Body Fluid-Independent Effects of Dietary Salt Consumption in Chronic Kidney Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Salt Intake in Relation to Renal Function in CKD Patients
3. New Insights in Sodium Homeostasis Set Light on Body Fluid-Independent Effects of High Salt
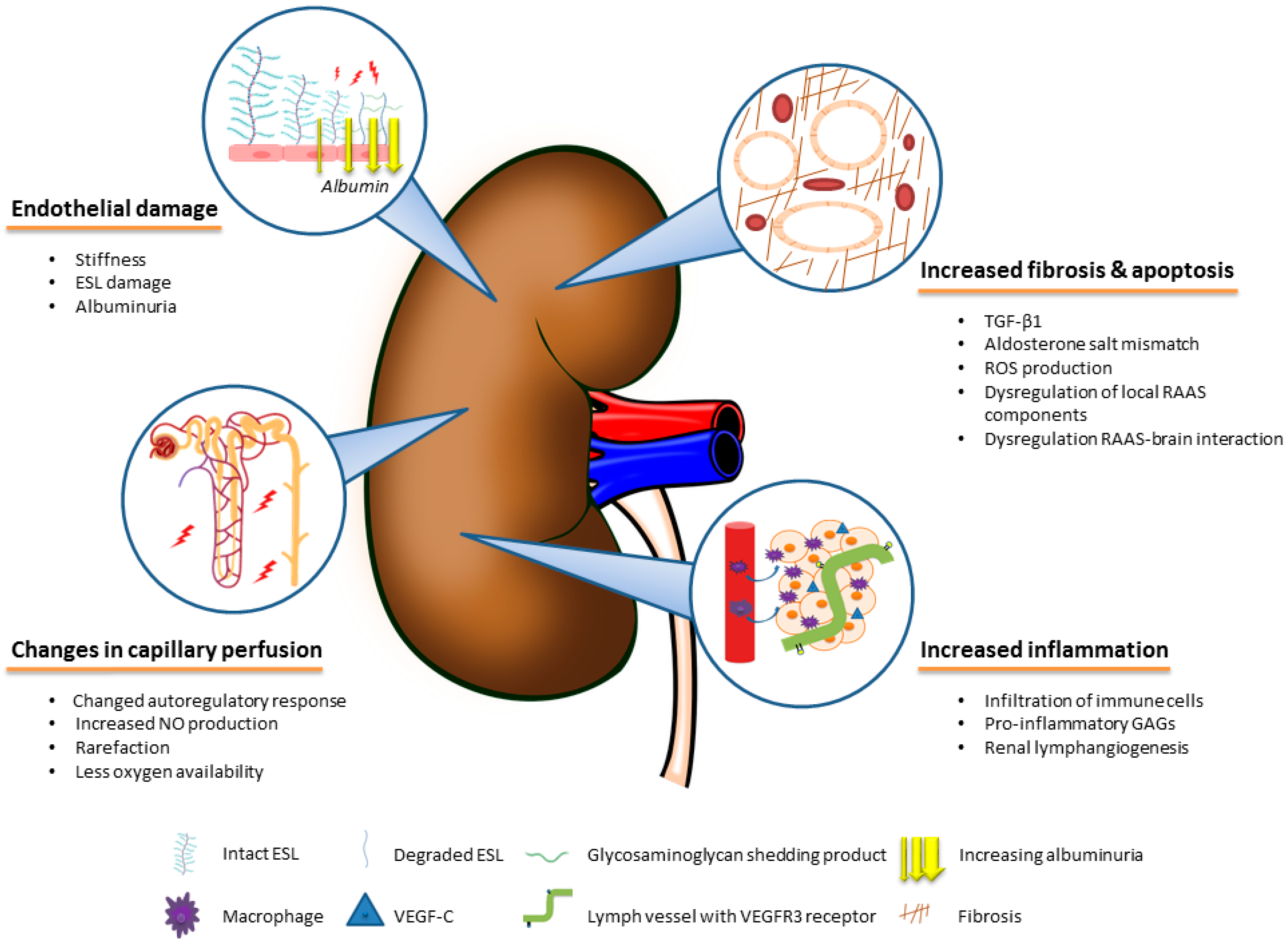
4. The Role of Body Fluid-Independent Effects of High Salt Intake on the Kidney
4.1. Direct Effects of Salt on Fibrotic Pathways in the Kidney
4.2. Effects of Salt on the Renal Vascular Microcirculation and Endothelium
4.3. Inflammatory Effects of Salt
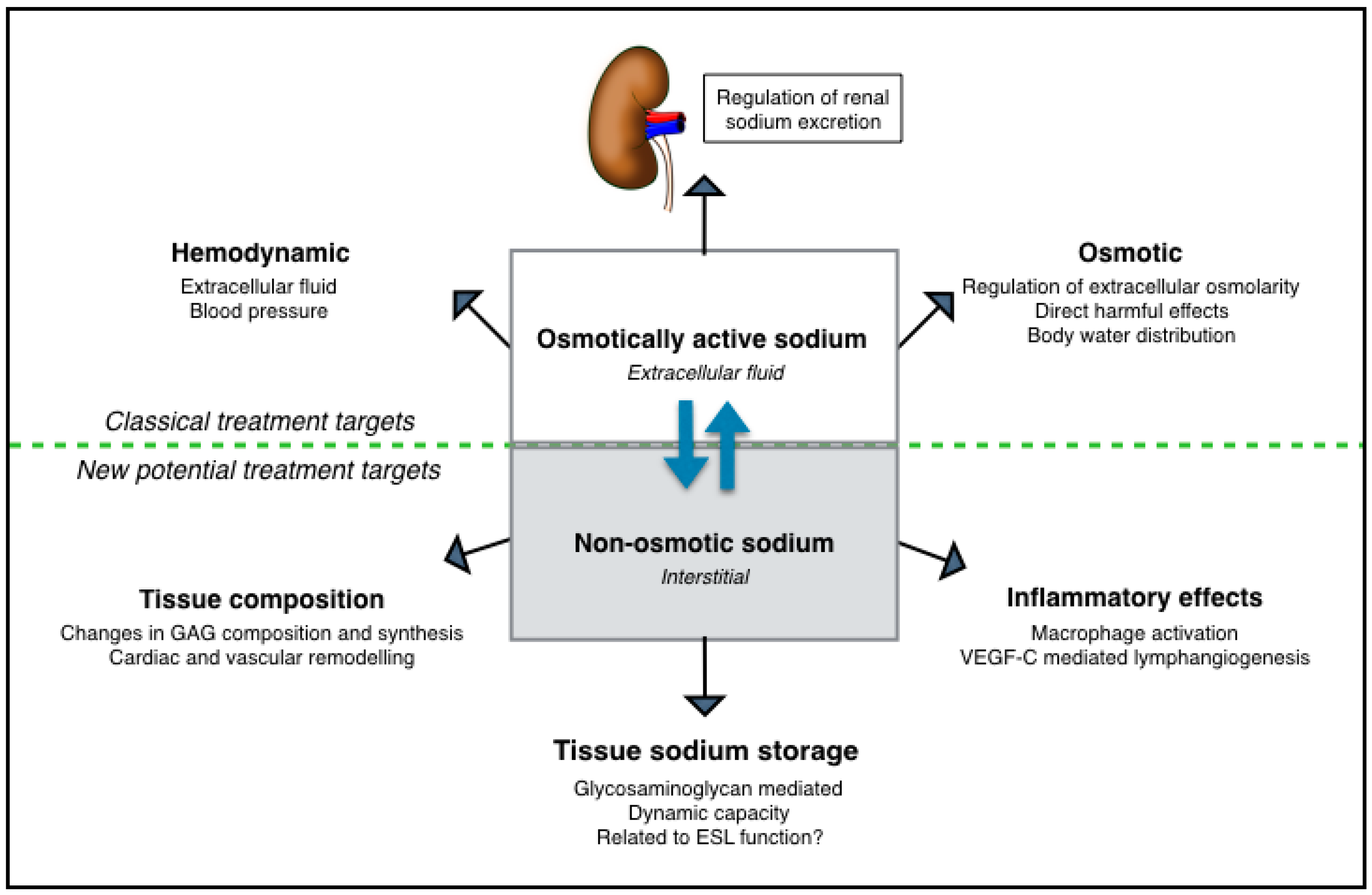
5. Extrarenal Tissue Sodium Storage in CKD
6. Non-Osmotic Sodium Buffering as Potential Treatment Target
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.M.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- McMahon, E.J.; Campbell, K.L.; Bauer, J.D.; Mudge, D.W. Altered dietary salt intake for people with chronic kidney disease. Cochrane Database Syst. Rev. 2015, 18, Cd010070. [Google Scholar] [CrossRef] [PubMed]
- Vogt, L.; Waanders, F. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J. Am. Soc. Nephrol. 2008, 19, 999–1007. [Google Scholar] [CrossRef]
- D’Elia, L.; Rossi, G. Meta-Analysis of the Effect of Dietary Sodium Restriction with or without Concomitant Renin-Angiotensin-Aldosterone System-Inhibiting Treatment on Albuminuria. Clin. J. Am. Soc. Nephrol. 2015, 10, 1542–1552. [Google Scholar] [CrossRef]
- Slagman, M.C.; Waanders, F. Moderate dietary sodium restriction added to angiotensin converting enzyme inhibition compared with dual blockade in lowering proteinuria and blood pressure: Randomised controlled trial. BMJ 2011, 343, d4366. [Google Scholar] [CrossRef]
- Buter, H.; Hemmelder, M.H. The blunting of the antiproteinuric efficacy of ACE inhibition by high sodium intake can be restored by hydrochlorothiazide. Nephrol. Dial Transpl. 1998, 13, 1682–1685. [Google Scholar] [CrossRef]
- Ekinci, E.I.; Thomas, G. Effects of salt supplementation on the albuminuric response to telmisartan with or without hydrochlorothiazide therapy in hypertensive patients with type 2 diabetes are modulated by habitual dietary salt intake. Diabetes Care 2009, 32, 1398–1403. [Google Scholar] [CrossRef]
- Vegter, S.; Perna, A. Sodium Intake, ACE Inhibition, and Progression to ESRD. J. Am. Soc. Nephrol. 2012, 23, 165. [Google Scholar] [CrossRef]
