
Cell Suspensions of Cannabis sativa (var. Futura): Effect of Elicitation on Metabolite Content and Antioxidant Activity

 , , , ,
, , , ,
Abstract
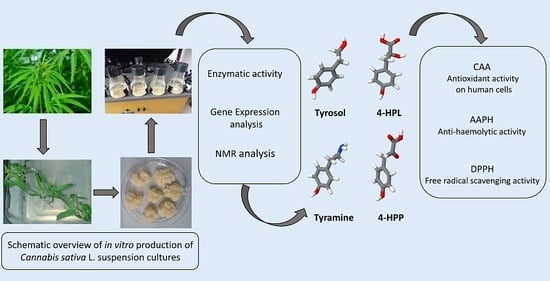
1. Introduction
2. Results
2.1. Cell Suspension Growth and Elicitor Treatments
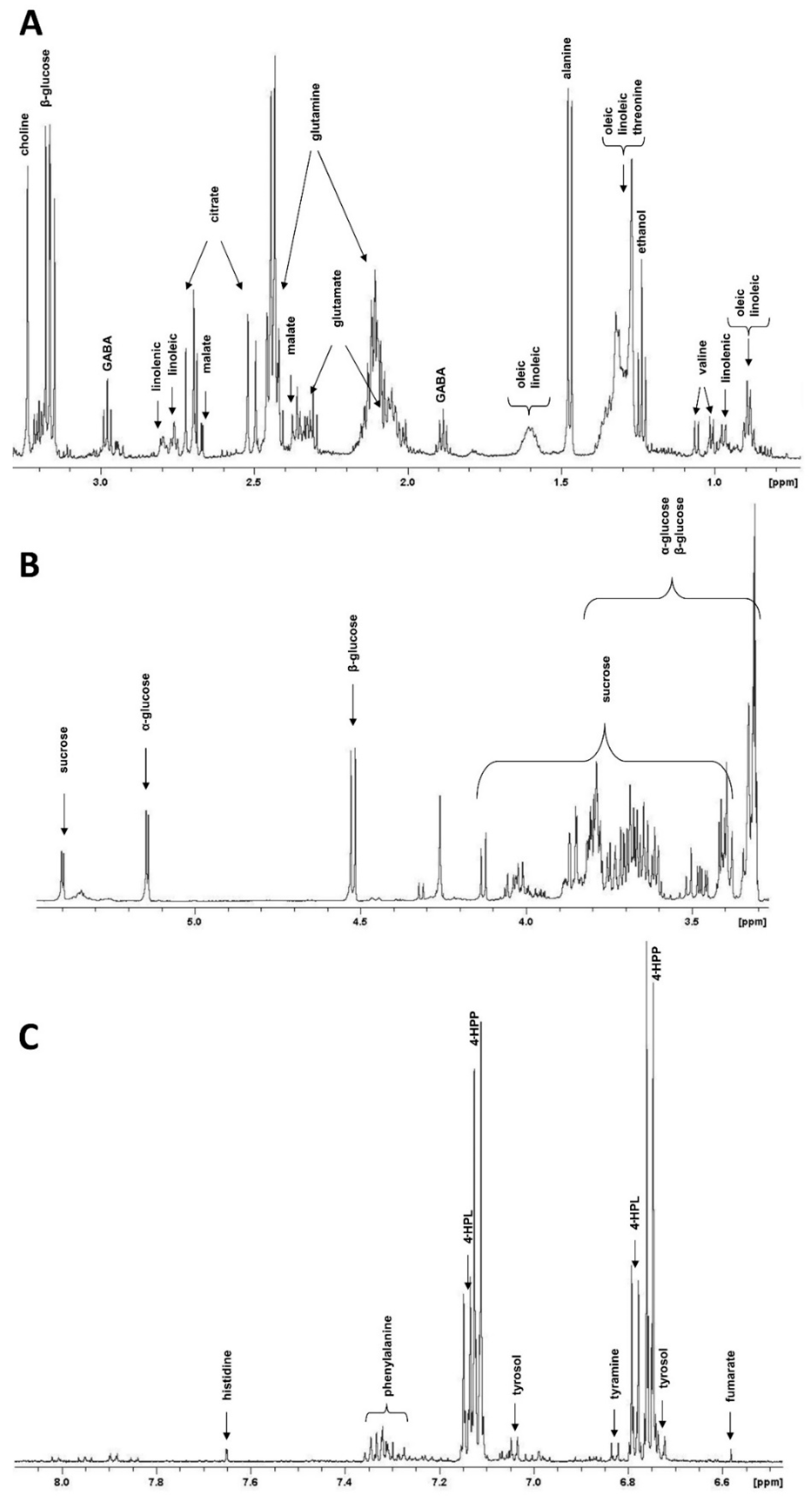
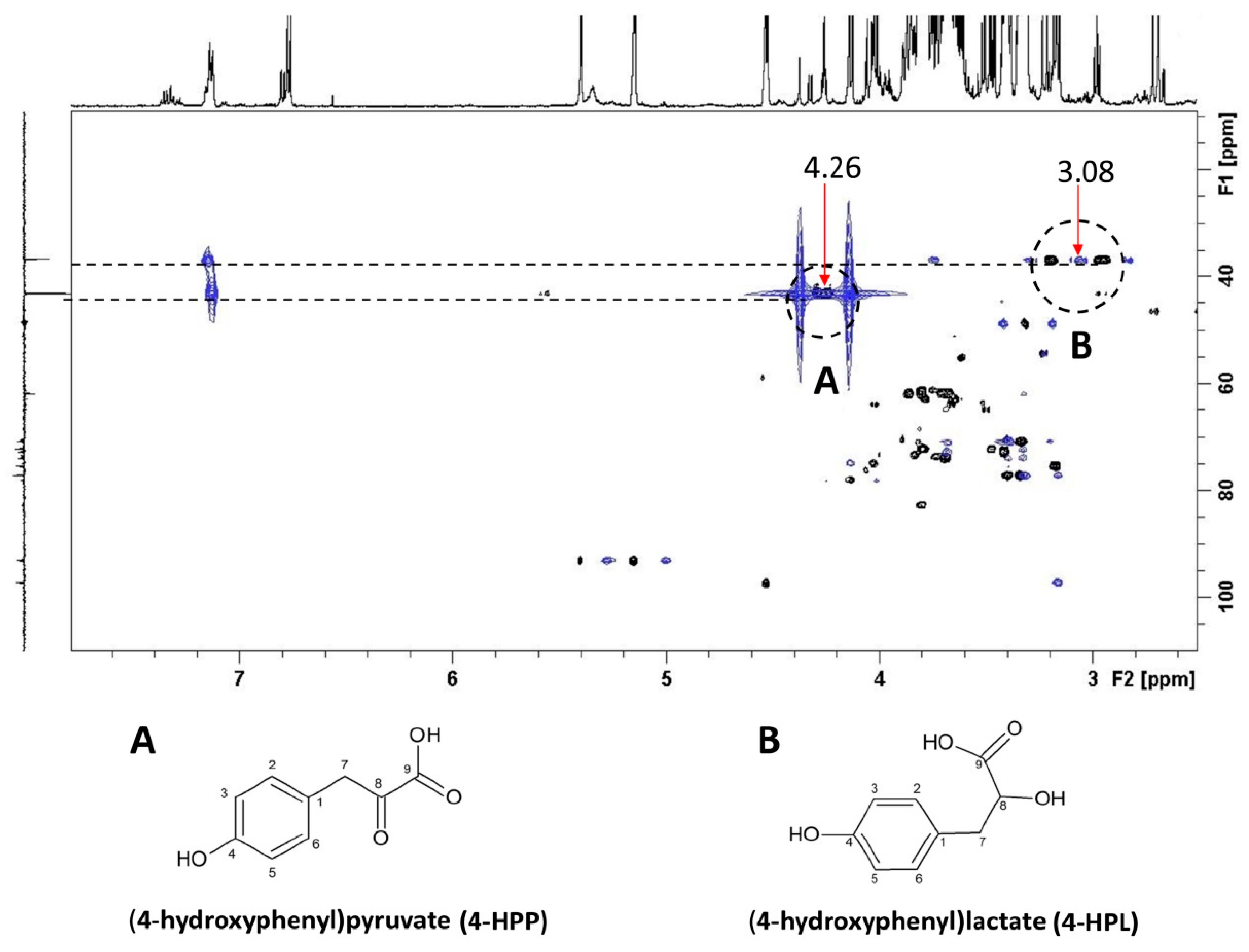
2.2. NMR Metabolite Profiling
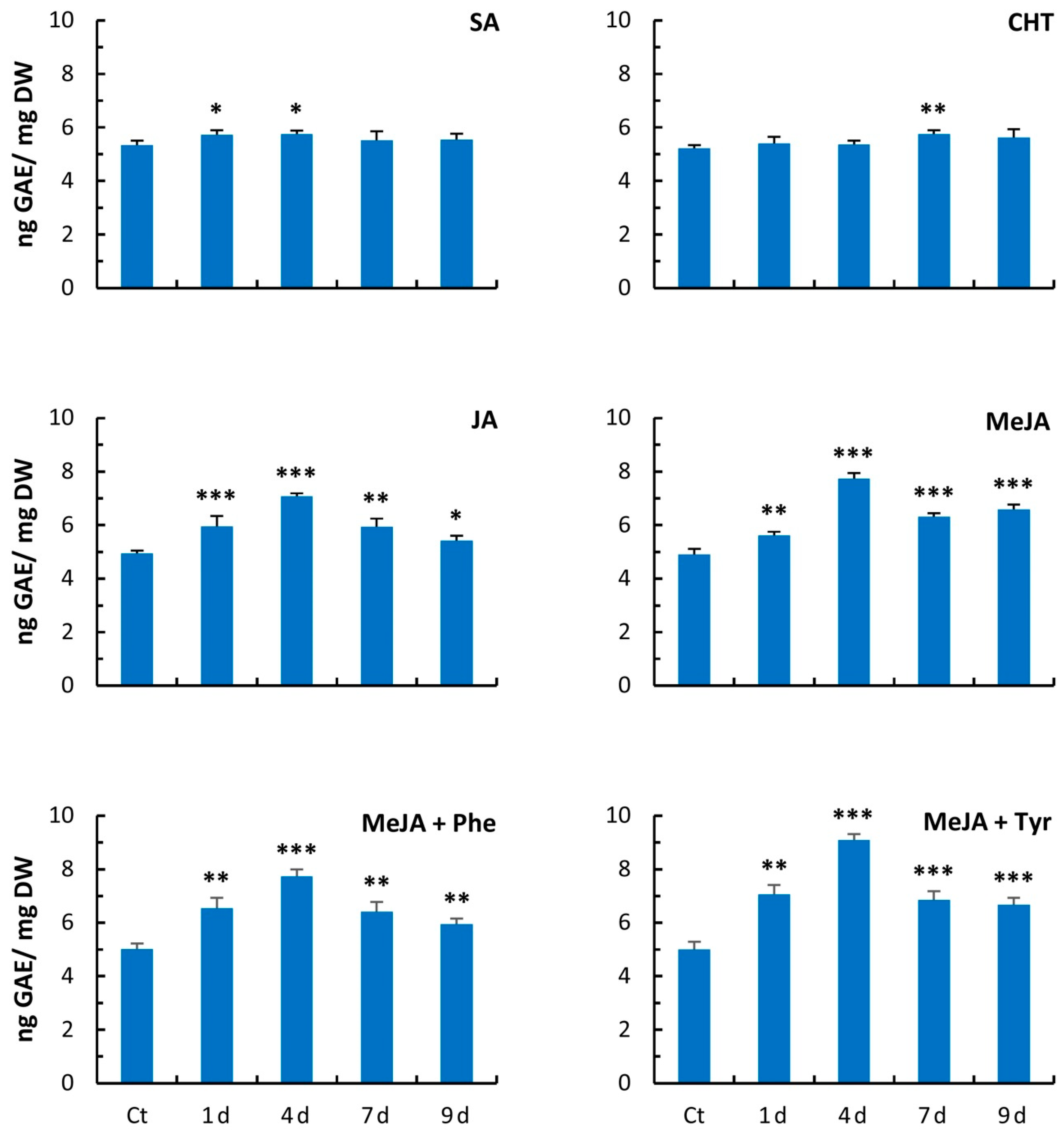
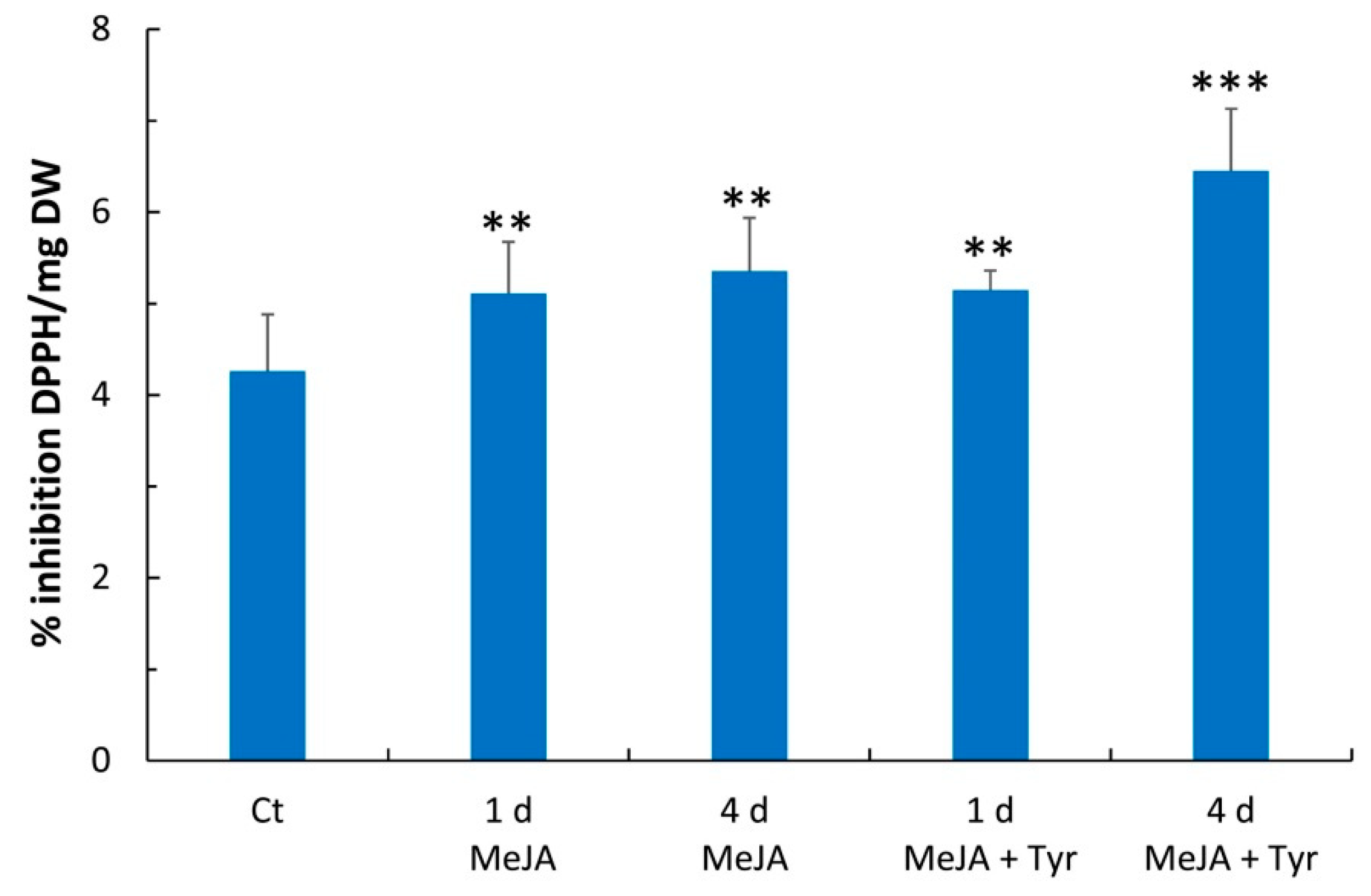
2.3. Free Radical Scavenging Activity
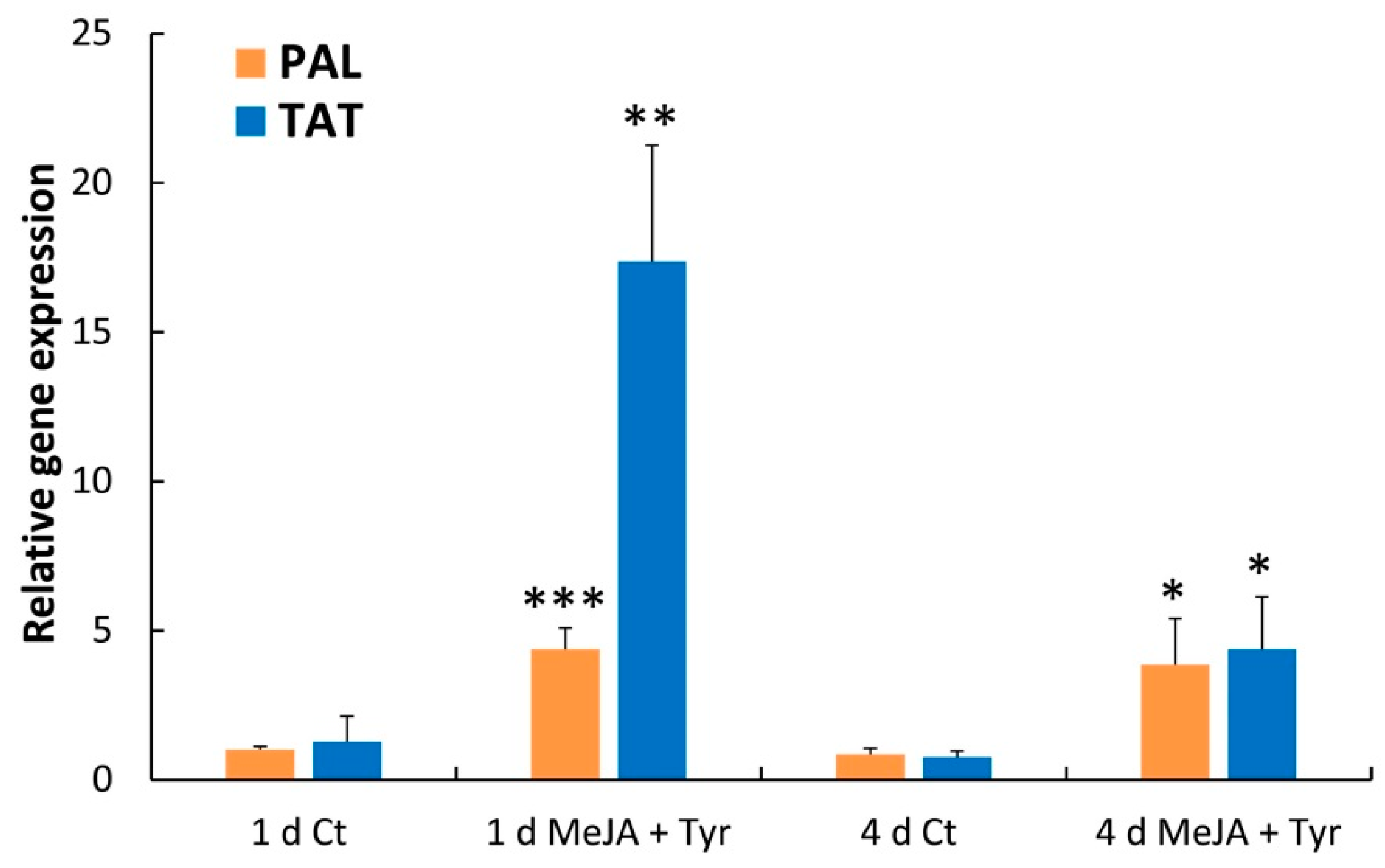
2.4. Effects of Elicitors on PAL and TAT Enzymatic Activities and Transcript Levels
3. Discussion
4. Materials and Methods
4.1. Plant Material and Cell Suspension Cultures
4.2. Elicitor Treatments
4.3. Cell Viability
4.4. Metabolite Extraction and NMR Measurements
4.5. Assay of Total Phenolics
4.6. DPPH Radical Scavenging Assay
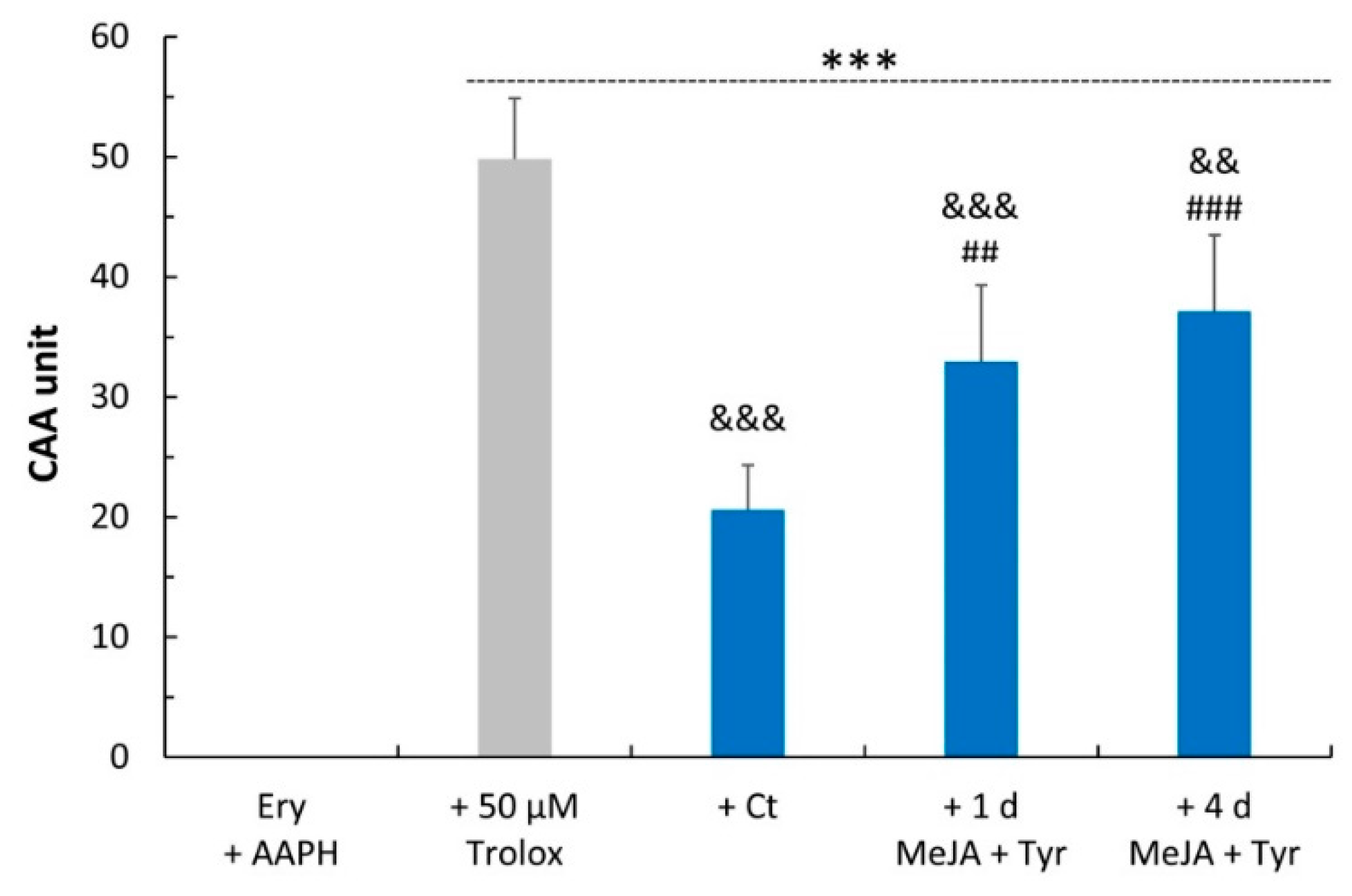
4.7. Cellular Antioxidant Activity (CAA) Assay in Red Blood Cells
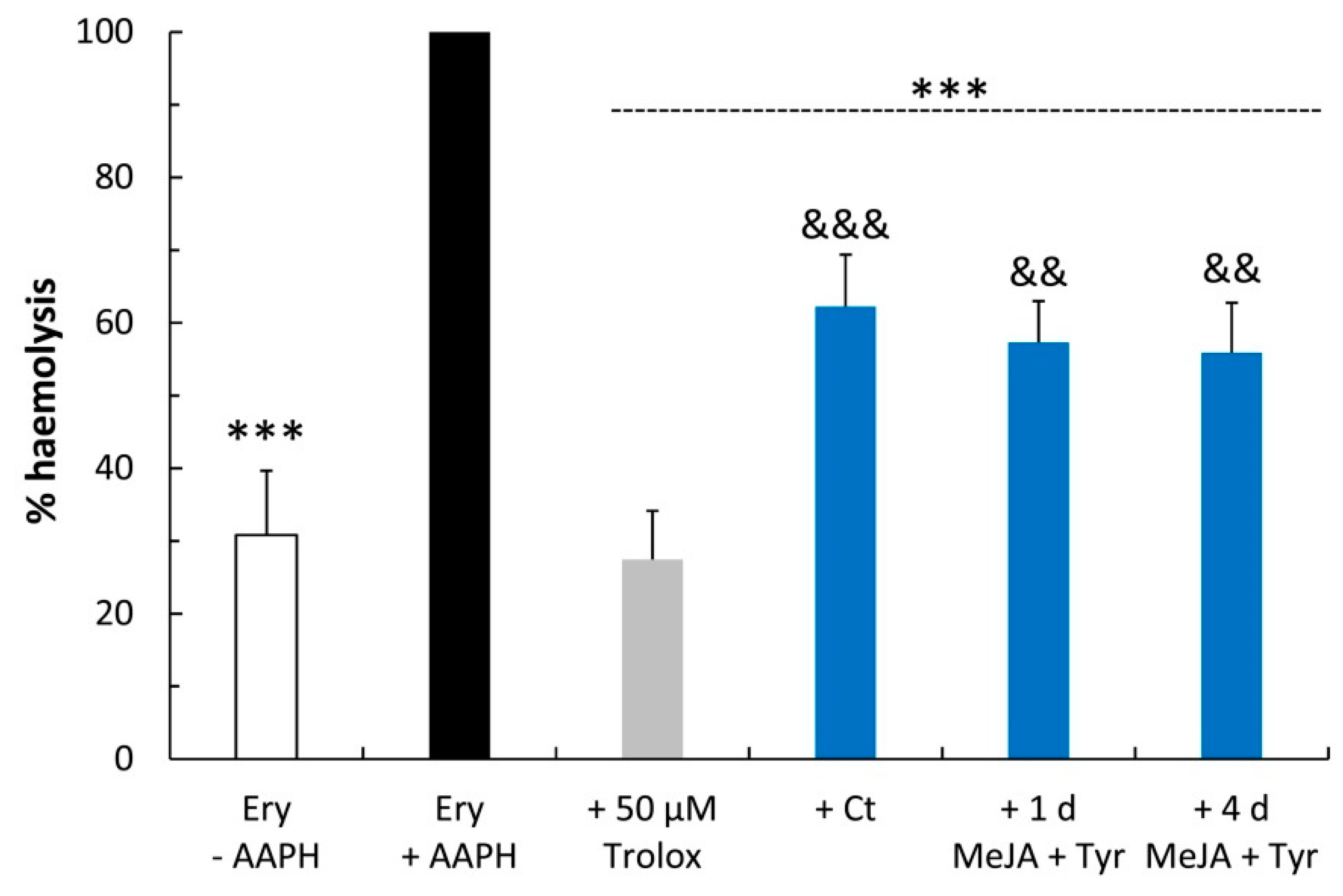
4.8. Erythrocyte Oxidative Hemolysis
4.9. PAL and TAT Enzymatic Assays
4.10. RNA Extraction and Real Time PCR
Author Contributions
Funding
Conflicts of Interest
References
- Turner, C.E.; Elsohly, M.A.; Boeren, E.G. Constituents of Cannabis sativa L. XVII. A review of the natural constituents. J. Nat. Prod. 1980, 43, 169–234. [Google Scholar] [CrossRef] [PubMed]
- Flores-Sanchez, I.J.; Verpoorte, R. Secondary metabolism in cannabis. Phytochem. Rev. 2008, 7, 615–639. [Google Scholar] [CrossRef]
- Broséus, J.; Anglada, F.; Esseiva, P. The differentiation of fibre-and drug type Cannabis seedlings by gas chromatography/mass spectrometry and chemometric tools. Forensic. Sci. Int. 2010, 200, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Chianese, G.; Taglialatela-Scafati, O. Cannabinoids: Occurrence and medicinal chemistry. Curr. Med. Chem. 2011, 18, 1085–1099. [Google Scholar] [CrossRef]
