Abstract
The epoxidation of the natural product α-euphol followed by cleavage of the obtained epoxide with BF3-etherate, provides 3β-hydroxy-eupholanost-8-en-24-one in satisfactory overall yield.
Introduction
Epoxides are among the most frequently occurring functional groups found in natural products and their synthetic analogues. They have been widely used because of their chemical reactivity [1,2], which can be explained by the ring strain of the small heterocycle. Conversion of epoxides to carbonyl compounds is a synthetically useful reaction, which is commonly achieved by BF3 and its etherate [3]. The present work discloses the application of this methodology to the synthesis of the C24 derivative of α-euphol, a reagent and intermediate in sterol biosynthesis.
Results and discussion
The tetracyclic triterpenes of the euphane class are distinguished from lanostane by C13, C14 and C17 stereochemical inversion. To our knowledge, the literature contains no report of a successful synthetic route to the C24 derivatives of α-euphol, but several papers and review articles [4,5,6,7] have appeared describing in great detail the synthesis of C24 derivatives of lanosterol in poor yields. Recently, Parish and al. [8] reported their progress on a conceptually simple synthesis of 24-keto-lanosterol from commercial lanosterol using a hydroboration-oxidation reaction which requires two supplementary steps (a preliminary protection of hydroxy group at C3 and a final deprotection to afford the desired product). Also, Zdzislaw and al [9] have described a BF3-Et2O catalysed rearrangement of C9 carbocations derived from 9,11-epoxylanostanes. As an application of this reaction on the major constituent of Moroccan Euphorbium species, we have focused our attention on the side chain of α-euphol (1). We describe herein a rapid and convenient chemical synthesis of 3 starting from natural α-euphol (1) obtained from dried Euphorbia resinifera latex in a manner described in [10]. The ketone 3 thus obtained can be used as an intermediate in synthesis of Δ7-24-alkylsterols which could serve as markers in the diagnosis and therapy of Pneumocystis carinii pneumonia (PcP) [11].
The treatment of α-euphol with 1 equivalent of metachloroperbenzoic acid (mCPBA) in methylene chloride at room temperature gave a mixture of two diastereoisomers of the epoxide 2 (78%) and all our attempts to separate the two isomers mixture failed. The resulting epoxide 2 was then smoothly converted to the corresponding ketone 3 in a satisfactory yield using BF3-etherate in dry benzene at –10°C.
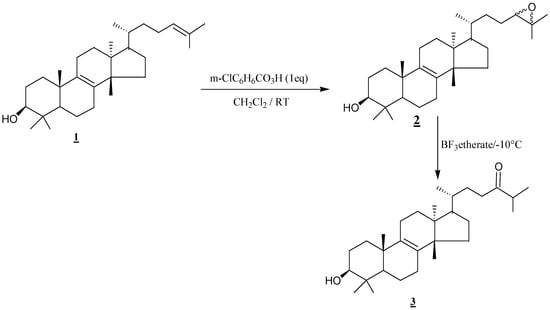
Scheme 1.
Conclusions
Epoxide cleavage effected by BF3-etherate work is a facile and convenient method for transforming α-euphol 1 to 3β-hydroxy-eupholanost-8-en-24-one (3). This protocol requires no preliminary protection of C3 hydroxy group. The easy work up procedure would also permit extension of the method to other triterpenes.
Experimental
General
Solvents were purified in the usual way. The IR spectra (KBr pellets) were recorded on a Perkin-Elmer Infracord 273 spectrophotometer.The 1H-NMR spectra (400 MHz in deuteriochloroform with Me4Si as the internal standard) and 13C-NMR spectra (100 MHz in CDCl3) were recorded using a Jeol JMS DX 300 spectrometer. Mass spectra were obtained on an AEI MS 50.instrument. The purity of compounds was checked by TLC (silica gel, ethyl acetate/hexane). Analytical plates were visualised by use of a UV light followed by an iodine chamber. Column chromatography was performed using E. Merck silica gel (230-400 mesh).
