
Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology


Abstract
1. Introduction
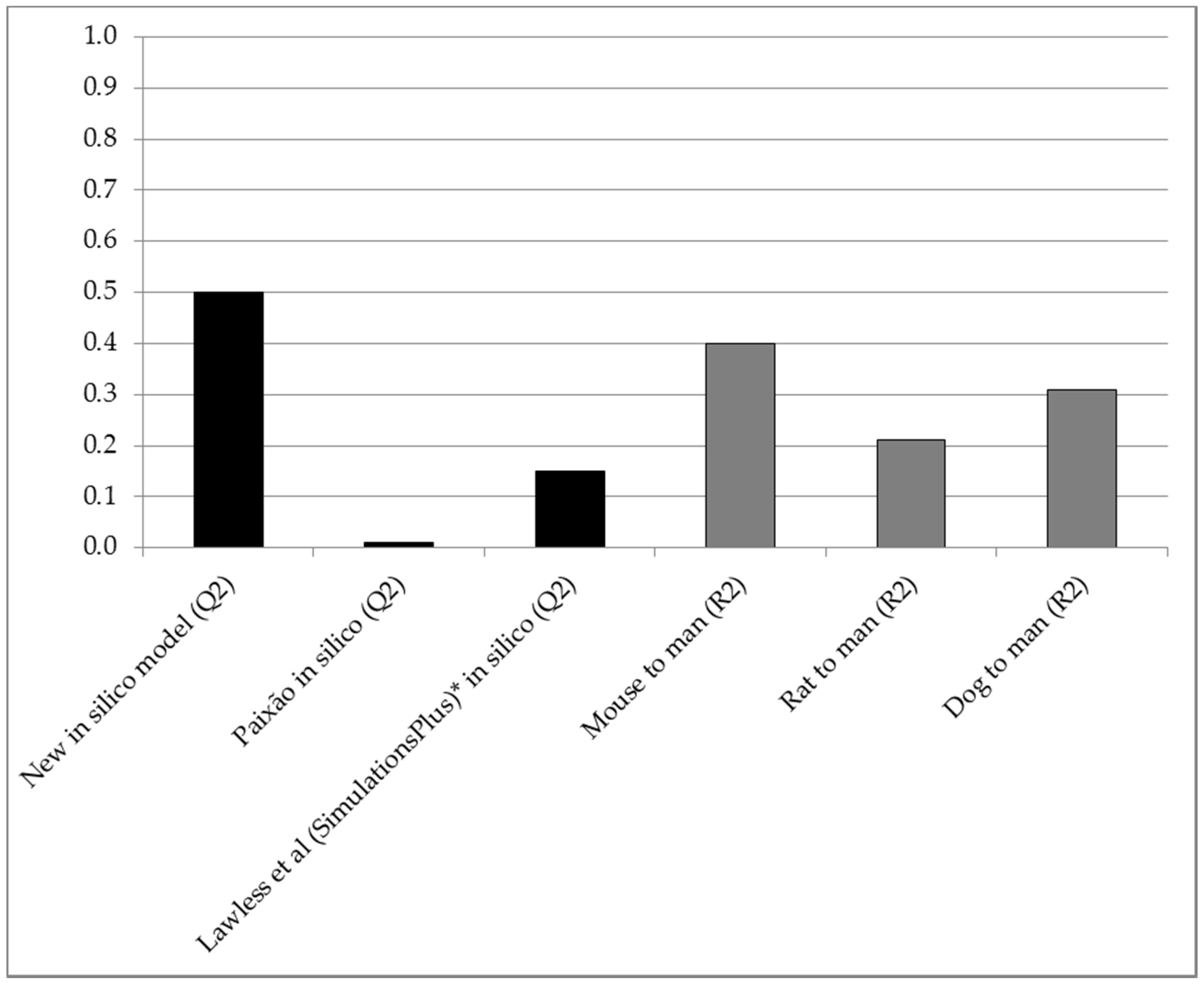
2. Results
3. Discussion
4. Materials and Methods
4.1. Feature Extraction
4.2. Quantitative Structure–Activity Relationship (QSAR)/Support Vector Machine (SVM) Model Development
4.3. Data Collection
4.4. Performance Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fuse, E.; Tanii, H.; Kurata, N.; Kobayashi, H.; Shimada, Y.; Tamura, T.; Sasaki, Y.; Tanigawara, Y.; Lush, R.D.; Headlee, D.; et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998, 58, 3248–3253. [Google Scholar]
- Obrezanova, O. Towards Improved Safety and Efficacy Profiles of Compounds by Predicting In Vivo Pharmacokinetics Using Machine Learning. In Silico Toxicology Network Meeting via Zoom. 30 September 2020. Available online: http://drugdiscovery.net/tox2020/ (accessed on 6 January 2021).