- Peschel, W.; Politi, M. 1H NMR and HPLC/DAD for Cannabis sativa L. chemotype distinction, extract profiling and specification. Talanta 2015, 140, 150–165. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Phytocannabinoids. Progress in the Chemistry of Organic Natural Products; Kinghorn, A., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Cham, Switzerland, 2017; Volume 103, pp. 1–36. [Google Scholar] [CrossRef]
- Docimo, T.; Consonni, R.; Coraggio, I.; Mattana, M. Early phenylpropanoid biosynthetic steps in Cannabis sativa: Link between genes and metabolites. Int. J. Mol. Sci. 2013, 14, 13626–13644. [Google Scholar] [CrossRef]
- Andre, C.M.; Hausman, J.F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef]
- Sanches-Silva, A.; Costa, D.; Albuquerque, T.G.; Buonocore, G.G.; Ramos, F.; Castilho, M.C.; Machado, A.V.; Costa, H.S. Trends in the use of natural antioxidants in active food packaging: A review. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2014, 31, 374–395. [Google Scholar] [CrossRef]
- Kiokias, S.; Varzakas, T. Activity of flavonoids and β-carotene during the auto-oxidative deterioration of model food oil-in water emulsions. Food Chem. 2014, 150, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Bourgaud, F.; Gravot, A.; Milesi, S.; Gontier, E. Production of plant secondary metabolites: A historical perspective. Plant Sci. 2001, 161, 839–851. [Google Scholar] [CrossRef]
- Wilson, S.A.; Roberts, S.C. Metabolic engineering approaches for production of biochemicals in food and medicinal plants. Curr. Opin. Biotechnol. 2014, 26, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.R.; Ravishankar, G.A. Plant cell cultures: Chemical factories of secondary metabolites. Biotechnol. Adv. 2002, 20, 101–153. [Google Scholar] [PubMed]
