
Tackling Resistance to Cancer Immunotherapy: What Do We Know?

 , , ,
, , , 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
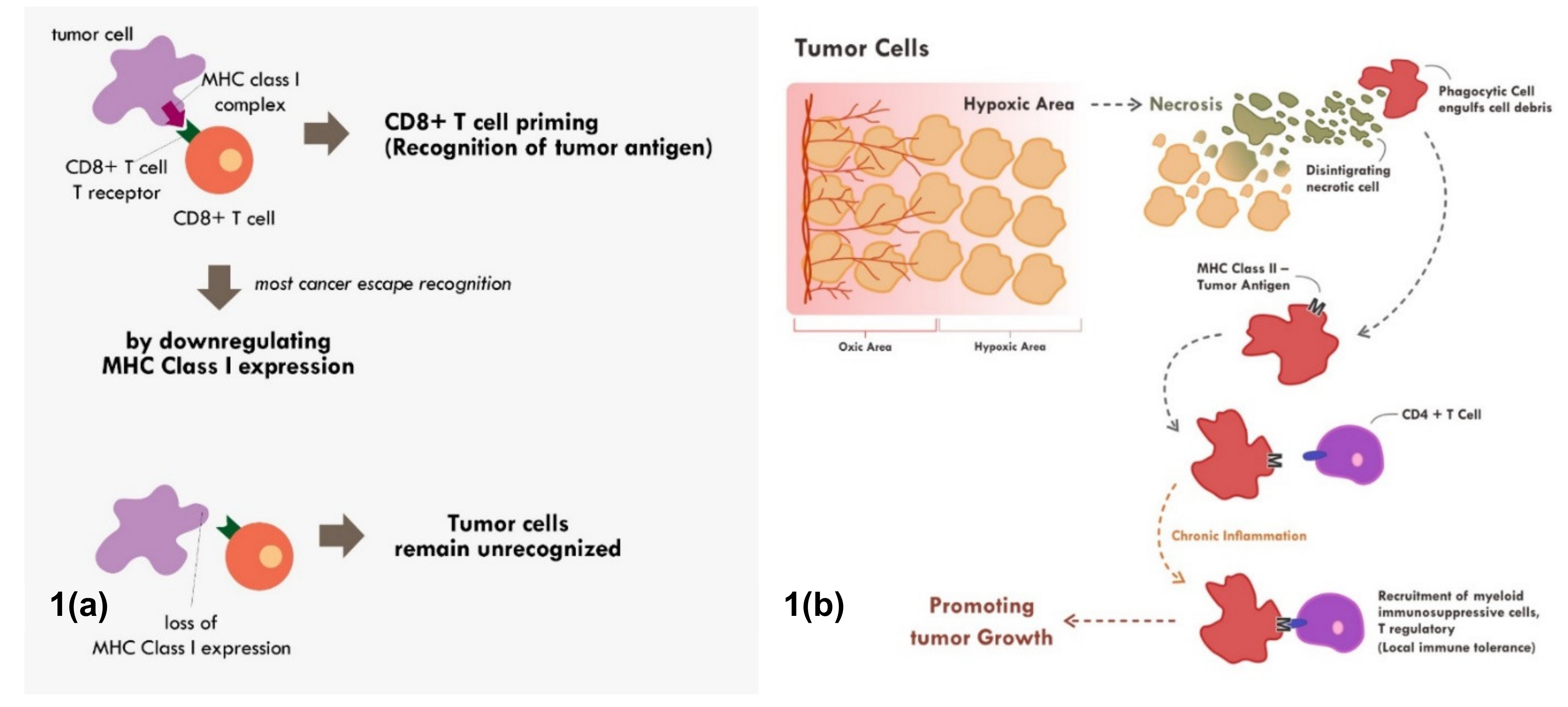
2. Major Histocompatibility Complex (MHC) Are Core Molecules in Immune Recognition
3. Recruitment and Priming of T Cells
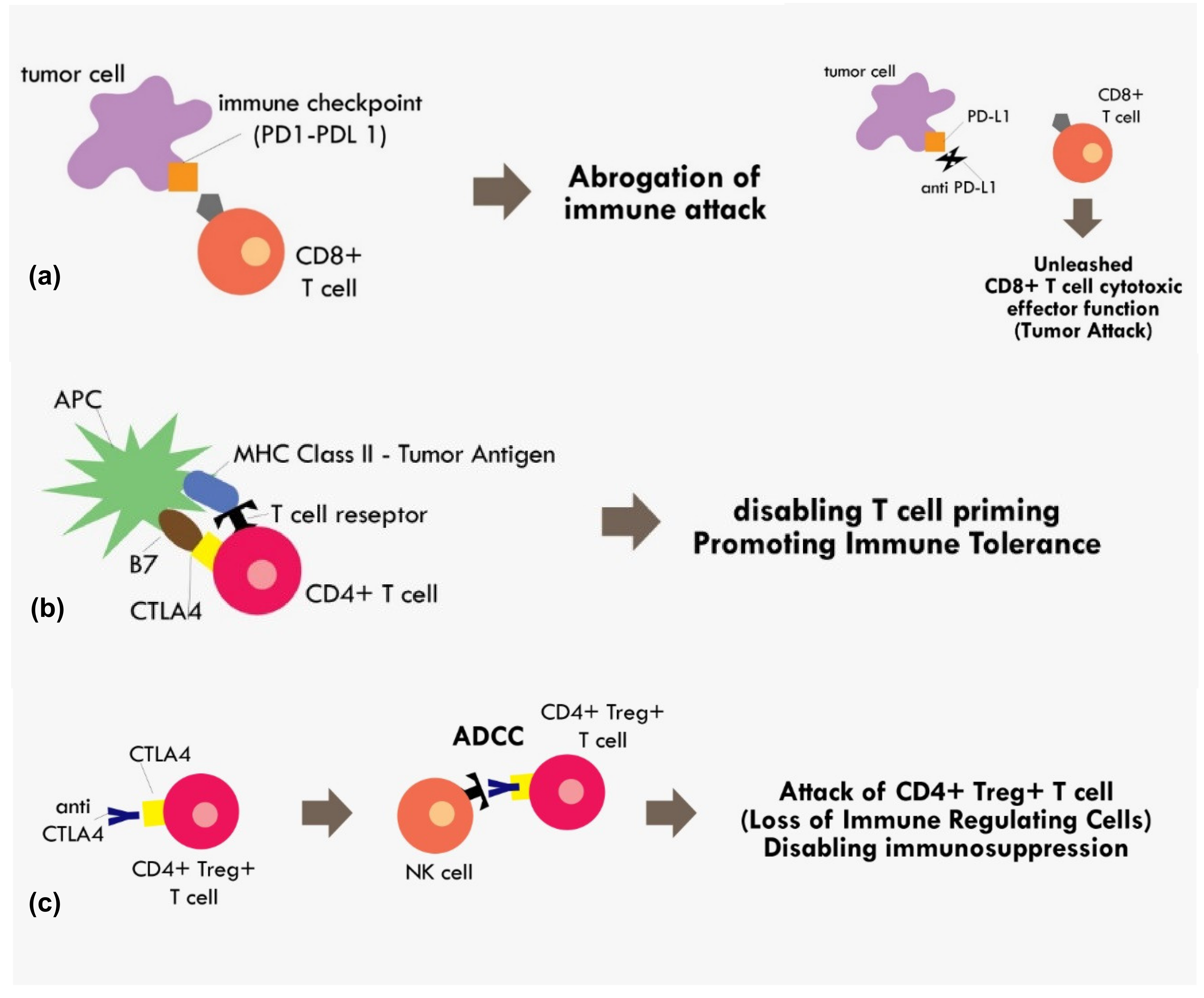
4. Immune Checkpoint Inhibition
5. Functional T Cells for Effective Immune Attack
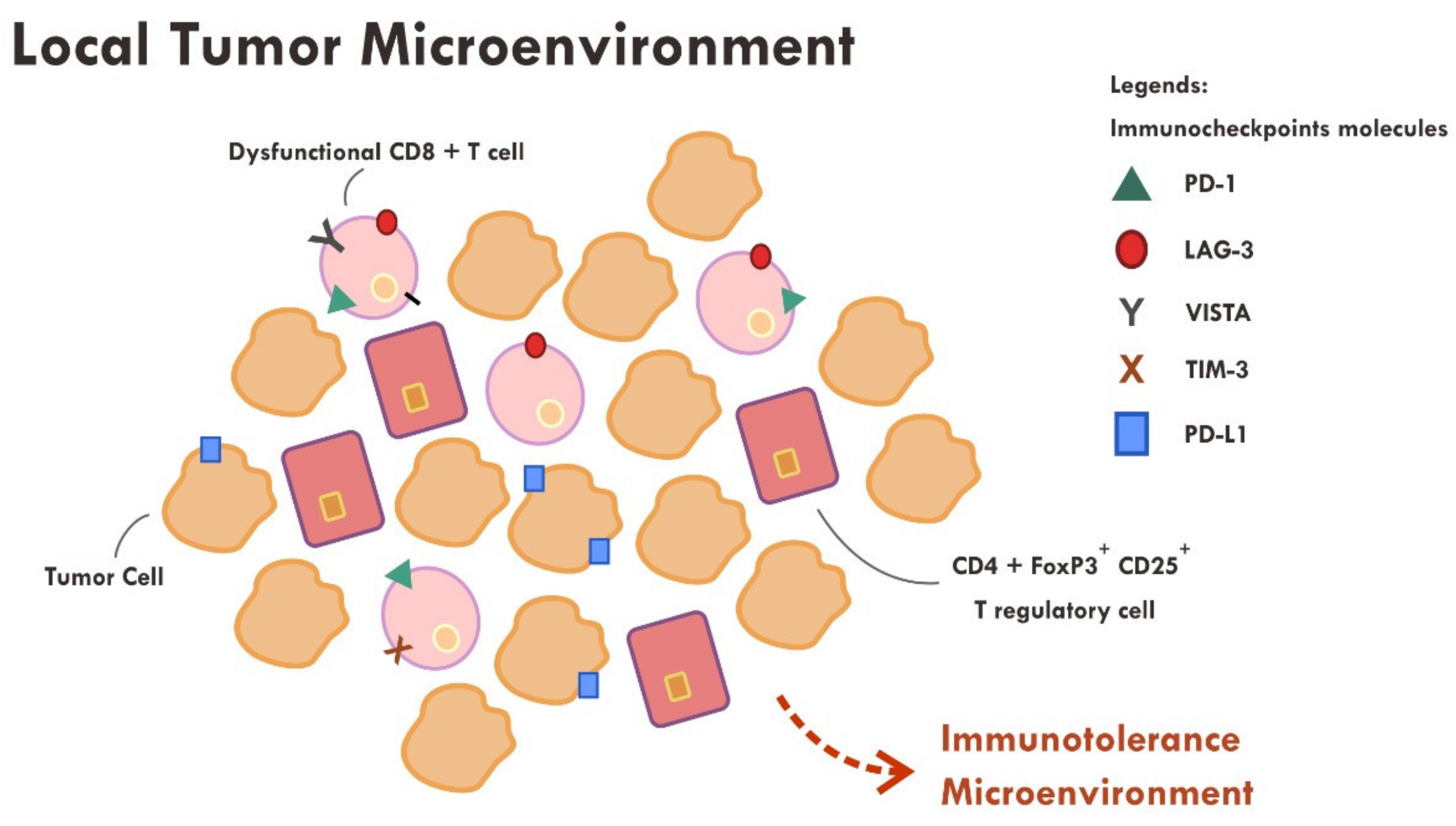
6. Hurdles of Immunotherapy
7. Hot Tumor Induction by Radiation Therapy
8. Synergistic Effect of Chemotherapy and Immunotherapy
9. Targeted Therapy Reverses Immunotherapy Tolerance
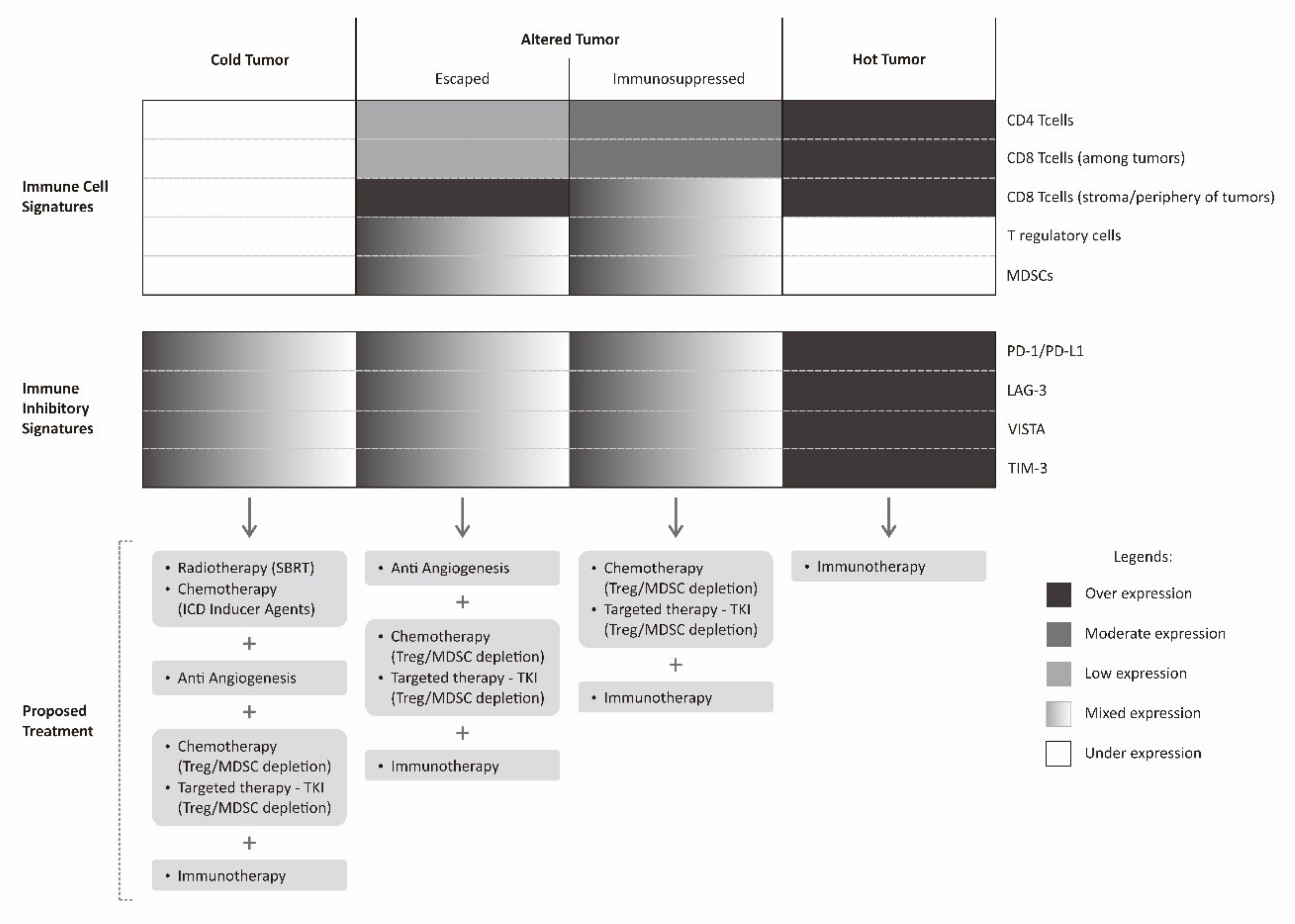
10. Treatment Based on Cancer Immune Landscape
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate Immunity, Inflammation and Tumour Progression: Double-Edged Swords. J. Intern. Med. 2019, 285, 524–532. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Ramia, E.; Tedeschi, A.; Forlani, G. CIITA-Driven MHC Class II Expressing Tumor Cells as Antigen Presenting Cell Performers: Toward the Construction of an Optimal Anti-Tumor Vaccine. Front. Immunol. 2019, 10, 1806. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor Neoantigens: From Basic Research to Clinical Applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA Methyltransferase Inhibition Upregulates MHC-I to Potentiate Cytotoxic T Lymphocyte Responses in Breast Cancer. Nat. Commun. 2018, 9, 248. [Google Scholar] [CrossRef]
- Kriegsman, B.A.; Vangala, P.; Chen, B.J.; Meraner, P.; Brass, A.L.; Garber, M.; Rock, K.L. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol. 2019, 203, 1999–2010. [Google Scholar] [CrossRef]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC Class I Transactivator Is a Target for Immune Evasion in Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef]
- van den Elsen, P.J.; Holling, T.M.; van der Stoep, N.; Boss, J.M. DNA Methylation and Expression of Major Histocompatibility Complex Class I and Class II Transactivator Genes in Human Developmental Tumor Cells and in T Cell Malignancies. Clin. Immunol. 2003, 109, 46–52. [Google Scholar] [CrossRef]
- Dan, H.; Zhang, S.; Zhou, Y.; Guan, Q. DNA Methyltransferase Inhibitors: Catalysts for Antitumour Immune Responses. Onco. Targets. Ther. 2019, 12, 10903–10916. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-Specific MHC-II Expression Represents a Tumour-Autonomous Phenotype and Predicts Response to Anti-PD-1/PD-L1 Therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Park, I.A.; Hwang, S.-H.; Song, I.H.; Heo, S.-H.; Kim, Y.