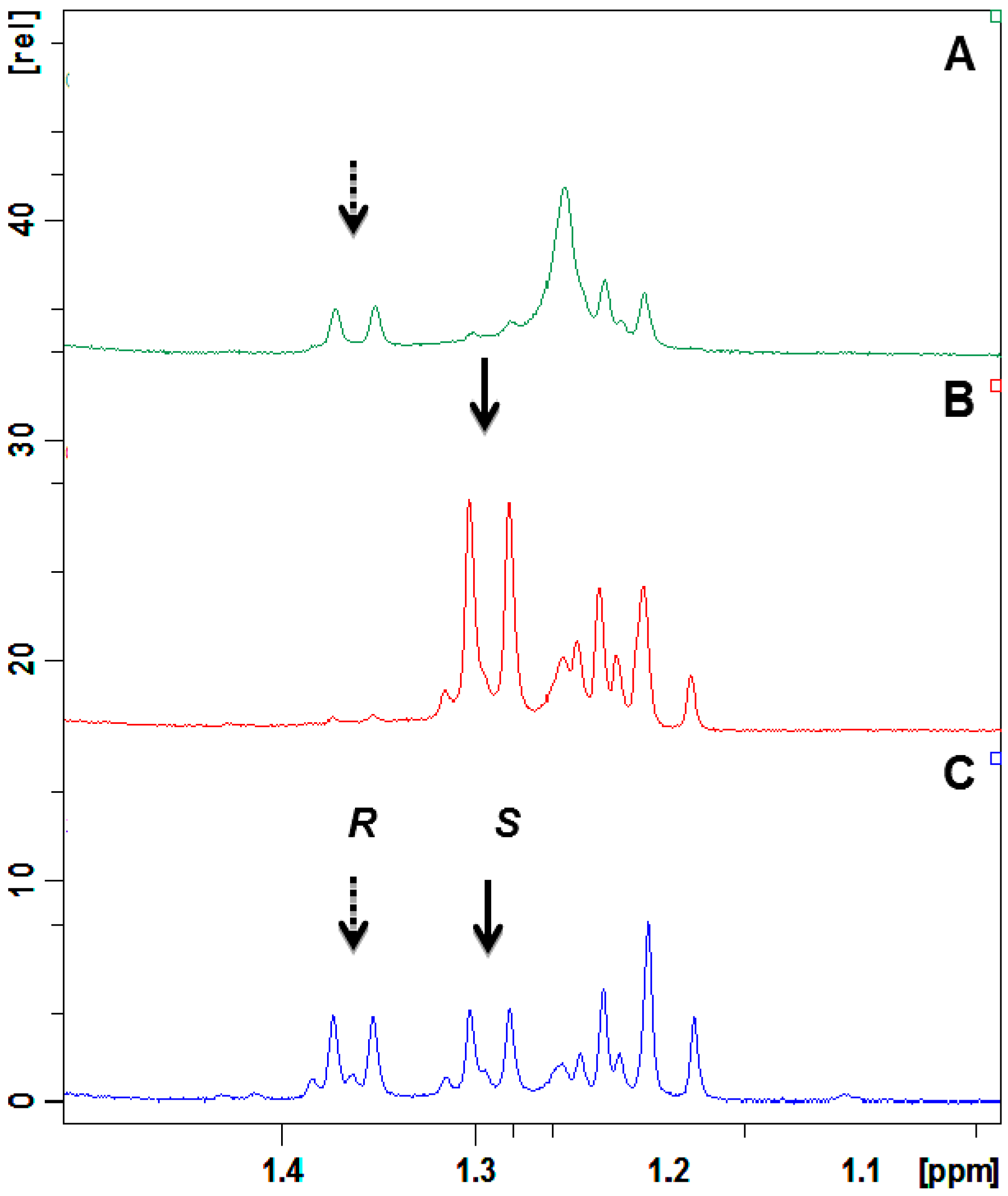
2.4. Absolute Configuration of the Obtained Alcohols 2
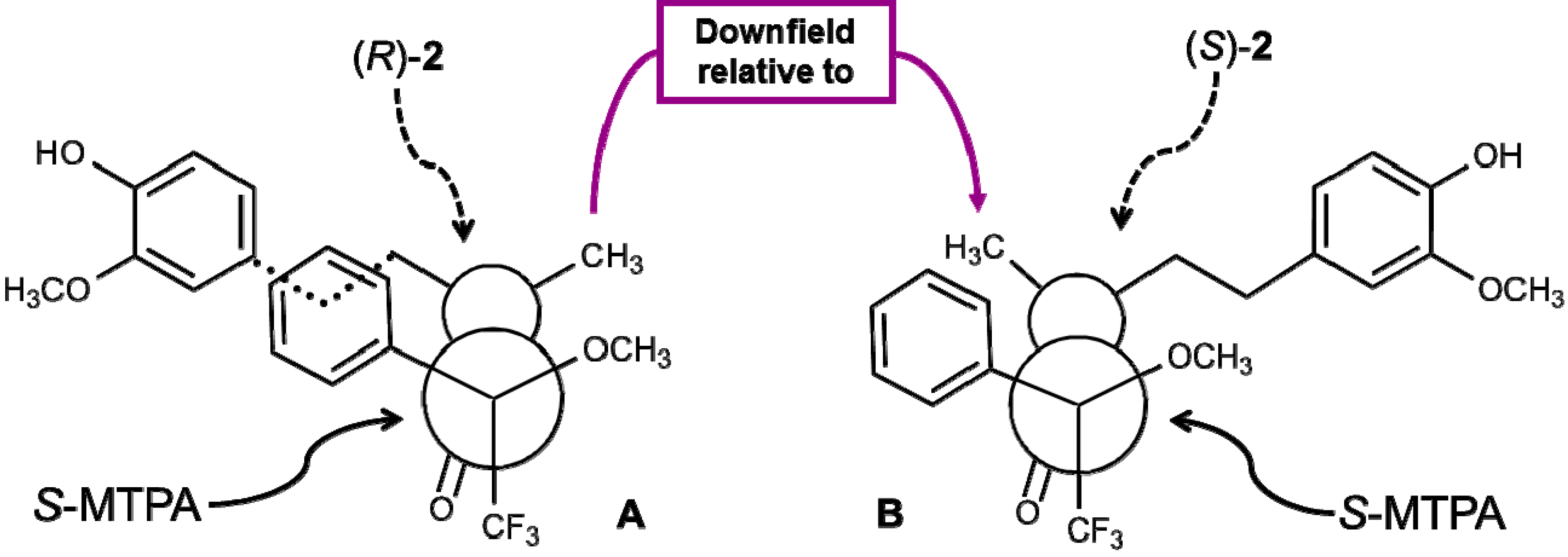
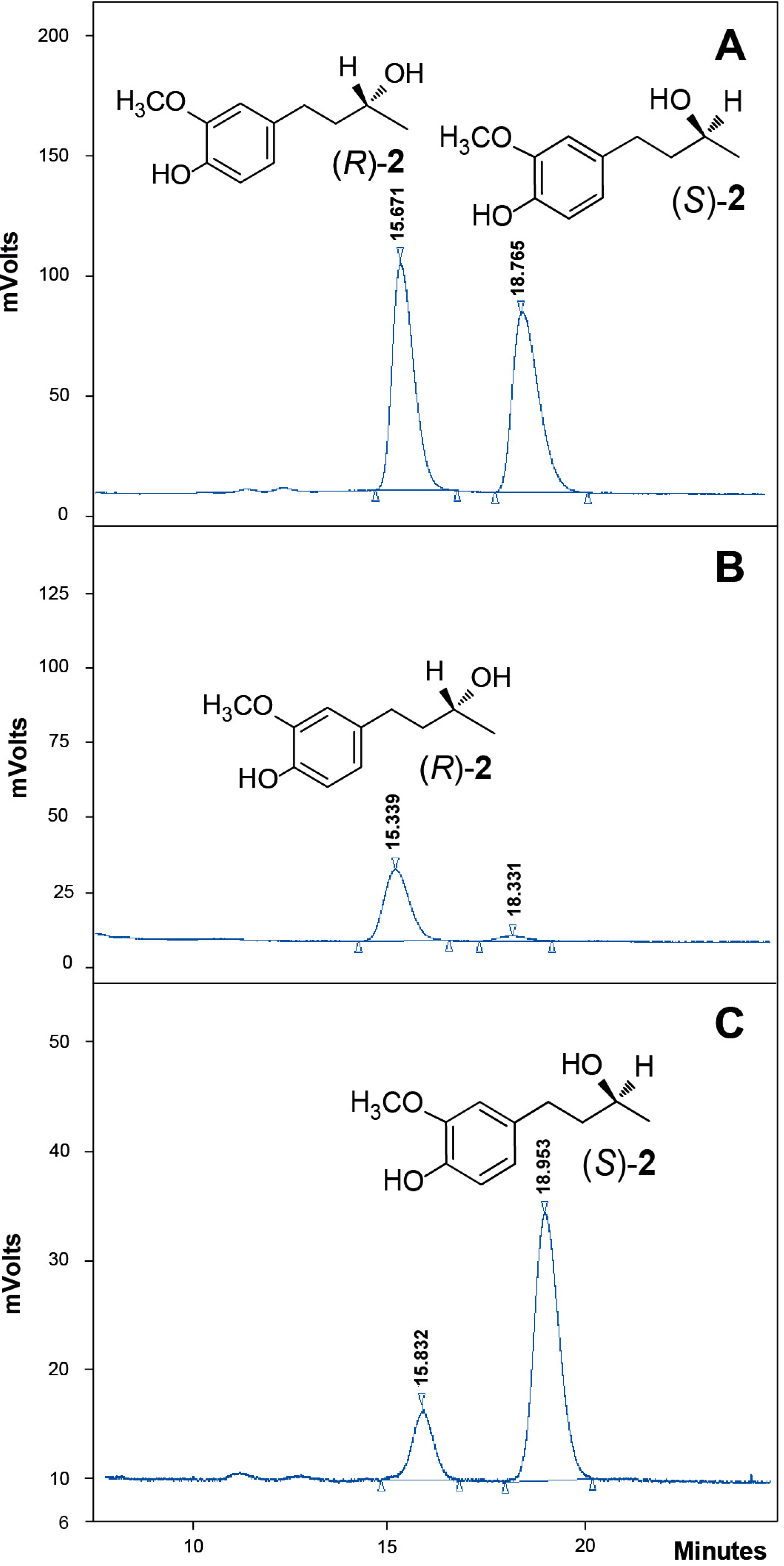
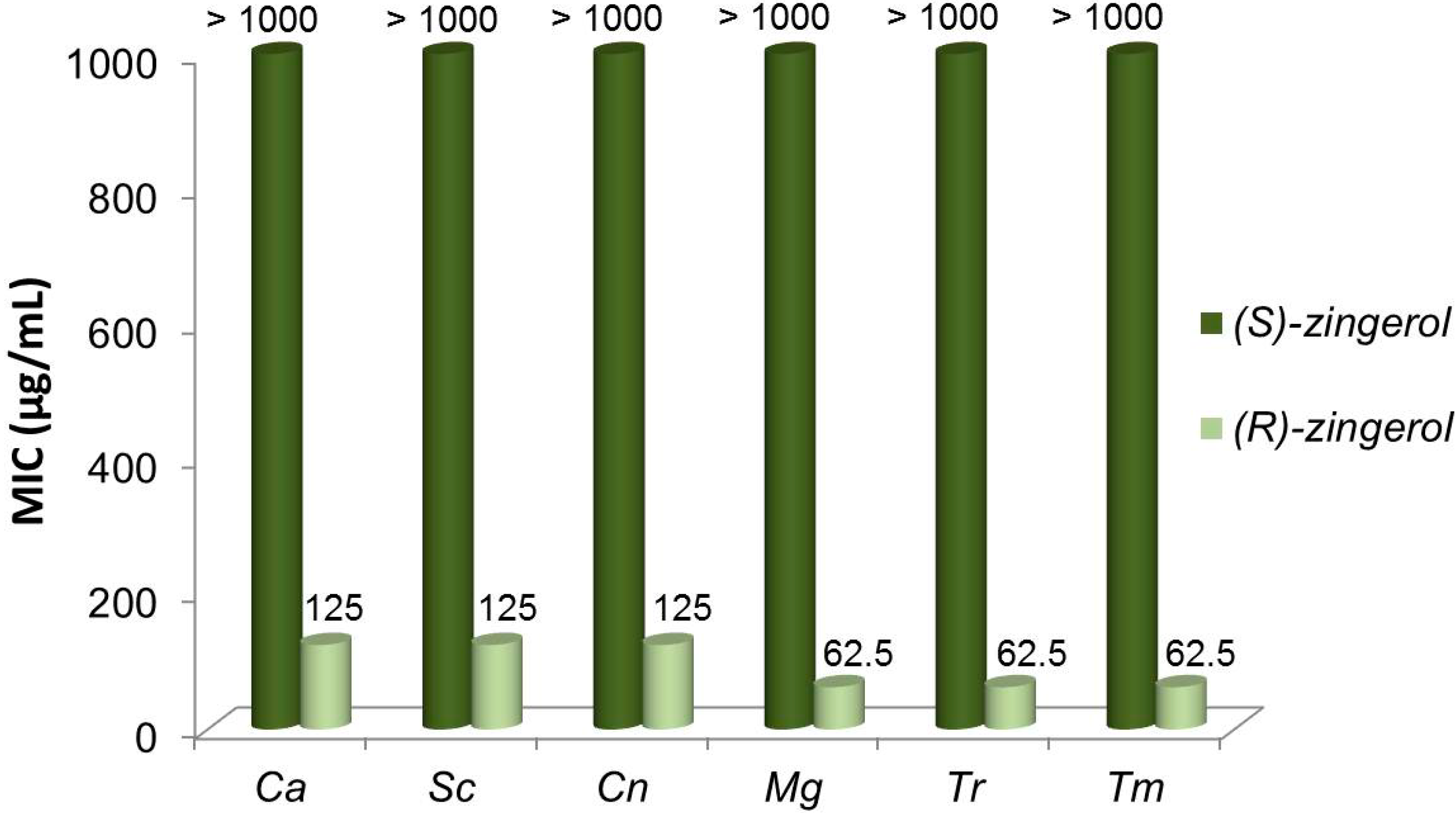
2.5. Antifungal Activity of (S)-, (R)- and rac-2
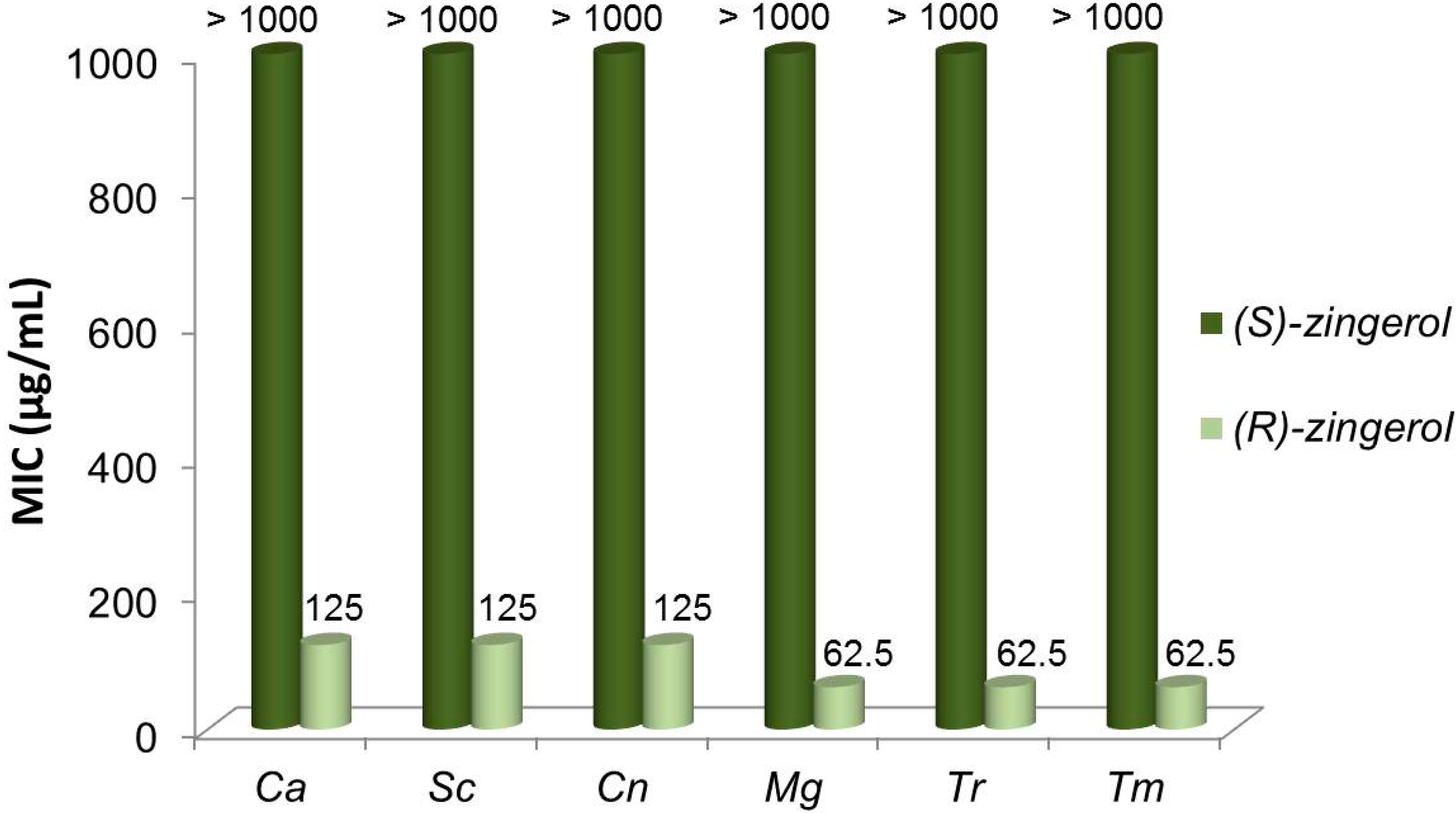
3. Discussion
4. Experimental Section
4.1. General Considerations
4.2. Preparation of 3
4.3. Fungal Strains Used for Biotransformation
4.4. Biotransformation Methodology
4.5. Analysis of the Product Mixtures with GC-MS
4.6. Preparation of MTPA Esters of (R)-, (S)- and rac-2
4.7. Synthesis of Ketone 1 for Comparative Purposes
4.8. Synthesis of rac-(±)-2 for Comparative Purposes
4.9. Synthesis of Allylic Alcohol 4 for Comparative Purposes
4.10. Chiral-HPLC Analyses
4.11. Antifungal Evaluation
4.11.1. Microorganisms and Media
4.11.2. Antifungal Susceptibility Testing
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krings, U.; Berger, R. Biotechnological production of flavours and fragrances. Appl. Microbiol. Biotechnol. 1998, 49, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Böker, A.; Fischer, M.; Berger, R. Raspberry ketone from submerged cultures cells of the basidiomycete Nidula tomentosa. Biotechnol. Prog. 2001, 17, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.A. Flavours: the legal framework. In Flavours and Fragrances; Günter Berger, R., Ed.; Springer Berlin Heidelberg: Heidelberg, Germanly, 2007; pp. 15–24. [Google Scholar]