- Paixão, P.J.P.A. In Silico Prediction of Human Oral Bioavailability. Artificial Neural Networks and Physiologically Based Models. Ph.D. Thesis, University of Lisboa, Lisboa, Portugal, 2010. [Google Scholar]
- Lawless, M.; DiBella, J.; Bolger, M.B.; Clark, R.D.; Huehn, E.; Waldman, M.; Zhang, J.; Lukacova, V.; Simulations Plus, Inc. In Silico Prediction of Oral Bioavailability. Available online: https://www.simulations-plus.com/assets/ASCPT-2016-San-Diego-In-silico-prediction-oral-bioavailability.pdf?utm_source=qsar_pbpk_brochure (accessed on 6 March 2021).
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.D.; Liu, B.; Rostami Hodjegan, A. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Houston, J.B.; Galetin, A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Met. Disp. 2011, 39, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Matsson, P.; Bergström, C.A.S.; Nagahara, N.; Tavelin, S.; Norinder, U.; Artursson, P. Exploring the role of different drug transport routes in permeability screening. J. Med. Chem. 2005, 48, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Skolnik, S.; Chen, X.; Wang, J. Attenuation of intestinal absorption by major efflux transporters: Quantitative tools and strategies using a Caco-2 model. Drug Met. Disp. 2011, 39, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, S.; Lin, X.; Wang, J.; Chen, X.-H.; He, T.; Zhang, B. Towards prediction of in vivo intestinal absorption using a 96-well Caco-2 assay. J. Pharm. Sci. 2010, 99, 3246–3265. [Google Scholar] [CrossRef] [PubMed]
- Sohlenius-Sternbeck, A.-K.; Afzelius, L.; Prusis, P.; Neelissen, J.; Hoogstraate, J.; Johansson, J.; Floby, E.; Bengtsson, A.; Gissberg, O.; Sternbeck, J.; et al. Evaluation of the human prediction of clearance from hepatocyte and microsome intrinsic clearance for 52 drug compounds. Xenobiot 2010, 40, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Stringer, R.; Nicklin, P.L.; Houston, J.B. Reliability of human cryopreserved hepatocytes and liver microsomes as in vitro systems to predict metabolic clearance. Xenobiot 2008, 38, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Poulin, P.; Jones, R.D.O.; Jones, H.M.; Yates, J.W.T. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, Part 5: Prediction of plasma concentration-time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J. Pharm. Sci. 2011, 100, 4127–4157. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, T.; Zanelli, U.; Gallemann, D.; Perrin, D.; Dolgos, H.; Petersson, C. Comparison of methods for the prediction of human clearance from hepatocyte intrinsic clearance for a set of reference compounds and an external evaluation set. Xenobiot 2017, 47, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Spjuth, O.; Helmus, T.; Willighagen, E.L.; Kuhn, S.; Eklund, M.; Wagener, J.; Murray-Rust, P.; Steinbeck, C.; Wikberg, J.E.S. Bioclipse: An open source workbench for chemo- and bioinformatics. BMC Bioinform 2007, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spjuth, O.; Alvarsson, J.; Berg, A.; Eklund, M.; Kuhn, S.; Mäsak, C.; Torrance, G.; Wagener, J.; Willighagen, E.L.; Steinbeck, C.; et al. Bioclipse 2: A scriptable integration platform for the life sciences. BMC Bioinform. 2009, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Faulon, J.L.; Churchwell, C.J.; Visco, D.P. The signature molecular descriptor. 2. Enumerating molecules from their extended valence sequences. J. Chem. Inf. Comp. Sci. 2003, 43, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Alvarsson, J.; Eklund, M.; Andersson, C.; Carlsson, L.; Spjuth, O.; Wikberg, J.E. Benchmarking study of parameter variation when using signature fingerprints together with support vector machines. J. Inf. Model. 2014, 54, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- SIMCA, Version 16, Sartorius Stedim Umetrics, Umeå, Sweden. 2019. Available online: https://www.sartorius.com/en/products/process-analytical-technology/data-analytics-software/mvda-software/simca (accessed on 22 March 2021).