-A.; Bang, W.S.; Park, H.S.; Lee, M.; Gong, G.; Lee, H.J. Expression of the MHC Class II in Triple-Negative Breast Cancer is Associated with Tumor-Infiltrating Lymphocytes and Interferon Signaling. PLoS ONE 2017, 12, e0182786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic Cell Death in Cancer Therapy: Present and Emerging Inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Inoue, H.; Tani, K. Multimodal Immunogenic Cancer Cell Death as a Consequence of Anticancer Cytotoxic Treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The Ins and Outs of MHC Class II-Mediated Antigen Processing and Presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef]
- Vakkila, J.; Lotze, M.T. Inflammation and Necrosis Promote Tumour Growth. Nature Rev. Immunol. 2004, 641–648. [Google Scholar] [CrossRef]
- Chou, S.-D.; Khan, A.N.H.; Magner, W.J.; Tomasi, T.B. Histone Acetylation Regulates the Cell Type Specific CIITA Promoters, MHC Class II Expression and Antigen Presentation in Tumor Cells. Int. Immunol. 2005, 17, 1483–1494. [Google Scholar] [CrossRef]
- Meissner, M.; Whiteside, T.L.; Kaufmann, R.; Seliger, B. CIITA versus IFN-Gamma Induced MHC Class II Expression in Head and Neck Cancer Cells. Arch. Dermatol. Res. 2009, 301, 189–193. [Google Scholar] [CrossRef]
- Chen, R.; Gong, Y.; Zou, D.; Wang, L.; Yuan, L.; Zhou, Q. Correlation between Subsets of Tumor-Infiltrating Immune Cells and Risk Stratification in Patients with Cervical Cancer. PeerJ 2019, 7, e7804. [Google Scholar] [CrossRef] [PubMed]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Holmich, E.R.; Raskov, H.; Gogenur, I. The Prognostic Value of Tumour-Infiltrating Lymphocytes in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Thike, A.A.; Li, H.; Yeong, J.; Koo, S.-L.; Dent, R.A.; Tan, P.H.; Iqbal, J. Increased CD4 and CD8-Positive T Cell Infiltrate Signifies Good Prognosis in a Subset of Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2016, 156, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Maskey, N.; Thapa, N.; Maharjan, M.; Shrestha, G.; Maharjan, N.; Cai, H.; Liu, S. Infiltrating CD4 and CD8 Lymphocytes in HPV Infected Uterine Cervical Milieu. Cancer Manag. Res. 2019, 11, 7647–7655. [Google Scholar] [CrossRef]
- Lechner, A.; Schlosser, H.; Rothschild, S.I.; Thelen, M.; Reuter, S.; Zentis, P.; Shimabukuro-Vornhagen, A.; Theurich, S.; Wennhold, K.; Garcia-Marquez, M.; et al. Characterization of Tumor-Associated T-Lymphocyte Subsets and Immune Checkpoint Molecules in Head and Neck Squamous Cell Carcinoma. Oncotarget 2017, 8, 44418–44433. [Google Scholar] [CrossRef]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A Reversed CD4/CD8 Ratio of Tumor-Infiltrating Lymphocytes and a High Percentage of CD4(+)FOXP3(+) Regulatory T Cells Are Significantly Associated with Clinical Outcome in Squamous Cell Carcinoma of the Cervix. Cell. Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef]