- Schrader, J.; Etschmann, M.; Sell, D.; Hilmer, J.; Rabenhorst, J. Applied biocatalysis for the synthesis of natural flavour compounds-current industrial processes and future prospects. Biotechnol. Lett. 2004, 26, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.N.; Archana, P.R.; Aithal, B.K.; Rao, B.S.S. Protective effect of zingerone, a dietary compound against radiation induced genetic damage and apoptosis in human lymphocites. Eur. J. Pharmacol. 2011, 657, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Harjai, K.; Chhibber, S. Recent update on multiple pharmacological benefits of zingerone: a quick review. Am. J. Phytomed. Clin. Ther. 2014, 2, 693–704. [Google Scholar]
- Rajan, I.; Narayanan, N.; Rabindran, R.; Jayasree, P.; Manish Kumar, P. Zingerone protects against stannous chloride-induced and hydrogen peroxide-induced oxidative DNA damage in vitro. Biol. Trace Elem. Res. 2013, 155, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Aeschbach, R.; Löliger, J.; Scott, B.; Murcia, A.; Butler, J.; Halliwell, B.; Aruoma, O. Antioxidant actions of thymol, carvacrol, 6-gingerol, zingerone and hydroxytyrosol. Food Chem. Toxicol. 1994, 32, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo Cañas, P.; García Romero, E.; Gómez Alonso, S.; Palop Herreros, M. Changes in the composition of Tempranillo wines during spontaneous malolactic fermentation. J. Food Compos. Anal. 2008, 21, 724–730. [Google Scholar]
- Fisher, C.; Scott, T. Flavour compounds. In Food Flavours, Biology and Chemistry; The Royal Society of Chemistry: Cambridge, UK, 1997; pp. 15–55. [Google Scholar]