- Sköld, C.; Winiwarter, S.; Wernevik, J.; Bergström, F.; Engström, L.; Allen, R.; Box, K.; Comer, J.; Mole, J.; Hallberg, A.; et al. Presentation of a structurally diverse and commercially available drug data set for correlation and benchmarking studies. J. Med. Chem. 2006, 49, 6660–6671. [Google Scholar] [CrossRef] [PubMed]
- Sietsema, W.K. The absolute oral bioavailability of selected drugs. Int. J. Clin. Pharmacol. Ther. Toxicol. 1989, 27, 179–211. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Comparison | All 156 Compounds | 28 Compounds with Mouse Data | 101 Compounds with Rat Data | 106 Compounds with Dog Data |
|---|---|---|---|---|
| In silico predictive accuracy (Q2) | 0.50 | 0.54 | 0.61 | 0.48 |
| Mouse vs. man correlation (R2) | - | 0.40 | - | - |
| Rat vs. man correlation (R2) | - | - | 0.21 | - |
| Dog vs. man correlation (R2) | - | - | - | 0.31 |
| Model | Predicted Property | Acronym | Model Type (Number of Components) | Description |
|---|---|---|---|---|
| M1 | Passive intestinal permeability-based fraction absorbed | fabs,p | QSAR 1/SVM 2 (6) | Predicts passive intestinal permeability-based fraction absorbed in vivo in man (not considering active transport, solubility or instability in gastrointestinal fluids) |
| M2 | Intrinsic hepatic metabolic clearance | CLint | QSAR/SVM (5) | Predicts intrinsic hepatic metabolic clearance in vivo in man (phase I metabolism and conjugation) |
| M3 | CYP3A4-specificity | QSAR/SVM (5) | Predicts the substrate specificity for CYP3A4 (yes/no) | |
| M4 | Fraction unbound in human plasma | fu | QSAR/PLS 3 (7) | Predicts in vitro fraction unbound in human plasma |
| M5 | Maximum in vivo solubility/dissolution potential | fdiss | QSAR/PLS (5) | Predicts the maximum solubility/dissolution potential in the human gastrointestinal tract in vivo following oral dosing |
| M6 | MDR-1-specificity | QSAR/PLS-DA 4 (4) | Predicts the substrate specificity for MDR-1 (yes/no) | |
| M7 | BCRP-specificity | QSAR/PLS-DA (5) | Predicts the substrate specificity for BCRP (yes/no) | |
| M8 | Biliary CL | CLbile | QSAR/PLS (6) | Predicts the biliary clearance in vivo in man |
| M9 | Blood-to-plasma ratio | Cbl/Cpl | QSAR/PLS (8) | Predicts the blood-to-plasma concentration ratio |
| M10 | Phenol detection | Structural alerts | Phenol groups are used for selecting a different method for prediction of gut-wall extraction | |
| M11 | Quinolones detection | Structural alerts | Quinolones generally require consideration of active intestinal uptake | |
| M12 | Beta-lactam antibiotics detection | Structural alerts | Beta-lactam antibiotics generally require consideration of active intestinal uptake | |
| M13 | Intestinal absorption and extraction in the gut-wall | PBPK 5 | Algorithms for integrating mechanisms involved in intestinal absorption and gut wall extraction (fabs, fdiss, active uptake, efflux by MDR1 and/or BCRP, degradation by CYP3A4 and/or conjugating mucosal enzymes) and prediction of fraction of dose absorbed across the intestinal mucosa | |
| M14 | Extraction in the liver | PBPK | Algorithms for integrating mechanisms involved in liver extraction (CLint, fu, Cbl/Cpl, CLbile, liver blood flow; well-stirred liver extraction model) and prediction of fraction extracted by the liver |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagerholm, U.; Hellberg, S.; Spjuth, O. Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology. Molecules 2021, 26, 2572. https://doi.org/10.3390/molecules26092572
Fagerholm U, Hellberg S, Spjuth O. Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology. Molecules. 2021; 26(9):2572. https://doi.org/10.3390/molecules26092572
Chicago/Turabian StyleFagerholm, Urban, Sven Hellberg, and Ola Spjuth. 2021. "Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology" Molecules 26, no. 9: 2572. https://doi.org/10.3390/molecules26092572
APA StyleFagerholm, U., Hellberg, S., & Spjuth, O. (2021). Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology. Molecules, 26(9), 2572. https://doi.org/10.3390/molecules26092572