- Loeser, S.; Loser, K.; Bijker, M.S.; Rangachari, M.; van der Burg, S.H.; Wada, T.; Beissert, S.; Melief, C.J.M.; Penninger, J.M. Spontaneous Tumor Rejection by Cbl-b-Deficient CD8+ T Cells. J. Exp. Med. 2007, 204, 879–891. [Google Scholar] [CrossRef]
- Steele, K.E.; Tan, T.H.; Korn, R.; Dacosta, K.; Brown, C.; Kuziora, M.; Zimmermann, J.; Laffin, B.; Widmaier, M.; Rognoni, L.; et al. Measuring Multiple Parameters of CD8+ Tumor-Infiltrating Lymphocytes in Human Cancers by Image Analysis. J. Immunother. Cancer 2018, 6, 20. [Google Scholar] [CrossRef]
- Catacchio, I.; Silvestris, N.; Scarpi, E.; Schirosi, L.; Scattone, A.; Mangia, A. Intratumoral, Rather than Stromal, CD8+ T Cells Could Be a Potential Negative Prognostic Marker in Invasive Breast Cancer Patients. Transl. Oncol. 2019, 12, 585–595. [Google Scholar] [CrossRef]
- Oelkrug, C.; Ramage, J.M. Enhancement of T Cell Recruitment and Infiltration into Tumours. Clin. Exp. Immunol. 2014, 178, 1–8. [Google Scholar] [CrossRef]
- Muller, W.A. Getting Leukocytes to the Site of Inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Rotman, J.; Heeren, A.M.; Gassama, A.A.; Lougheed, S.M.; Pocorni, N.; Stam, A.G.M.; Bleeker, M.C.G.; Zijlmans, H.J.M.A.A.; Mom, C.H.; Kenter, G.G.; et al. Adenocarcinoma of the Uterine Cervix Shows Impaired Recruitment of CDC1 and CD8+ T Cells and Elevated Beta-Catenin Activation Compared to Squamous Cell Carcinoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-Vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen Cross-Presentation and T-Cell Cross-Priming in Cancer Immunology and Immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28 (Suppl. 12), xii44–xii55. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-Derived Factors Modulating Dendritic Cell Function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Hellstrom, K.E.; Hellstrom, I. From the Hellstrom Paradox toward Cancer Cure. Prog. Mol. Biol. Transl. Sci. 2019, 164, 1–24. [Google Scholar] [CrossRef]
- Yoneda, A.; Jinushi, M. T Cell Immunoglobulin Domain and Mucin Domain-3 as an Emerging Target for Immunotherapy in Cancer Management. ImmunoTargets Ther. 2013, 2, 135–141. [Google Scholar] [CrossRef][Green Version]
- He, Y.; Rivard, C.J.; Rozeboom, L.; Yu, H.; Ellison, K.; Kowalewski, A.; Zhou, C.; Hirsch, F.R. Lymphocyte-Activation Gene-3, an Important Immune Checkpoint in Cancer. Cancer Sci. 2016, 107, 1193–1197. [Google Scholar] [CrossRef]