- Chang, Y.P.; Liu, C.H.; Wu, C.C.; Chiang, C.M.; Lian, J.L.; Hsieh, S.L. Dietary administration of zingerone to enhance growth, non-specific immune response, and resistance to Vibrio alginolyticus in Pacific white shrimp (Litopenaus vannamei) juveniles. Fish Shellfish Immunol. 2012, 32, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Ji, Y.; Hae, Y.; Jeong, J. Zingerone as antioxidant against peroxynitrile. J. Agric. Food Chem. 2005, 53, 7617–7622. [Google Scholar] [CrossRef] [PubMed]
- Kubra, I.; Jaganmohanrao, L. An overview on inventions related to ginger processing and products for food and pharmaceutical applications. Recent Pat. Food Nutr. Agric. 2012, 4, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Iwami, M.; Shiina, T.; Hirayama, H.; Shimizu, Y. Intraluminal administration of zingerol, a non-pungent analogue of zingerone, inhibits colonic motility in rats. Biomed. Res. 2011, 32, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Takhi, M.; Sampath Kumar, H.; Srinivas, K.; Yadav, J. Stereochemistry of 4-aryl-2-butanols from Himalayan Taxus baccata. Phytochemistry 1993, 33, 697–699. [Google Scholar] [CrossRef]
- Tan, K. H.; Nishida, R. Mutual reproductive benefits between a wild orchid, Bulbophyllum patens and Bactrocera fruit flies via a floral synomone. J. Chem. Ecol. 2000, 26, 533–546. [Google Scholar] [CrossRef]
- Lu, J.H.; Liu, Y.; Tu, G.Z.; Zhao, Y.Y. Phenolic glucosides from Oxytropis myriophylla. J. Asian Nat. Prod. Res. 2001, 4, 43–46. [Google Scholar] [CrossRef]
- Otsuka, H.; Yu, Q.; Matsunami, K. Bumaldosides A, B and C from the leaves of Staphylea bumalda. Heterocycles 2010, 80, 339–348. [Google Scholar] [CrossRef]