- Guyton, A.C.; Coleman, T.G. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am. J. Med. 1972, 52, 584–594. [Google Scholar] [CrossRef]
- Borst, J.G.; Borst-De Geus, A. Hypertension explained by Starling’s theory of circulatory homoeostasis. Lancet 1963, 1, 677–682. [Google Scholar] [CrossRef]
- Zhang, W.C.; Zheng, X.J. High salt primes a specific activation state of macrophages, M(Na). Cell Res. 2015, 25, 893–910. [Google Scholar] [CrossRef]
- Hijmans, R.S.; van Londen, M. Dermal tissue remodeling and non-osmotic sodium storage in kidney patients. J. Transl. Med. 2019, 17, 88. [Google Scholar] [CrossRef]
- Kempner, W. Treatment of heart and kidney disease and of hypertensive and arteriosclerotic vascular disease with the rice diet. Ann. Intern. Med. 1949, 31, 821–856. [Google Scholar] [CrossRef]
- Garofalo, C.; Borrelli, S. Dietary Salt Restriction in Chronic Kidney Disease: A Meta-Analysis of Randomized Clinical Trials. Nutrients 2018, 10, 732. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Lambers Heerspink, H.J. Microalbuminuria: Target for renoprotective therapy PRO. Kidney Int. 2014, 86, 40–49. [Google Scholar] [CrossRef]
- He, J.; Mills, K.T. Urinary Sodium and Potassium Excretion and CKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1202–1212. [Google Scholar] [CrossRef]
- Mills, K.T.; Chen, J. Sodium Excretion and the Risk of Cardiovascular Disease in Patients With Chronic Kidney Disease. JAMA 2016, 315, 2200–2210. [Google Scholar] [CrossRef]
- Mente, A.; O’Donnell, M. Urinary sodium excretion, blood pressure, cardiovascular disease, and mortality: A community-level prospective epidemiological cohort study. Lancet 2018, 392, 496–506. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.G.; van den Hoek, T.C. Use of a Single Baseline Versus Multiyear 24-Hour Urine Collection for Estimation of Long-Term Sodium Intake and Associated Cardiovascular and Renal Risk. Circulation 2017, 136, 917–926. [Google Scholar] [CrossRef]
- He, F.J.; Markandu, N.D. Plasma sodium: Ignored and underestimated. Hypertension 2005, 45, 98–102. [Google Scholar] [CrossRef]
- Kuwabara, M.; Hisatome, I. Increased Serum Sodium and Serum Osmolarity Are Independent Risk Factors for Developing Chronic Kidney Disease; 5 Year Cohort Study. PLoS ONE 2017, 12, e0169137. [Google Scholar] [CrossRef]
- Titze, J.; Maillet, A. Long-term sodium balance in humans in a terrestrial space station simulation study. Am. J. Kidney Dis. 2002, 40, 508–516. [Google Scholar] [CrossRef]
- Rakova, N.; Juttner, K. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab. 2013, 17, 125–131. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.; Rorije, N.M. Quantification of nonosmotic sodium storage capacity following acute hypertonic saline infusion in healthy individuals. Kidney Int. 2017, 91, 738–745. [Google Scholar] [CrossRef]
- Wouda, R.D.; Dekker, S.E.I. Effects of Water Loading on Observed and Predicted Plasma Sodium, and Fluid and Urine Cation Excretion in Healthy Individuals. Am. J. Kidney Dis. 2019, 74, 320–327. [Google Scholar] [CrossRef]
- Machnik, A.; Neuhofer, W. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Wiig, H.; Schroder, A. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Investig. 2013, 123, 2803–2815. [Google Scholar] [CrossRef]
- Hammon, M.; Grossmann, S. 23Na Magnetic Resonance Imaging of the Lower Leg of Acute Heart Failure Patients during Diuretic Treatment. PLoS ONE 2015, 10, e0141336. [Google Scholar] [CrossRef]
- Dahlmann, A.; Dorfelt, K. Magnetic resonance-determined sodium removal from tissue stores in hemodialysis patients. Kidney Int. 2015, 87, 434–441. [Google Scholar] [CrossRef]
- Kopp, C.; Linz, P. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef]
- Helle, F.; Karlsen, T.V. High-salt diet increases hormonal sensitivity in skin pre-capillary resistance vessels. Acta Physiol. (Oxf.) 2013, 207, 577–581. [Google Scholar] [CrossRef]
- Schmidlin, O.; Forman, A. Salt sensitivity in blacks: Evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension 2011, 58, 380–385. [Google Scholar] [CrossRef]
- Ganguli, M.; Tobian, L.; Iwai, J. Cardiac output and peripheral resistance in strains of rats sensitive and resistant to NaCl hypertension. Hypertension 1979, 1, 3–7. [Google Scholar] [CrossRef]
- Titze, J.; Lang, R. Osmotically inactive skin Na+ storage in rats. Am. J. Physiol. Ren. Physiol. 2003, 285, F1108–F1117. [Google Scholar] [CrossRef]
- Titze, J.; Shakibaei, M. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H203–H208. [Google Scholar] [CrossRef]
- Schafflhuber, M.; Volpi, N. Mobilization of osmotically inactive Na+ by growth and by dietary salt restriction in rats. Am. J. Physiol. Ren. Physiol. 2007, 292, F1490–F1500. [Google Scholar] [CrossRef]