- Muller, S.; Victoria Lai, W.; Adusumilli, P.S.; Desmeules, P.; Frosina, D.; Jungbluth, A.; Ni, A.; Eguchi, T.; Travis, W.D.; Ladanyi, M.; et al. V-Domain Ig-Containing Suppressor of T-Cell Activation (VISTA), a Potentially Targetable Immune Checkpoint Molecule, is Highly Expressed in Epithelioid Malignant Pleural Mesothelioma. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2020, 33, 303–311. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A Reappraisal of CTLA-4 Checkpoint Blockade in Cancer Immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.A.G.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Ha, S.-J. Re-Defining T-Cell Exhaustion: Subset, Function, and Regulation. Immune Netw. 2020, 20, e2. [Google Scholar] [CrossRef]
- McGoverne, I.; Dunn, J.; Batham, J.; Tu, W.J.; Chrisp, J.; Rao, S. Epitherapy and Immune Checkpoint Blockade: Using Epigenetic Reinvigoration of Exhausted and Dysfunctional T Cells to Reimburse Immunotherapy Response. BMC Immunol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in Cancer Immunosuppression - Implications for Anticancer Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab Strongly Impacts Effector/ Activated Regulatory T Cells and CD8(+) T Cells in the Tumor Microenvironment. J. Immunother. Cancer 2018, 6, 106. [Google Scholar] [CrossRef]
- Togashi, Y.; Kamada, T.; Sasaki, A.; Nakamura, Y.; Fukuoka, S.; Tada, Y.; Kawazoe, A.; Shitara, K.; Nishikawa, H. Clinicopathological, Genomic and Immunological Features of Hyperprogressive Disease during PD-1 Blockade in Gastric Cancer Patients. J. Clin. Oncol. 2018, 36 (Suppl. 15), 4106. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in Cancer Immunotherapy 2019—Latest Trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer Immunotherapy: The Beginning of the End of Cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene Laherparepvec: First in Class Oncolytic Virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Medrano, R.F.V.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and Antitumor Effects of Type I Interferons and Their Application in Cancer Therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015, 36, 725–737. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.-X. Targeting the Tumor Microenvironment with Interferon-β Bridges Innate and Adaptive Immune Responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef]
- Fujimura, T.; Hidaka, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Tono, H.; Tsukada, A.; Haga, T.; Hashimoto, A.; Morimoto, R.; et al. Phase I Study of Nivolumab Combined with IFN-β for Patients with Advanced Melanoma. Oncotarget 2017, 8, 71181–71187. [Google Scholar] [CrossRef][Green Version]