- Suzuki, A.; Ochiai, R.; Tokimitsi, I.U.S. Agents for preventing, improving or treating hypertension. U.S. Patent 7,351,436 B2, 1 April 2008. [Google Scholar]
- Isobe, T.; Iwase, T.; Kayane, S.; Miura, Y. Throat care agents. U.S. Patent 20,030,170,322 A1, 11 September 2003. [Google Scholar]
- Kayane, S.; Isobe, T.; Iwase, T.; Miura, Y. Throat are Agents. EP Patent 1,293,131 A1, 19 March 2003. [Google Scholar]
- Plourde, G.L. Studies towards the diastereoselective spiroannulation of phenolic derivatives. Tetrahedron Lett. 2002, 43, 3597–3599. [Google Scholar] [CrossRef]
- Banno, K.; Mukaiyama, T. A new synthesis of the pungent principles of ginger-zingerone, gingerol and shogaol. Bull. Chem. Soc. Jpn. 1976, 49, 1453–1454. [Google Scholar] [CrossRef]
- Vijendra Kumar, N.; Murthy, T.; Manjunatha, J.; Bettadaiah, B. Synthesis and quorum sensing inhibitory activity of key phenolic compounds of ginger and their derivatives. Food Chem. 2014, 159, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Prelog, V. Specification of the stereospecificity of some oxido-reductases by diamond lattice sections. Pure Appl. Chem. 1964, 9, 119–130. [Google Scholar] [CrossRef]
- Shimoda, K.; Harada, T.; Hamada, H.; Nakajima, N.; Hamada, H. Biotransformation of raspberry ketone and zingerone by cultured cells of Phytolacca americana. Phytochemistry 2007, 68, 487–492. [Google Scholar] [CrossRef]
- Kitayama, T.; Isomori, S.; Nakamura, K. Asymmetric synthesis of enantiomerically pure zingerols by lipase-catalyzed transesterification and efficient synthesis of their analogues. Tetrahedron Asymmetry 2013, 24, 621–627. [Google Scholar] [CrossRef]