- Namdeo, A.G. Plant cell elicitation for production of secondary metabolites: A review. Pharmacogn. Rev. 2007, 1, 69–79. [Google Scholar]
- Wilson, S.A.; Roberts, S.C. Recent advances towards development and commercialization of plant cell culture processes for the synthesis of biomolecules. Plant Biotechnol. J. 2012, 10, 249–268. [Google Scholar] [CrossRef]
- Wrobel, T.; Dreger, M.; Wielgus, K.; Stomski, R. The application of plant in vitro cultures in cannabinoid production. Biotechnol. Lett. 2018, 40, 445–454. [Google Scholar] [CrossRef]
- Flores-Sanchez, I.J.; Peč, J.; Fei, J.; Choi, Y.H.; Dušek, J.; Verpoorte, R. Elicitation studies in cell suspension cultures of Cannabis sativa L. J. Biotechnol. 2009, 143, 157–168. [Google Scholar] [CrossRef]
- Peč, J.; Flores-Sanchez, I.J.; Choi, Y.H.; Verpoorte, R. Metabolic analysis of elicited cell suspension cultures of Cannabis sativa L. by 1H-NMR spectroscopy. Biotechnol. Lett. 2010, 32, 935–941. [Google Scholar] [CrossRef]
- Zhao, J.L.; Zhou, L.G.; Wu, J.Y. Effects of biotic and abiotic elicitors on cell growth and tanshinone accumulation in Salvia miltiorrhiza cell cultures. Appl. Microbiol. Biotechnol. 2010, 87, 137–144. [Google Scholar] [CrossRef]
- Ouyang, J.; Wang, X.D.; Zhao, B.; Wang, Y.C. Enhanced production of phenylethanoid glycosides by precursor feeding to cell culture of Cistanche deserticola. Process Biochem. 2005, 40, 3480–3484. [Google Scholar] [CrossRef]
- Wang, J.W.; Wu, J.Y. Effective Elicitors and Process Strategies for Enhancement of Secondary Metabolite Production in Hairy Root Cultures. In Biotechnology of Hairy Root Systems. Advances in Biochemical Engineering/Biotechnology; Doran, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 134, pp. 55–89. [Google Scholar]
- Baldi, A.; Dixit, V.K. Yield enhancement strategies for artemisinin production by suspension cultures of Artemisia annua. Bioresour. Technol. 2008, 99, 4609–4614. [Google Scholar] [CrossRef]
- Zhang, Y.; Turner, J.G. Wound-induced endogenous jasmonates stunt plant growth by inhibiting mitosis. PLoS ONE 2008, 3, e3699. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Yang, Y.; Xie, F.; Wen, J.F.; Wu, J.; Wilson, I.W.; Tang, Q.; Liu, H.; Qiu, D. Deep sequencing reveals transcriptome re-programming of Taxus × media cells to the elicitation with methyl jasmonate. PLoS ONE 2013, 8, e62865. [Google Scholar] [CrossRef]
- Patil, R.A.; Lenka, S.K.; Normanly, J.; Walker, E.L.; Roberts, S.C. Methyl jasmonate represses growth and affects cell cycle progression in cultured Taxus cells. Plant Cell Rep. 2014, 33, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Zare, N.; Farjaminezhad, R.; Asghari-Zakaria, R.; Farjaminezhad, M. Enhanced thebaine production in Papaver bracteatum cell suspension culture by combination of elicitation and precursor feeding. Nat. Prod. Res. 2014, 28, 711–717. [Google Scholar] [CrossRef]
- Wang, M.; Toda, K.; Maeda, H.A. Biochemical properties and subcellular localization of tyrosine aminotransferases in Arabidopsis thaliana. Phytochemistry 2016, 132, 16–25. [Google Scholar] [CrossRef]
- Sivanandhan, G.; Selvaraj, N.; Ganapathi, A.; Manickavasagam, M. Enhanced biosynthesis of withanolides by elicitation and precursor feeding in cell suspension culture of Withania somnifera L. Dunal in shake-flask culture and bioreactor. PLoS ONE 2014, 49, e104005. [Google Scholar] [CrossRef]
- Chu, M.; Pedreño, M.A.; Alburquerque, N.; Faize, L.; Burgos, L.; Almagro, L. A new strategy to enhance the biosynthesis of trans-resveratrol by overexpressing stilbene synthase gene in elicited Vitis vinifera cell cultures. Plant Physiol. Biochem. 2017, 113, 141–148. [Google Scholar] [CrossRef]
- Riewe, D.; Koohi, M.; Lisec, J.; Pfeiffer, M.; Lippmann, R.; Schmeichel, J.; Willmitzer, L.; Altmann, T. A tyrosine aminotransferase involved in tocopherol synthesis in Arabidopsis. Plant J. 2012, 71, 850–859. [Google Scholar] [CrossRef]
- Bedewitz, M.A.; Góngora-Castillo, E.; Uebler, J.B.; Gonzales-Vigil, E.; Wiegert-Rininger, K.E.; Childs, K.L.; Hamilton, J.P.; Vaillancourt, B.; Yeo, Y.-S.; Chappell, J.; et al. A root-expressed L-phenylalanine: 4-hydroxyphenylpyruvate aminotransferase is required for tropane alkaloid biosynthesis in Atropa belladonna. Plant Cell 2014, 26, 3745–3762. [Google Scholar] [CrossRef]
- Schenck, C.A.; Maeda, H.A. Tyrosine biosynthesis, metabolism, and catabolism in plants. Phytochemistry 2018, 149, 82–102. [Google Scholar] [CrossRef]
- Petersen, M.; Simmonds, M.S. Rosmarinic acid. Phytochemistry 2003, 62, 121–125. [Google Scholar] [CrossRef]
- Ru, M.; Wang, K.; Bai, Z.; Peng, L.; He, S.; Wang, Y.; Liang, Z. A tyrosine aminotransferase involved in rosmarinic acid biosynthesis in Prunella vulgaris L. Sci. Rep. 2017, 7, 4892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Ma, L.Q.; Yu, H.S.; Zhang, H.; Wang, H.T.; Qin, Y.F.; Shi, G.L.; Wang, Y.N. A tyrosine decarboxylase catalyzes the initial reaction of the salidroside biosynthesis pathway in Rhodiola sachalinensis. Plant Cell Rep. 2011, 30, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Widhalm, J.R.; Qian, Y.; Maeda, H.; Cooper, B.R.; Jannasch, A.S.; Gonda, I.; Lewinsohn, E.; Rhodes, D.; Dudareva, N. An alternative pathway contributes to phenylalanine biosynthesis in plants via a cytosolic tyrosine:phenylpyruvate aminotransferase. Nat. Commun. 