- Davar, D.; Wang, H.; Chauvin, J.-M.; Pagliano, O.; Fourcade, J.J.; Ka, M.; Menna, C.; Rose, A.; Sander, C.; Borhani, A.A.; et al. Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, JCO1800632. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.J.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus Chemotherapy for Previously Untreated, PD-L1-Expressing, Locally Advanced or Metastatic Non-Small-Cell Lung Cancer (KEYNOTE-042): A Randomised, Open-Label, Controlled, Phase 3 Trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C. Toxicities Associated With Adoptive T-Cell Transfer for Cancer. Cancer J. 2015, 21, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, J.; Jabbarzadeh Kaboli, P.; Shen, J.; Wu, X.; Zhao, Y.; Ji, H.; Du, F.; Zhou, Y.; Wang, Y.; et al. Natural Killer Cells as a Double-Edged Sword in Cancer Immunotherapy: A Comprehensive Review from Cytokine Therapy to Adoptive Cell Immunotherapy. Pharmacol. Res. 2020, 155, 104691. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Lee, J.-H.; Kwack, K.; Choi, S.-W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers 2019, 11, 1534. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.P.; Villa-Álvarez, M.; Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez, S. NK Cells in the Treatment of Hematological Malignancies. J. Clin. Med. 2019, 8, 1557. [Google Scholar] [CrossRef]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing NK Cells for Cancer Treatment. Front. Immunol. 2019, 10, 2836. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Ӧhrling, K.; Kaufman, H.L. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III-IV Melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol. Ther. Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of Intratumoral Immune Reaction and Human Colorectal Cancer Recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Gondhowiardjo, S.A.; Handoko Adham, M.; Rachmadi, L.; Kodrat, H.; Tobing, D.L.; Haryoga, I.M.; Dwiyono, A.G.; Kristian, Y.A.; Mayang Permata, T.B. Tumor Microenvironment Predicts Local Tumor Extensiveness in PD-L1 Positive Nasopharyngeal Cancer. PLoS ONE 2020, 15, e0230449. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; McBride, W.H. T Lymphocytes and Normal Tissue Responses to Radiation. Front. Oncol. 2012, 2, 119. [Google Scholar] [CrossRef] [PubMed]
- Goans, R.E.; Waselenko, J.K. Medical Management of Radiological Casualties. Health Phys. 2005, 89, 505–512. [Google Scholar] [CrossRef]