- Rodrigues, J.; Moran, P.; Conceiçao, G.; Fardelone, L. Recent advances in the biocatalytic asymmetric reduction of acetophenones and α,β-unsaturated carbonyl compounds. Food Technol. Biotechnol. 2004, 42, 295–303. [Google Scholar]
- Sortino, M.; Cechinel Filho, V.; Zacchino, S. Highly enantioselective reduction of the C–C double bond of N-phenyl-2-methyl- and N-phenyl-2,3-dimethyl- maleimides by fungal strains. Tetrahedron Asymmetry 2009, 20, 1106–1108. [Google Scholar] [CrossRef]
- Kosjek, B.; Stampfer, W.; van Deursen, R.; Faber, K.; Kroutil, W. Efficient production of raspberry ketone via green biocatalytic oxidation. Tetrahedron 2003, 59, 9517–9521. [Google Scholar] [CrossRef]
- Buchbauer, G.; Jäger, W.; Gruber, A.; Dietrich, H. R-(+)- and S-(−)-carvone: Influence of chirality on locomotion activity in mice. Flavour Frag. J. 2005, 20, 686–689. [Google Scholar] [CrossRef]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.; Damu, A.; Cherng, C.; Jeng, J.; Teng, C.; Wu, T. Isolation of a natural antioxidant, dehydrozingerone from Zingiber officinale and synthesis of its analogues for recognition of effective antioxidant and antityrosinase agents. Arch. Pharm. Res. 2005, 28, 518–528. [Google Scholar] [CrossRef]
- Chartrain, M.; Sturr, M. Fungal bioconversions: Applications to the manufacture of pharmaceuticals. In Handbook of Industrial Mycology; An, Z., Ed.; Marcel Dekker: New York, NY, USA, 2005; pp. 563–590. [Google Scholar]
- Fronza, G.; Fuganti, C.; Mendozza, M.; Rallo, R.; Ottolins, G.; Joulain, D. Stereochemistry of the doube bonbd saturation in the formation in Baker’s yeast of 4-(4-hydroxyphenyl)-2-butanone (raspberry ketone). Tetrahedron 1996, 52, 4041–4052. [Google Scholar] [CrossRef]