Epoxidation of α-euphol
Metachloroperbenzoic acid (mCPBA, 0.57g, 2.34 mmol) was added to a stirred solution of compound 1 (1g, 2.34 mmol) in methylene chloride (40mL) at room temperature The solution was stirred for 3 hours and then washed successively with 10% aqueous sodium bisulphite (3 x 20mL) and saturated sodium bicarbonate (3x20mL). The organic layer was dried over sodium sulphate. After filtration and evaporation, the crude product was purified by chromatography over SiO2 (elution with 5% ethyl acetate in hexane) to give 0.81g (78%) of the epoxide 2. Spectral data: FABMS: m/z = 443 ([M+1]+), 426([M+1]-OH), 409, 270, 241, 203; 1H-NMR (CDCl3) (ppm): 3.2 (dd, J1=12 Hz, J2=4 Hz, C3-H), 2.7 (t, J=10 Hz, C24-H),1.24 (s, C26-H3), 1.28 (s, C27-H3), 0.74 (s, C18-H3), 0.77 (s, C28-H3), 0.85 (s, C19-H3), 0.92 (d, J=6Hz, C21-H3), 0.97 (s, C29-H3), 0.98 (s, C30-H3); 13C-NMR (CDCl3) (ppm): 78.5 (C3), 134.1 (C8), 133.4 (C9), 64.08 (C24), 64.23 (C25); IR (KBr) (cm-1): 3400 (OH), 1287 (epoxide C-O).
Rearrangement of euphol monoepoxide (2) with boron trifluoride etherate in benzene
Boron trifluoride etherate (1mL, freshly distilled under reduced pressure) was added via syringe over a 10 minute period to a stirred solution of 1g of the monoepoxide 2 in anhydrous benzene (30mL), cooled in an ice bath and maintained under a N2 atmosphere. The ice-bath was then removed and stirring was continued for 1 hour during which time the solution became cloudy and reddish in colour. The reaction mixture was poured into cooled water and ether. After shaking, the layers were separated, the organic layer was treated with saturated sodium bicarbonate (3x30mL), dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 95:5 hexane/ethyl acetate to afford 3β-hydroxy-eupholanost-8-en-24-one (3) in 60% yield. Spectral data: MS: m/z = 443 ([M+1]+), 425 ([M+1]-H2O), 409, 297, 260, 247; 1H-NMR (CDCl3) (ppm): 3.18 (dd, J1=12 Hz, J2=4 Hz, C3-H), 2.21-2.40 (m, C23-H2), 2.45-2.60 (m, C25-H), 1.09 (d, J=6Hz, C26-H3), 1.07 (d, J=6Hz, C27-H3), 0.69 (s, C18-H3), 0.73 (s, C28-H3), 0.78 (s, C19-H3), 0.82 (d, J=6Hz, C21-H3), 0.90 (s, C29-H3), 0.94 (s, C30-H3); 13C-NMR (CDCl3) (ppm): 201.28 (C24), 133.40, 133.15 (C8; C9), 78.96 (C3), 50.96, 50.02 (C5; C17), 28.07 (C29), 29.07 (C16), 29.74 (C23), 30.77, 31.21(C12, C15), 35.22 (C1), 35.56(C16), 37.08 (C10), 37.68 (C22), 38.94 (C4), 40.86 (C25), 44.12 (C13), 15.55 (C30), 18.24 (C6), 18.51, 18.57 (C26, C27), 19.2 (C21), 20.14 (C19), 21.52 (C11), 24.46 (C2), 24.62 (C28), 27.66 (C7), 49.59 (C14), 16.81 (C18); IR (KBr) (cm-1): 3400 (OH), 1720 ( C=O), 1700 (C=C), 1200.
References
- Smith, J. G. Synthesis 1984, 629.
- Rao, A.S; Pakinkar, S.K.; Kirtane, J. G. Tetrahedron 1983, 39, 2323.
- Rickborn, B. Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: New York, 1991; vol.3, pp. 733–775. [Google Scholar]
- Barton, D. H. R.; Harrison, D.M.; Moss, G.P. Chem. Commun. 1996, 595.
- Bloch, K.; Urech, J. The Purification of Lanosterol. Biochem. Prep. 1958, 96, 32. [Google Scholar]
- Briggs, L. H.; Bartelly, J. P.; Rutledge, P. S. J. Chem. Soc., Perkin 1 1973, 806.
- Raab, K. H.; De Souza, N. J.; Nes, W .R. Biochem. Biophys. Acta 1968, 152, 742. [PubMed]
- Parish, E. J.; Kizito, S. A; Sun, H. J. Chem. Research (S). 1997, 64.
- Zdzislaw, P.; Martynow, J. J. Chem. Soc., Perkin Trans. 1. 1995, 201.
- Benharref, A.; Lavergne, J.-P. Bull. Soc. Chim. Fr. 1985, 965.
- Kaneshiro, E. S.; Amit, Z.; Swonger, M. M.; Kreishman, G. P.; Brooks, E. E.; Kreishman, M.; Jayasimhulu, K.; Parish, E. J.; Sun, H.; Kizito, S. A.; Beach, D. H. Biochemistry. 1999, 96, 97.
- Sample Availability: Samples of compounds 2 and 3 are available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes