Anti-Inflammatory, Anti-Diabetic, and Anti-Alzheimer’s Effects of Prenylated Flavonoids from Okinawa Propolis: An Investigation by Experimental and Computational Studies

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
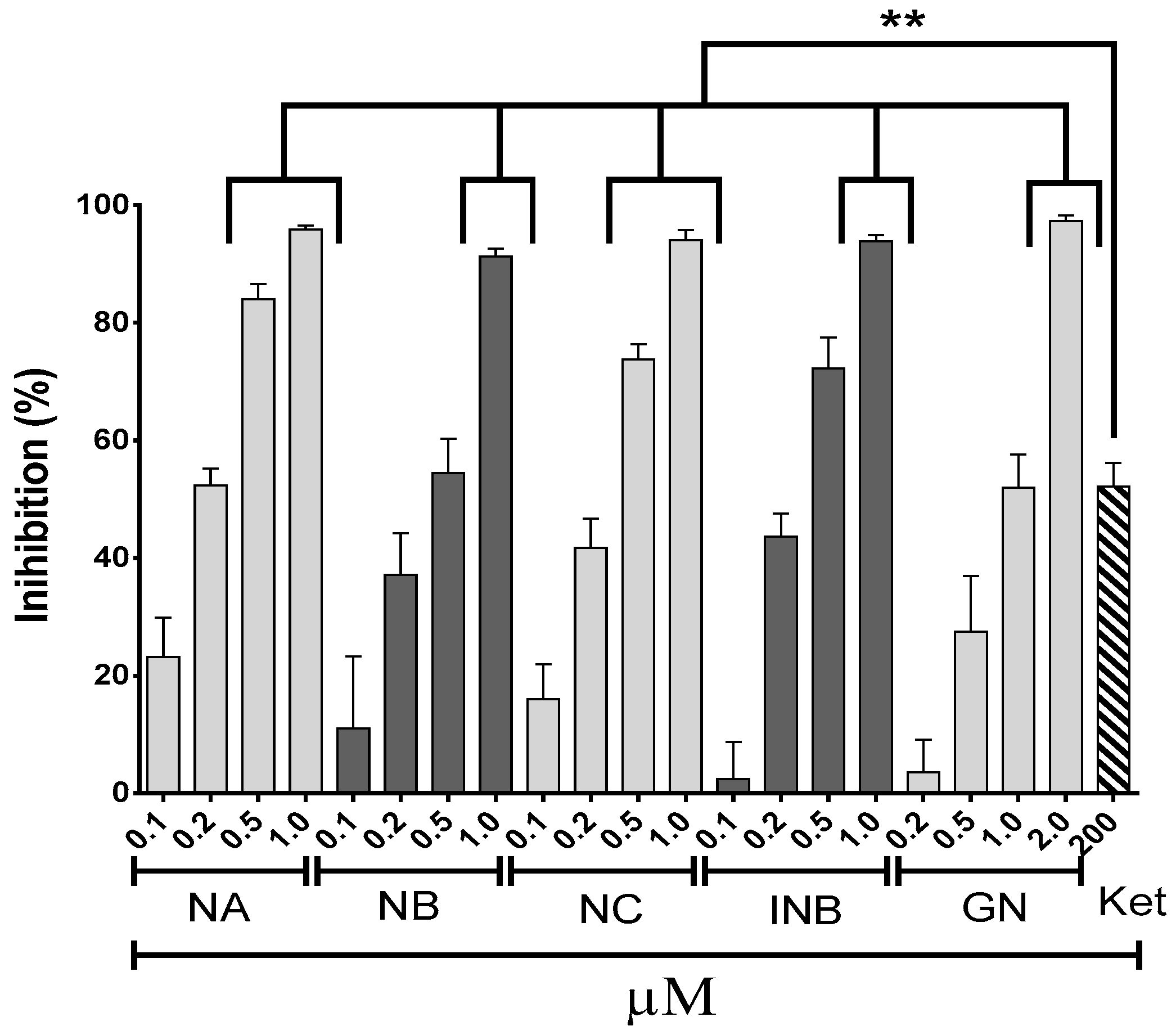
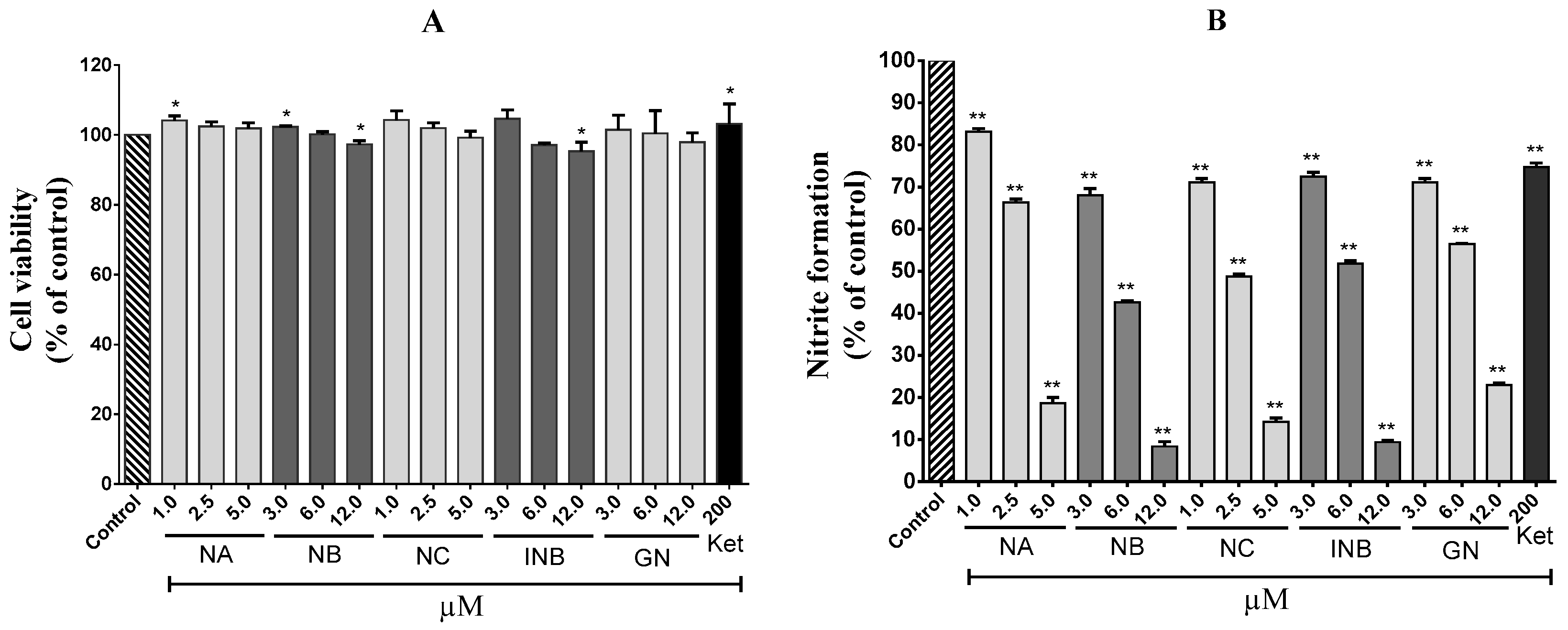
2.1. Anti-Inflammatory Effects
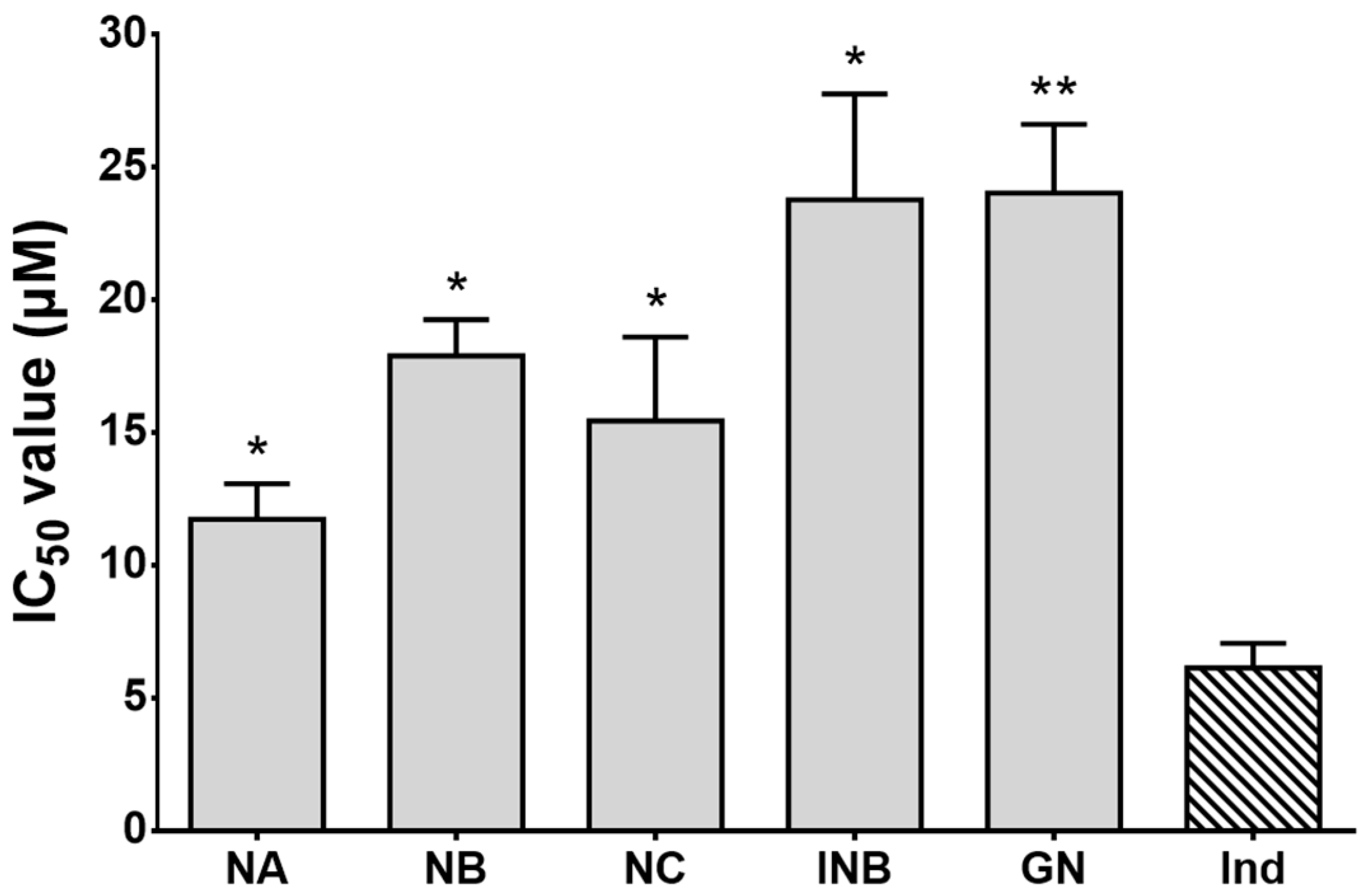
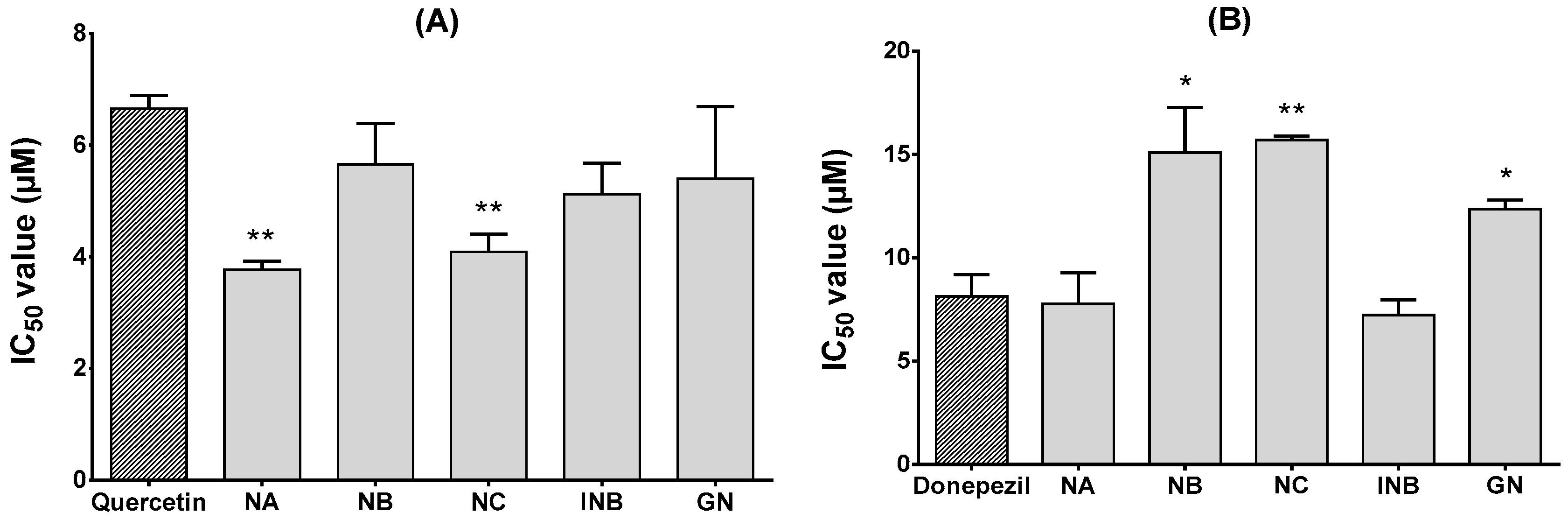
2.2. α-Glucosidase and Acetylcholinesterase (AChE) Inhibitory Effects
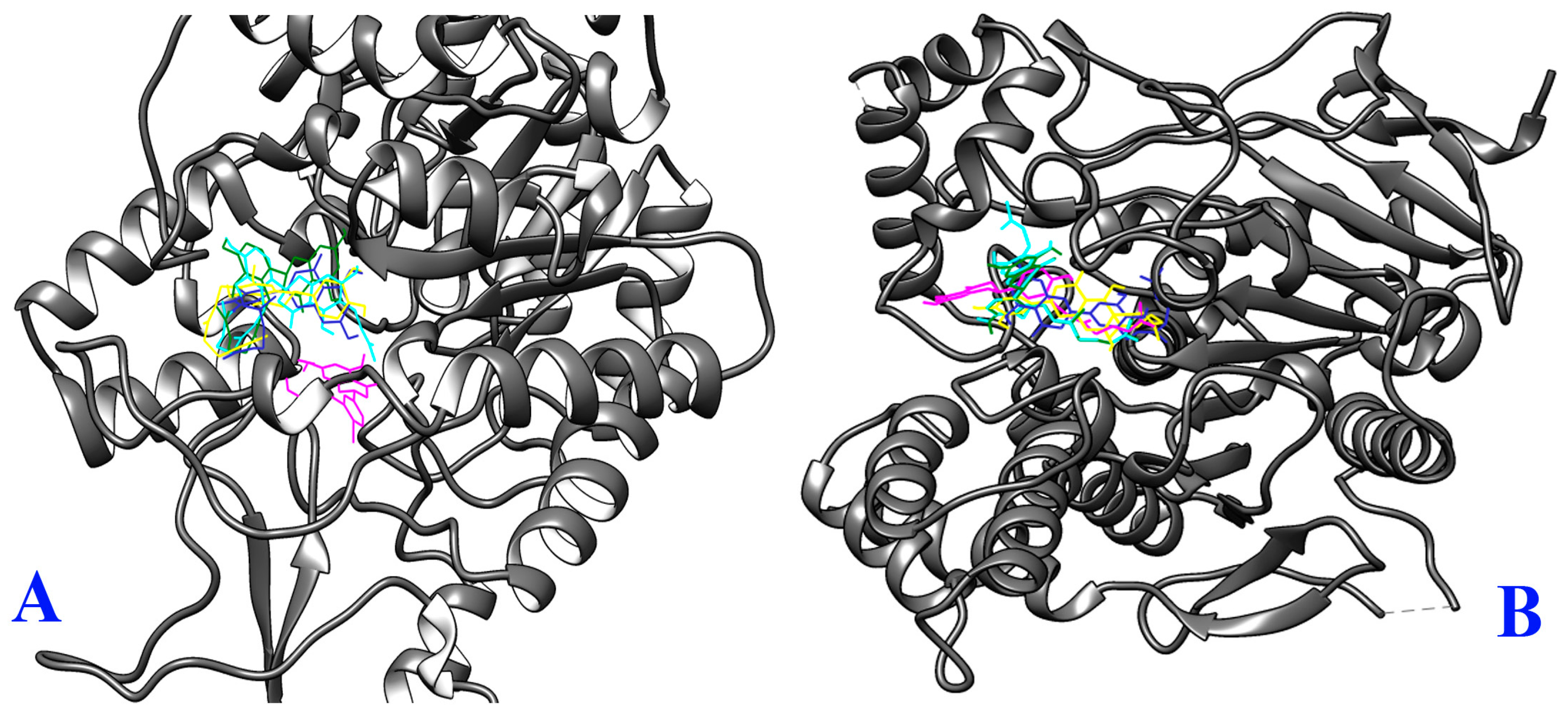
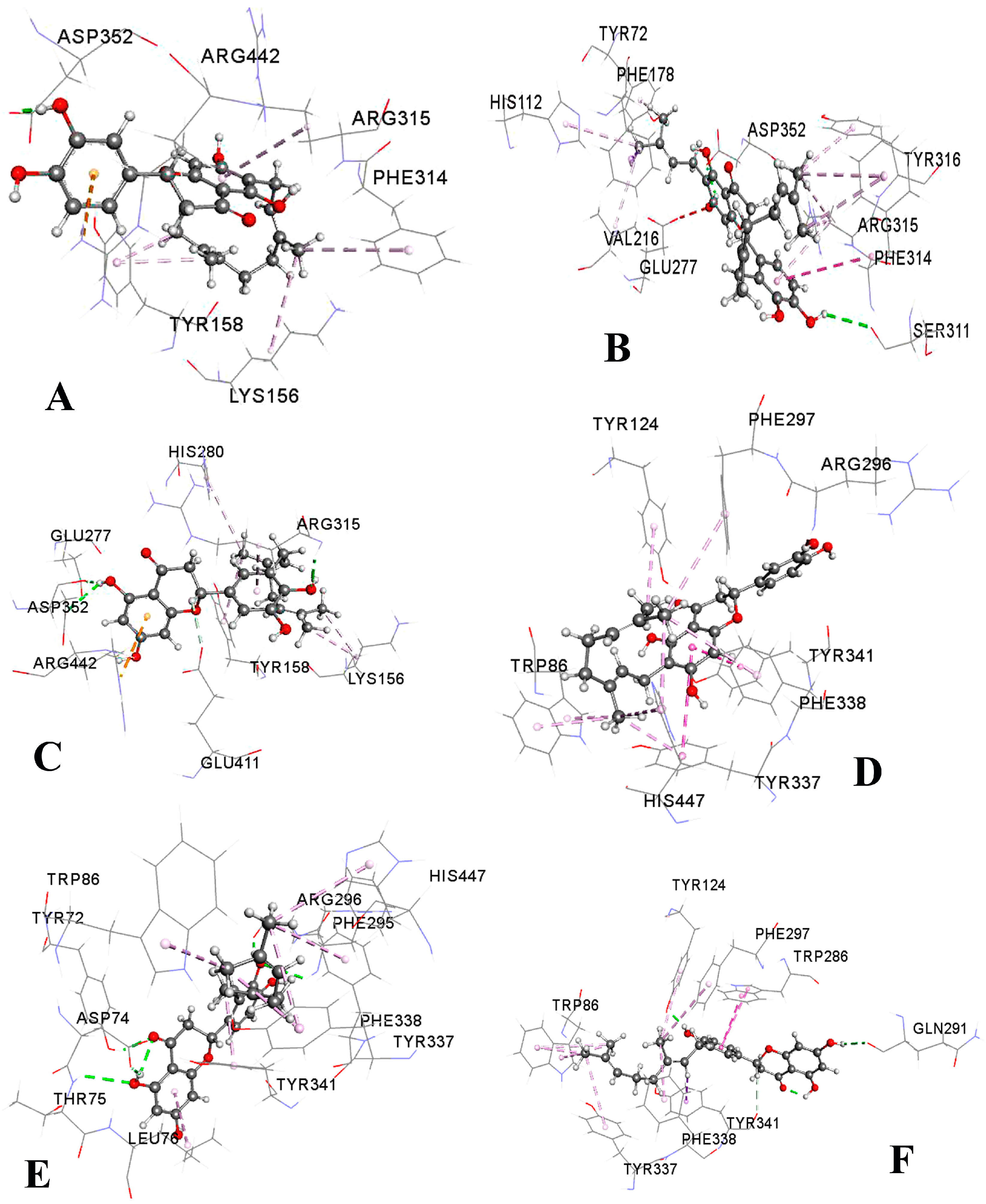
2.3. Molecular Docking Study
2.4. Pharmacokinetic and Toxicological Properties
2.5. Density Functional Theory (DFT)-Based Computations
3. Materials and Methods
3.1. Chemicals and Reagents
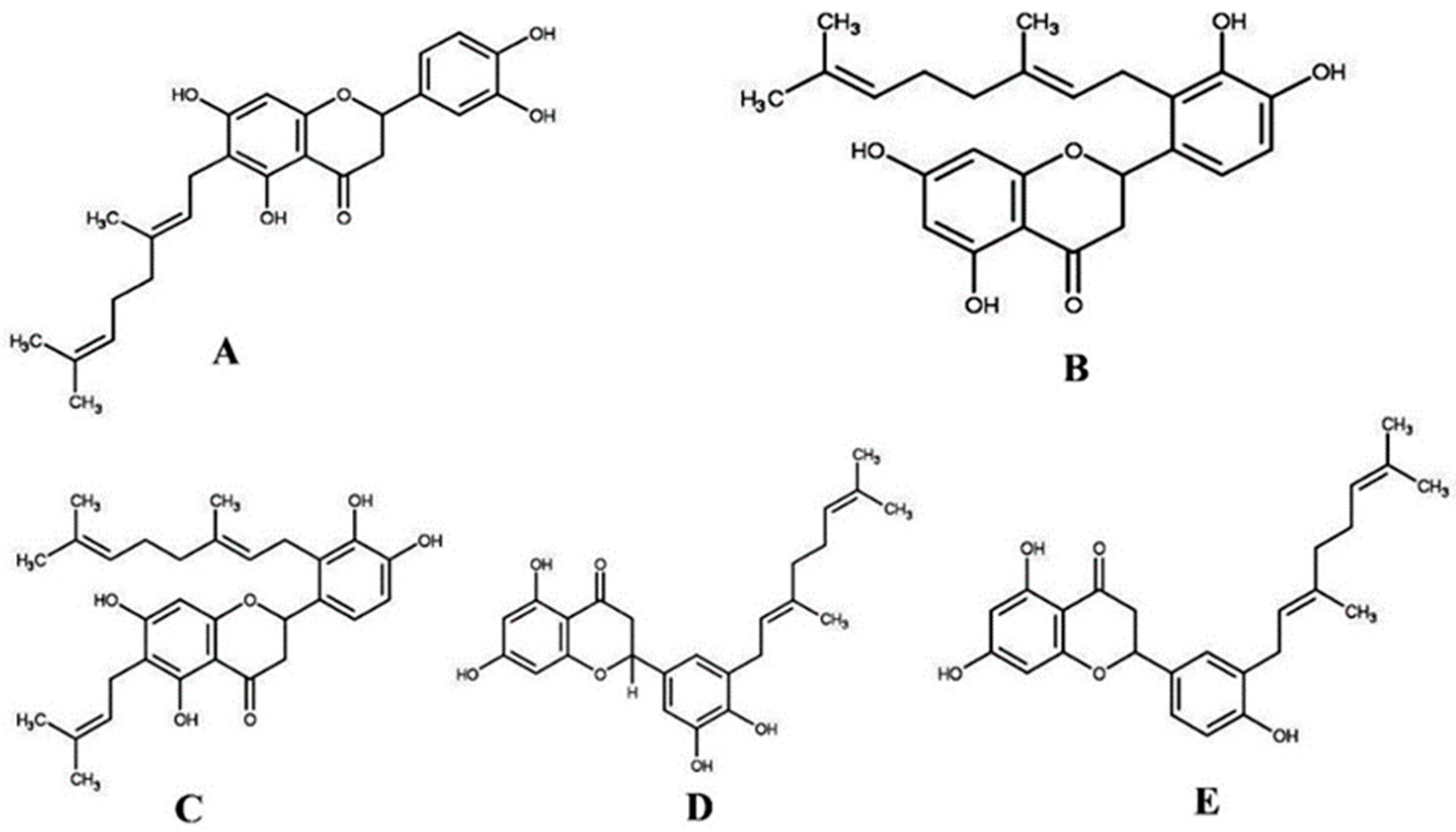
3.2. Sample Collection and Purification of Compounds
3.3. Albumin Denaturation Inhibition Assay
3.4. Cell Viability Assay
3.5. Nitrite Inhibition Assay
3.6. Cell-Based Assay for COX-2 Inhibition
3.7. α-Glucosidase Inhibition Assay
3.8. Acetylcholinesterase Inhibition Assay
3.9. Molecular Docking
3.9.1. Preparation of Receptors
3.9.2. Preparation of Ligands
3.9.3. Docking Simulation
3.9.4. Analysis and Visualization of Docking Results
3.10. Calculation of Pharmacokinetic Parameters
3.11. Prediction of Toxicological Properties
3.12. Density Functional Theory (DFT) Calculation
S = 1/η.
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, J. Vitamin D: Anti-inflammatory effects to prevent and treat diseases. In Vitamin D: Oxidative Stress, Immunity, and Aging; Gombart, A.F., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 307–316. [Google Scholar]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diabetes Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Moreira, P.I. Alzheimer’s disease and diabetes: An integrative view of the role of mitochondria, oxidative stress, and insulin. J. Alzheimers Dis. 2012, 30, S199–S215. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Islam, A.; Haryo Sasongko, T.; Hua Gan, S. Type 2 diabetes mellitus and Alzheimer’s disease: Bridging the pathophysiology and management. Curr. Pharm. Des. 2016, 22, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar] [PubMed]
- Gasparini, L.; Netzer, W.J.; Greengard, P.; Xu, H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol. Sci. 2002, 23, 288–293. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Gorjao, R.; Vinolo, M.A.; Rodrigues, A.C.; Nachbar, R.T.; Curi, R. Molecular targets related to inflammation and insulin resistance and potential interventions. BioMed Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Commentary on “a roadmap for the prevention of dementia: The inaugural Leon Thal Symposium.” An impending prevention clinical trial for Alzheimer’s disease: Roadmaps and realities. Alzheimers Dement. 2008, 4, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Taira, N.; Nguyen, B.C.; Be Tu, P.T.; Tawata, S. Effect of Okinawa propolis on PAK1 activity, Caenorhabditis elegans longevity, melanogenesis, and growth of cancer cells. J. Agric. Food Chem. 2016, 64, 5484–5489. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, S.; Murase, M.; Momose, N.; Fukumoto, S. Analysis of antioxidant prenylflavonoids in different parts of Macaranga tanarius, the plant origin of Okinawan propolis. Asian Pac. J. Trop. Med. 2014, 7, 16–20. [Google Scholar] [CrossRef]
- Kumazawa, S.; Ueda, R.; Hamasaka, T.; Fukumoto, S.; Fujimoto, T.; Nakayama, T. Antioxidant prenylated flavonoids from propolis collected in Okinawa, Japan. J. Agric. Food Chem. 2007, 55, 7722–7725. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.C.Q.; Yoshimura, K.; Kumazawa, S.; Tawata, S.; Maruta, H. Frondoside A from sea cucumber and nymphaeols from Okinawa propolis: Natural anti-cancer agents that selectively inhibit PAK1 in vitro. Drug Discov. Ther. 2017, 11, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Maruta, H. Herbal therapeutics that block the oncogenic kinase PAK1: A practical approach towards PAK1-dependent diseases and longevity. Phytother. Res. 2014, 28, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Khan, R.; Khan, H.; Tokuda, H. Cytotoxic, antitumour-promoting and inhibition of protein denaturation effects of flavonoids, isolated from Potentilla evestita Th. Wolf. Nat. Prod. Res. 2015, 29, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Shin, E.M.; Guo, L.Y.; Youn, U.J.; Bae, K.; Kang, S.S.; Zou, L.B.; Kim, Y.S. Anti-inflammatory activity of 4-methoxyhonokiol is a function of the inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages via NF-kappaB, JNK and p38 MAPK inactivation. Eur. J. Pharmacol. 2008, 586, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Kacem, M.; Simon, G.; Leschiera, R.; Misery, L.; ElFeki, A.; Lebonvallet, N. Antioxidant and anti-inflammatory effects of Ruta chalepensis L. extracts on LPS-stimulated RAW 264.7 cells. In Vitro Cell Dev. Biol. Anim. 2015, 51, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.C.Q.; Takahashi, H.; Uto, Y.; Shahinozzaman, M.D.; Tawata, S.; Maruta, H. 1,2,3-Triazolyl ester of Ketorolac: A“click chemistry”-based highly potent PAK1-blocking cancer-killer. Eur. J. Med. Chem. 2017, 126, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Park, E.H.; Hur, G.M.; Ryu, Y.S.; Lee, Y.S.; Lee, J.Y.; Kim, Y.M.; Jin, C. Caffeic acid phenethyl ester inhibits nitric oxide synthase gene expression and enzyme activity. Cancer Lett. 2002, 175, 53–66. [Google Scholar] [CrossRef]
- Kasturi, S.; Surarapu, S.; Uppalanchi, S.; Anireddy, J.S.; Dwivedi, S.; Anantaraju, H.S.; Perumal, Y.; Sigalapalli, D.K.; Babu, B.N.; Ethiraj, K.S. Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorg. Med. Chem. Lett. 2017, 27, 2818–2823. [Google Scholar] [CrossRef] [PubMed]
- Benalla, W.; Bellahcen, S.; Bnouham, M. Antidiabetic medicinal plants as a source of alpha glucosidase inhibitors. Curr. Diabetes Rev. 2010, 6, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.P.; Carvalho, L.R.; Young, M.C.; Cardoso-Lopes, E.M.; Centeno, D.C.; Zambotti-Villela, L.; Colepicolo, P.; Yokoya, N.S. Evaluation of acetylcholinesterase inhibitory activity of Brazilian red macroalgae organic extracts. Rev. Bras. Farmacogn. 2015, 25, 657–662. [Google Scholar] [CrossRef]
- Khan, H.; Amin, S.; Kamal, M.A.; Patel, S. Flavonoids as acetylcholinesterase inhibitors: Current therapeutic standing and future prospects. Biomed. Pharmacother. 2018, 101, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Amer, A.M.; Abulifa, A.R.; Elzwawi, M.M. Ginger components as new leads for the design and development of novel multi-targeted anti-Alzheimer’s drugs: A computational investigation. Drug Des. Dev. Ther. 2014, 8, 2045. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Mohamed, N.; Alhussen, F. Molecular interaction studies of green tea catechins as multitarget drug candidates for the treatment of Parkinson’s disease: Computational and structural insights. Netw. Comput. Neural Syst. 2016, 26, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Wang, P.; Ashok, P.; Yang, L.M.; Zheng, Y.T.; Murugesan, S. Rational design, synthesis, anti-HIV-1 RT and antimicrobial activity of novel 3-(6-methoxy-3, 4-dihydroquinolin-1 (2H)-yl)-1-(piperazin-1-yl) propan-1-one derivatives. Bioorg. Chem. 2016, 67, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Sulpizi, M.; Folkers, G.; Rothlisberger, U.; Carloni, P.; Scapozza, L. Applications of density functional theory-based methods in medicinal chemistry. Quant. Struct.-Act. Relat. 2002, 21, 173–181. [Google Scholar] [CrossRef]
- Lien, E.J.; Guo, Z.R.; Li, R.L.; Su, C.T. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J. Pharm. Sci. 1982, 71, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.M.; Shawon, J.; Halim, M.A. Multiple receptor conformers based molecular docking study of fluorine enhanced ethionamide with mycobacterium enoyl ACP reductase (InhA). J. Mol. Graph. Model. 2017, 77, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn. 2017, 36, 2099–2117. [Google Scholar] [CrossRef] [PubMed]
- Osman, N.I.; Sidik, N.J.; Awal, A.; Adam, N.A.; Rezali, N.I. In vitro xanthine oxidase and albumin denaturation inhibition assay of Barringtonia racemosa L. and total phenolic content analysis for potential anti-inflammatory use in gouty arthritis. J. Intercult. Ethnopharmacol. 2016, 5, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Shahinozzaman, M.; Ishii, T.; Takano, R.; Halim, M.; Hossain, M.; Tawata, S. Cytotoxic desulfated saponin from Holothuria atra predicted to have high binding affinity to the oncogenic kinase PAK1: A combined in vitro and in silico study. Sci. Pharm. 2018, 86, 32. [Google Scholar] [CrossRef] [PubMed]
- Stanikunaite, R.; Khan, S.I.; Trappe, J.M.; Ross, S.A. Cyclooxygenase-2 inhibitory and antioxidant compounds from the truffle Elaphomyces granulatus. Phytother. Res. 2009, 23, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Afrapoli, F.M.; Asghari, B.; Saeidnia, S.; Ajani, Y.; Mirjani, M.; Malmir, M.; Baza, R.D.; Hadjiakhoondi, A.; Salehi, P.; Hamburger, M.; et al. In vitro α-glucosidase inhibitory activity of phenolic constituents from aerial parts of Polygonum hyrcanicum. J. Pharm. Sci. 2012, 20, 1–6. [Google Scholar]
- Fawole, O.A.; Amoo, S.O.; Ndhlala, A.R.; Light, M.E.; Finnie, J.F.; Van Staden, J. Antiinflammatory, anticholinesterase, antioxidant and phytochemical properties of medicinal plants used for pain-related ailments in South Africa. J. Ethnopharmacol. 2010, 127, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Taha, M.; Shah, S.A.; Afifi, M.; Imran, S.; Sultan, S.; Rahim, F.; Khan, K.M. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 2018, 77, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision A. 02; Gaussian. Inc.: Wallingford, CT, USA, 2009; p. 200. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Nitrite Inhibition (IC50 Value, µM) |
|---|---|
| NA | 3.2 |
| NB | 5.4 |
| NC | 2.4 |
| INB | 6.2 |
| GN | 7.0 |
| Compound | Binding Affinity (kcal/mol) | Hydrogen Bonds | Hydrophobic Bonds | Electrostatic Bonds | ||||
|---|---|---|---|---|---|---|---|---|
| π–σ | π–π Stacked | π–Alkyl | Alkyl | Amide–π Stacked | ||||
| 3A4A | ||||||||
| NA | −9.3 | Asp352 (1.92) | Tyr158 (3.56) | Arg315 (4.88) Tyr158 (4.11) Phe314 (4.97) | Lys156 (5.05) | Arg442 (4.67) | ||
| NB | −8.7 | Gln353 (2.20) Arg315 (3.50) | Arg315 (3.98) | Phe303 (4.62) | Tyr158 (4.36) Phe314 (5.10) Phe314 (4.89) | Arg315 (5.18) Arg315 (4.42) | ||
| NC | −9.9 | Ser311 (2.82) Asp352 (2.17) | Phe178 (3.71) | Arg315 (4.51) Tyr72 (4.51) His112 (4.90) Phe314 (5.00) Phe314 (4.96) Tyr316 (5.02) | Arg315 (4.36) Arg315 (4.37) Val216 (4.33) | Phe314 (5.83) Arg315 (5.83) | ||
| INB | −9.2 | Glu277 (2.24) Asp352 (2.60) Arg315 (2.21) | Arg315 (3.85) Tyr158 (4.73) His280 (5.32) | Lys156 (3.47) Lys156 (4.03) | Arg442 (4.07) | |||
| GN | −7.1 | Lys466 (2.44) Pro467 (2.31) Trp36 (2.19) | Trp36 (5.38) | Phe469 (4.97) Tyr470 (4.63) | ||||
| 4EY7 | ||||||||
| NA | −11.5 | Trp86 (3.97) | Tyr337 (5.41) Tyr341 (4.00) | Trp86 (4.05) Tyr124 (4.93) Phe297 (5.23) Tyr337 (3.95) Phe338 (4.79) His447 (5.43) His447 (4.46) | ||||
| NB | −11.2 | Asp74 (2.02) Arg296 (1.86) Tyr72 (2.05) Thr75 (2.77) Phe295 (2.77) | Tyr341 (3.67) | Leu76 (4.61) Trp86 (4.57) Tyr337 (4.02) Tyr337 (4.52) Phe338 (5.36) His447 (4.60) | ||||
| NC | −11.0 | Ser293 (2.17) Tyr72 (2.07) Tyr72 (2.02) Phe295 (2.69) Arg296 (2.31) Arg296 (2.29) | Tyr341 (3.73) | Leu76 (4.86) Trp86 (4.63) Tyr337 (4.41) Tyr337 (4.02) Phe338 (5.29) His447 (4.57) | ||||
| INB | −11.0 | Asp74 (2.00) Phe295 (1.88) | Trp86 (3.89) | Tyr337 (5.10) Trp286 (4.01) Tyr341 (4.72) Trp286 (5.32) | Trp86 (4.57) Trp86 (5.07) Tyr337 (5.39) His447 (4.71) | |||
| GN | −11.3 | Gln291 (2.29) Tyr124 (1.89) | Tyr341 (3.75) Trp86 (3.78) Trp86 (3.88) Trp86 (3.94) | Trp286 (3.92) Trp286 (4.98) | Trp86 (5.04) Tyr124 (5.35) Phe297 (5.36) Tyr337 (4.91) Phe338 (4.64) | |||
| Compound | % ABS a | TPSA (Å2) b | MW c | MiLogP d | HBD e | HBA f | n-ROTB g | Lipinski’s Violation |
|---|---|---|---|---|---|---|---|---|
| Rule | - | - | ˂500 | ≤5 | ˂5 | ˂10 | ≤10 | ≤1 |
| NA | 72.01 | 107.22 | 424.49 | 5.52 | 4 | 6 | 6 | 1 |
| NB | 72.01 | 107.22 | 424.49 | 5.49 | 4 | 6 | 6 | 1 |
| NC | 72.01 | 107.22 | 492.61 | 7.53 | 4 | 6 | 8 | 1 |
| INB | 72.01 | 107.22 | 424.49 | 5.49 | 4 | 6 | 6 | 1 |
| GN | 78.99 | 86.99 | 408.49 | 6.21 | 3 | 5 | 6 | 1 |
| Parameters | Compound | ||||
|---|---|---|---|---|---|
| NA | NB | NC | INB | GN | |
| Ames toxicity | Non Ames toxic | Non Ames toxic | Non Ames toxic | Non Ames toxic | Non Ames toxic |
| Carcinogens | Non-carcinogenic | Non-carcinogenic | Non-carcinogenic | Non-carcinogenic | Non-carcinogenic |
| Acute oral toxicity | III | III | III | III | III |
| Rat acute toxicity | 3.1399 | 3.1399 | 3.1399 | 3.1399 | 3.1399 |
| hERG a | Weak inhibitor | Weak inhibitor | Weak inhibitor | Weak inhibitor | Weak inhibitor |
| Carcinogenicity (Three-class) | Not required | Not required | Not required | Not required | Not required |
| Compound | Electronic Energy (Hartree) | Enthalpy (Hartree) | Gibbs Free Energy (Hartree) | Dipole Moment (Debye) | εHOMO (Hartree) | εLUMO (Hartree) | Gap (Hartree) | η | S |
|---|---|---|---|---|---|---|---|---|---|
| NA | −1420.38 | −1420.38 | −1420.48 | 3.337 | −0.2097 | −0.0465 | 0.1632 | 0.08161 | 12.2526 |
| NB | −1420.37 | −1420.37 | −1420.47 | 3.168 | −0.2107 | −0.0502 | 0.1605 | 0.08028 | 12.4564 |
| NC | −1615.60 | −1615.60 | −1615.72 | 5.228 | −0.2090 | −0.0489 | 0.1600 | 0.08004 | 12.4929 |
| INB | −1420.38 | −1420.38 | −1420.47 | 2.235 | −0.2135 | −0.0536 | 0.1599 | 0.07995 | 12.5078 |
| GN | −1345.16 | −1345.16 | −1345.25 | 4.959 | −0.2207 | −0.0522 | 0.1684 | 0.08423 | 11.8715 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahinozzaman, M.; Taira, N.; Ishii, T.; Halim, M.A.; Hossain, M.A.; Tawata, S. Anti-Inflammatory, Anti-Diabetic, and Anti-Alzheimer’s Effects of Prenylated Flavonoids from Okinawa Propolis: An Investigation by Experimental and Computational Studies. Molecules 2018, 23, 2479. https://doi.org/10.3390/molecules23102479
Shahinozzaman M, Taira N, Ishii T, Halim MA, Hossain MA, Tawata S. Anti-Inflammatory, Anti-Diabetic, and Anti-Alzheimer’s Effects of Prenylated Flavonoids from Okinawa Propolis: An Investigation by Experimental and Computational Studies. Molecules. 2018; 23(10):2479. https://doi.org/10.3390/molecules23102479
Chicago/Turabian StyleShahinozzaman, Md, Nozomi Taira, Takahiro Ishii, Mohammad A. Halim, Md Amzad Hossain, and Shinkichi Tawata. 2018. "Anti-Inflammatory, Anti-Diabetic, and Anti-Alzheimer’s Effects of Prenylated Flavonoids from Okinawa Propolis: An Investigation by Experimental and Computational Studies" Molecules 23, no. 10: 2479. https://doi.org/10.3390/molecules23102479
APA StyleShahinozzaman, M., Taira, N., Ishii, T., Halim, M. A., Hossain, M. A., & Tawata, S. (2018). Anti-Inflammatory, Anti-Diabetic, and Anti-Alzheimer’s Effects of Prenylated Flavonoids from Okinawa Propolis: An Investigation by Experimental and Computational Studies. Molecules, 23(10), 2479. https://doi.org/10.3390/molecules23102479