- Morrissey, I.; Hoshino, K.; Sato, K.; Yoshida, A.; Hayakawa, I.; Bures, M.; Shen, L. Mechanism of differential activities of ofloxacin enantiomers. Antimicrob. Agents Chemother. 1996, 40, 1775–1784. [Google Scholar] [PubMed]
- CLSI, Clinical and Laboratory Standards Institute. Reference method for broth dilution antifungal susceptibility testing for filamentous fungi; Approved standard, M38A2, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008; Volume 28, No. 16; pp. 1–35. [Google Scholar]
- CLSI, Clinical and Laboratory Standards Institute. Reference methods for broth dilution antifungal susceptibility testing for yeasts; Approved standard M27A3, 3rd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008; Volume 28, No. 14; pp. 1–25. [Google Scholar]
- Andrade, L.; Keppler, A.; Schoenlein-Crusius, I.; Porto, A.; Comasseto, J. Evaluation of acetophenone monooxygenase and alcohol dehydrogenase activities in different fungal strains by biotransformation of acetophenone derivatives. J. Mol. Catal. B Enzym. 2004, 31, 129–135. [Google Scholar] [CrossRef]
- Zagozda, M.; Plenkiewicz, J. Enantioselective reduction of α,β-unsaturated ketones by Geotrichum candidum, Mortierella isabelina and Rhodotorula rubra yeast. Tetrahedron Asymmetry 2006, 17, 1958–1962. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.; Holtman, D. Enzymatic reductions for the chemist. Green Chem. 2011, 13, 2285–2313. [Google Scholar] [CrossRef]
- Weitzman, I.; Summerbell, R.C. The dermatophytes. Clin. Microbiol. Rev. 1995, 8, 240–259. [Google Scholar]