- Hildebrandt, G.; Seed, M.P.; Freemantle, C.N.; Alam, C.A.; Colville-Nash, P.R.; Trott, K.R. Mechanisms of the Anti-Inflammatory Activity of Low-Dose Radiation Therapy. Int. J. Radiat. Biol. 1998, 74, 367–378. [Google Scholar] [CrossRef]
- Sekarutami, S.M.; Handoko. The Future of Radiotherapy and Immunotherapy Concomitantly in Cancer Management. Med. J. Indones 2019, 28, 391–395. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation Modulates the Peptide Repertoire, Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- de Carvalho, H.A.; Villar, R.C. Radiotherapy and Immune Response: The Systemic Effects of a Local Treatment. Clinics 2018, 73 (Suppl. 1), e557s. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.-X. Radiotherapy and Immunotherapy: A Beneficial Liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation Therapy and Anti-Tumor Immunity: Exposing Immunogenic Mutations to the Immune System. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The CGAS-CGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Dutta, T.; Wang, L.; Song, L.; Gu, L.; Qian, L.; Benitez, A.; Ning, S.; Malhotra, A.; Deutscher, M.P.; et al. Human DNA Exonuclease TREX1 is Also an Exoribonuclease That Acts on Single-Stranded RNA. J. Biol. Chem. 2015, 290, 13344–13353. [Google Scholar] [CrossRef] [PubMed]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STING- and Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.-C.; Liu, L.F. Chemotherapeutics and Radiation Stimulate MHC Class I Expression through Elevated Interferon-Beta Signaling in Breast Cancer Cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef]
- Menon, H.; Ramapriyan, R.; Cushman, T.R.; Verma, V.; Kim, H.H.; Schoenhals, J.E.; Atalar, C.; Selek, U.; Chun, S.G.; Chang, J.Y.; et al. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front. Immunol. 2019, 10, 193. [Google Scholar] [CrossRef]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory Microenvironment Remodelling by Tumour Cells after Radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-Induced CXCL16 Release by Breast Cancer Cells Attracts Effector T Cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Ditsworth, D.; Fan, Y.; Zhao, F.; Crawford, H.C.; Zong, W.-X. Chemotherapy Induces Tumor Clearance Independent of Apoptosis. Cancer Res. 2008, 68, 9595–9600. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; Johnston, B. Concepts and Mechanisms Underlying Chemotherapy Induced Immunogenic Cell Death: Impact on Clinical Studies and Considerations for Combined Therapies. Oncotarget 2015, 6, 41600–41619. [Google Scholar] [CrossRef]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.-L.; et al. Molecular Mechanisms of ATP Secretion during Immunogenic Cell Death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Gato, M.; Caliendo, F.; Kochan, G.; Blanco-Luquin, I.; Van der Jeught, K.; Arce, F.; Guerrero-Setas, D.; Fernandez-Irigoyen, J.; et al. A Highly Efficient Tumor-Infiltrating MDSC Differentiation System for Discovery of Anti-Neoplastic Targets, Which Circumvents the Need for Tumor Establishment in Mice. Oncotarget 2014, 5, 7843–7857. [Google Scholar] [CrossRef]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-Treatment with Chemotherapy Can Enhance the Antigenicity and Immunogenicity of Tumours by Promoting Adaptive Immune Responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef]
- Kol, A.; Terwisscha van Scheltinga, A.; Pool, M.; Gerdes, C.; de Vries, E.; de Jong, S. ADCC Responses and Blocking of EGFR-Mediated Signaling and Cell Growth by Combining the Anti-EGFR Antibodies Imgatuzumab and Cetuximab in NSCLC Cells. Oncotarget 2017, 8, 45432–45446. [Google Scholar] [CrossRef] [PubMed]
- Fornasier, G.; Francescon, S.; Baldo, P. An Update of Efficacy and Safety of Cetuximab in Metastatic Colorectal Cancer: A Narrative Review. Adv. Ther. 2018, 35, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF Targets the Tumour Cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Takeshita, T.; Yan, L.; Asaoka, M.; Rashid, O.; Takabe, K. Late Recurrence of Breast Cancer Is Associated with Pro-Cancerous Immune Microenvironment in the Primary Tumor. Sci. Rep. 2019, 9, 16942. [Google Scholar] [CrossRef]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK Cell-Cancer Cycle: Advances and New Challenges in NK Cell-Based Immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.-C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the Local Tumor Microenvironment in Recurrent Cancers May Explain the Failure of Vaccines after Surgery. Proc. Natl. Acad. Sci. USA 2013, 110, E415–E424. [Google Scholar] [CrossRef] [PubMed]
- Nuryadi, E.; Sasaki, Y.; Hagiwara, Y.; Permata, T.B.M.; Sato, H.; Komatsu, S.; Yoshimoto, Y.; Murata, K.; Ando, K.; Kubo, N.; et al. Mutational Analysis of Uterine Cervical Cancer That Survived Multiple Rounds of Radiotherapy. Oncotarget 2018, 9, 32642–32652. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the Dynamic Tumor Microenvironment in Controversies Regarding Immune Checkpoint Inhibitors for the Treatment of Non-Small Cell Lung Cancer (NSCLC) with EGFR Mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Isambert, N.; Hervieu, A.; Zanetta, S.; Guion, J.-F.; Hennequin, A.; Rederstorff, E.; Bertaut, A.; Ghiringhelli, F. Phase Ib/II Trial Evaluating the Safety, Tolerability and Immunological Activity of Durvalumab (MEDI4736) (Anti-PD-L1) plus Tremelimumab (Anti-CTLA-4) Combined with FOLFOX in Patients with Metastatic Colorectal Cancer. ESMO Open 2018, 3, e000375. [Google Scholar] [CrossRef]