2013, 4, 2833. [Google Scholar] [CrossRef]
- Wong, C.C.; Li, H.B.; Cheng, K.W.; Chen, F. A systematic survey of antioxidant activity of 30 Chinese medicinal plants using the ferric reducing antioxidant power assay. Food Chem. 2006, 97, 705–711. [Google Scholar] [CrossRef]
- Dudonne, S.; Vitrac, X.; Coutiere, P.; Woillez, M.; Merillon, J.M. Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef]
- Moudache, M.; Nerín, C.; Colon, M.; Zaidi, F. Antioxidant effect of an innovative active plastic film containing olive leaves extract on fresh pork meat and its evaluation by Raman spectroscopy. Food Chem. 2017, 229, 98–103. [Google Scholar] [CrossRef]
- Ashrafi, A.; Jokar, M.; Nafchi, A.M. Preparation and characterization of biocomposite film based on chitosan and kombucha tea as active food packaging. Int. J. Biol. Macromol. 2018, 108, 444–454. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Lata, H.; Chandra, S.; Khan, I.; ElSohly, M.A. Thidiazuron-induced high-frequency direct shoot organogenesis of Cannabis sativa L. In Vitro Cell Dev. Biol. Plant. 2009, 45, 12–19. [Google Scholar] [CrossRef]
- Blom, T.J.M.; Kreis, W.; Van Iren, F.; Libbenga, K.R. A non-invasive method for the routine-estimation of fresh weight of cells grown in batch suspension cultures. Plant Cell Rep. 1992, 11, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Iriti, M.; Sironi, M.; Gomarasca, S.; Casazza, A.P.; Soave, C.; Faoro, F. Cell death-mediated antiviral effect of chitosan in tobacco. Plant Physiol. Biochem. 2006, 44, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomics analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, E.A.; Gillespie, K.M. Estimation of total phenolic content and other oxidation substrates in plant tissues using Folin-Ciocalteu reagent. Nat. Protoc. 2007, 2, 875–877. [Google Scholar] [CrossRef]
- Cheng, Z.; Moore, J.; Yu, L. High-throughput relative DPPH radical scavenging capacity assay. J. Agric. Food Chem. 2006, 54, 7429–7436. [Google Scholar] [CrossRef]
- Frassinetti, S.; Gabriele, M.; Caltavuturo, L.; Longo, V.; Pucci, L. Antimutagenic and antioxidant activity of a selected lectin-free common bean (Phaseolus vulgaris L.) in two cell-based models. Plant Foods Hum. Nutr. 2015, 70, 35–41. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef]
- Mikstacka, R.; Rimando, A.M.; Ignatowicz, E. Antioxidant effect of trans-resveratrol, pterostilbene, quercetin and their combinations in human erythrocytes in vitro. Plant Foods Hum. Nutr. 2010, 65, 57–63. [Google Scholar] [CrossRef]
- Lopukhina, A.; Dettenberg, M.; Weiler, E.W.; Hollander-Czytko, H. Cloning and characterization of a coronatine-regulated tyrosine aminotransferase from Arabidopsis. Plant Physiol. 2001, 126, 1678–1687. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.M.; Boubakir, Z.; Ambrose, S.J.; Purves, R.W.; Page, J.E. The hexanoyl-CoA precursor for cannabinoid biosynthesis is formed by an acyl-activating enzyme in Cannabis sativa trichomes. Plant J. 2012, 71, 353–365. [Google Scholar] [CrossRef]
Sample Availability: Samples of the Cannabis sativa extracts are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ct (µg/mg DW) | 4 d MeJA + Tyr (µg/mg DW) | Fold Increase |
|---|---|---|---|
| 4-HPP | 14.47 | 41.08 | 2.84 |
| 4-HPL | 5.05 | 13.37 | 2.65 |
| Tyrosol | 0.66 | 2.19 | 3.32 |
| Tyramine | 1.02 | 1.80 | 1.77 |
| Sample | PAL Specific Activity (nmol mg−1 min−1) | TAT Specific Activity (nmol mg−1 min−1) |
|---|---|---|
| Ct | 3.25 ± 0.80 | 0.58 ± 0.06 |
| 1 d MeJA | 5.87 ± 0.71 | 0.86 ± 0.11 |
| 4 d MeJA | 7.26 ± 0.33 | 1.81 ± 0.22 |
| 1 d MeJA + Phe | 6.15 ± 0.20 | 1.30 ± 0.14 |
| 4 d MeJA + Phe | 7.79 ± 0.39 | 2.59 ± 0.13 |
| 1 d MeJA + Tyr | 8.90 ± 0.33 | 2.16 ± 0.24 |
| 4 d MeJA + Tyr | 11.83 ± 0.75 | 4.80 ± 0.43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabotti, D.; Locatelli, F.; Cusano, E.; Baldoni, E.; Genga, A.; Pucci, L.; Consonni, R.; Mattana, M. Cell Suspensions of Cannabis sativa (var. Futura): Effect of Elicitation on Metabolite Content and Antioxidant Activity. Molecules 2019, 24, 4056. https://doi.org/10.3390/molecules24224056
Gabotti D, Locatelli F, Cusano E, Baldoni E, Genga A, Pucci L, Consonni R, Mattana M. Cell Suspensions of Cannabis sativa (var. Futura): Effect of Elicitation on Metabolite Content and Antioxidant Activity. Molecules. 2019; 24(22):4056. https://doi.org/10.3390/molecules24224056
Chicago/Turabian StyleGabotti, Damiano, Franca Locatelli, Erica Cusano, Elena Baldoni, Annamaria Genga, Laura Pucci, Roberto Consonni, and Monica Mattana. 2019. "Cell Suspensions of Cannabis sativa (var. Futura): Effect of Elicitation on Metabolite Content and Antioxidant Activity" Molecules 24, no. 22: 4056. https://doi.org/10.3390/molecules24224056
APA StyleGabotti, D., Locatelli, F., Cusano, E., Baldoni, E., Genga, A., Pucci, L., Consonni, R., & Mattana, M. (2019). Cell Suspensions of Cannabis sativa (var. Futura): Effect of Elicitation on Metabolite Content and Antioxidant Activity. Molecules, 24(22), 4056. https://doi.org/10.3390/molecules24224056