- Ramachandra, M.S.; Subbaraju, G.V. Synthesis and bioactivity of novel caffeic acid esters from Zuccagnia punctata. J. Asian Nat. Prod. Res. 2006, 8, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Elias, G.; Rao, M. Synthesis and anti-inflammatory activity of substituted (E)-4-phenyl-3-buten-2-ones. Eur. J. Med. Chem. 1988, 23, 379–380. [Google Scholar] [CrossRef]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; Vander Jagt, D.L. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.A.; Mosher, H.S. Nuclear Magnetic Resonance enantiomer reagents. Configurational correlations via Nuclear Magnetic Resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Zacchino, S.; Badano, H. Enantioselective synthesis and absolute configuration assignment of erythro-(3,4-methylenedioxy-7-hydroxy-1’-allyl-3’,5’-dimethoxy)-8.0.4’-neolignan and its acetate, isolated from nutmeg (Myristica fragrans). J. Nat. Prod. 1991, 54, 155–160. [Google Scholar] [CrossRef]
- Denniff, P.; Macloed, D.; Whiting, D. Syntheses of the (±)-[n]-gingerols (pungent principles of ginger) and related compounds through regioselective aldol condensations: relative pungency assays. J. Chem. Soc. Perkin Trans. 1981, 1, 82–87. [Google Scholar] [CrossRef]
- Saha, S.; Smith, R.; Lenz, E.; Wilson, I. Analysis of a ginger extract by high-performance liquid chromatography coupled to nuclear magnetic resonance spectroscopy using superheated deuterium oxide as the mobile phase. J. Chromatogr. 2003, 991, 143–150. [Google Scholar] [CrossRef]