- Lieverse, R.I.Y.; Van Limbergen, E.J.; Oberije, C.J.G.; Troost, E.G.C.; Hadrup, S.R.; Dingemans, A.-M.C.; Hendriks, L.E.L.; Eckert, F.; Hiley, C.; Dooms, C.; et al. Stereotactic Ablative Body Radiotherapy (SABR) Combined with Immunotherapy (L19-IL2) versus Standard of Care in Stage IV NSCLC Patients, ImmunoSABR: A Multicentre, Randomised Controlled Open-Label Phase II Trial. BMC Cancer 2020, 20, 557. [Google Scholar] [CrossRef]
- Kyte, J.A.; Røssevold, A.; Falk, R.S.; Naume, B. ALICE: A Randomized Placebo-Controlled Phase II Study Evaluating Atezolizumab Combined with Immunogenic Chemotherapy in Patients with Metastatic Triple-Negative Breast Cancer. J. Transl. Med. 2020, 18, 252. [Google Scholar] [CrossRef]
- Levy, B.; Paz-Ares, L.; Bennouna, J.; Felip, E.; Abreu, D.R.; Isla, D.; Barlesi, F.; Molinier, O.; Madelaine, J.; Audigier-Valette, C.; et al. Afatinib With Pembrolizumab for Treatment of Patients With Locally Advanced/Metastatic Squamous Cell Carcinoma of the Lung: The LUX-Lung IO/KEYNOTE 497 Study Protocol. Clin. Lung Cancer 2019, 20, e407–e412. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gondhowiardjo, S.A.; Handoko; Jayalie, V.F.; Apriantoni, R.; Barata, A.R.; Senoaji, F.; Utami, I.J.W.; Maubere, F.; Nuryadi, E.; Giselvania, A. Tackling Resistance to Cancer Immunotherapy: What Do We Know? Molecules 2020, 25, 4096. https://doi.org/10.3390/molecules25184096
Gondhowiardjo SA, Handoko, Jayalie VF, Apriantoni R, Barata AR, Senoaji F, Utami IJW, Maubere F, Nuryadi E, Giselvania A. Tackling Resistance to Cancer Immunotherapy: What Do We Know? Molecules. 2020; 25(18):4096. https://doi.org/10.3390/molecules25184096
Chicago/Turabian StyleGondhowiardjo, Soehartati A., Handoko, Vito Filbert Jayalie, Riyan Apriantoni, Andreas Ronald Barata, Fajar Senoaji, IGAA Jayanthi Wulan Utami, Ferdinand Maubere, Endang Nuryadi, and Angela Giselvania. 2020. "Tackling Resistance to Cancer Immunotherapy: What Do We Know?" Molecules 25, no. 18: 4096. https://doi.org/10.3390/molecules25184096
APA StyleGondhowiardjo, S. A., Handoko, Jayalie, V. F., Apriantoni, R., Barata, A. R., Senoaji, F., Utami, I. J. W., Maubere, F., Nuryadi, E., & Giselvania, A. (2020). Tackling Resistance to Cancer Immunotherapy: What Do We Know? Molecules, 25(18), 4096. https://doi.org/10.3390/molecules25184096