- Gemal, A.; Luche, J.L. Lanthanoids in organic synthesis. 6. The reduction of α-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103, 5454–5459. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svetaz, L.A.; Di Liberto, M.G.; Zanardi, M.M.; Suárez, A.G.; Zacchino, S.A. Efficient Production of the Flavoring Agent Zingerone and of both (R)- and (S)-Zingerols via Green Fungal Biocatalysis. Comparative Antifungal Activities between Enantiomers. Int. J. Mol. Sci. 2014, 15, 22042-22058. https://doi.org/10.3390/ijms151222042
Svetaz LA, Di Liberto MG, Zanardi MM, Suárez AG, Zacchino SA. Efficient Production of the Flavoring Agent Zingerone and of both (R)- and (S)-Zingerols via Green Fungal Biocatalysis. Comparative Antifungal Activities between Enantiomers. International Journal of Molecular Sciences. 2014; 15(12):22042-22058. https://doi.org/10.3390/ijms151222042
Chicago/Turabian StyleSvetaz, Laura A., Melina G. Di Liberto, María M. Zanardi, Alejandra G. Suárez, and Susana A. Zacchino. 2014. "Efficient Production of the Flavoring Agent Zingerone and of both (R)- and (S)-Zingerols via Green Fungal Biocatalysis. Comparative Antifungal Activities between Enantiomers" International Journal of Molecular Sciences 15, no. 12: 22042-22058. https://doi.org/10.3390/ijms151222042
APA StyleSvetaz, L. A., Di Liberto, M. G., Zanardi, M. M., Suárez, A. G., & Zacchino, S. A. (2014). Efficient Production of the Flavoring Agent Zingerone and of both (R)- and (S)-Zingerols via Green Fungal Biocatalysis. Comparative Antifungal Activities between Enantiomers. International Journal of Molecular Sciences, 15(12), 22042-22058. https://doi.org/10.3390/ijms151222042