- Titze, J. Sodium balance is not just a renal affair. Curr. Opin. Nephrol. Hypertens. 2014, 23, 101–105. [Google Scholar] [CrossRef]
- Fischereder, M.; Michalke, B. Sodium storage in human tissues is mediated by glycosaminoglycan expression. Am. J. Physiol. Ren. Physiol. 2017, 313, F319–F325. [Google Scholar] [CrossRef]
- Yi, B.; Titze, J. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: A longitudinal study. Transl. Res. 2015, 166, 103–110. [Google Scholar] [CrossRef]
- Binger, K.J.; Gebhardt, M. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J. Clin. Investig. 2015, 125, 4223–4238. [Google Scholar] [CrossRef]
- Collins, L.E.; Troeberg, L. Heparan sulfate as a regulator of inflammation and immunity. J. Leukoc. Biol. 2019, 105, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.B.; Brunn, G.J. Cutting Edge: An Endogenous Pathway to Systemic Inflammatory Response Syndrome (SIRS)-Like Reactions through Toll-Like Receptor 4. J. Immunol. 2004, 172, 20–24. [Google Scholar] [CrossRef] [PubMed]
- De Wardener, H.E.; MacGregor, G.A. Harmful effects of dietary salt in addition to hypertension. J. Hum. Hypertens. 2002, 16, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Burrell, L.M. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 1998, 98, 2621–2628. [Google Scholar] [CrossRef]
- Gilbert, R.E.; Cox, A. Expression of transforming growth factor-beta1 and type IV collagen in the renal tubulointerstitium in experimental diabetes: Effects of ACE inhibition. Diabetes 1998, 47, 414–422. [Google Scholar] [CrossRef]
- Garcia-Sanchez, O.; Lopez-Hernandez, F.J. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010, 77, 950–955. [Google Scholar] [CrossRef]
- Ying, W.Z.; Sanders, P.W. Dietary salt modulates renal production of transforming growth factor-β in rats. Am. J. Physiol. Ren. Physiol. 1998, 274, F635–F641. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Marney, A.M.; Brown, N.J. Aldosterone and end-organ damage. Clin. Sci. 2007, 113, 267. [Google Scholar] [CrossRef]
- Greene, E.L.; Kren, S. Role of aldosterone in the remnant kidney model in the rat. J. Clin. Investig. 1996, 98, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Blasi, E.R.; Rocha, R. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003, 63, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Matsui, H. Salt-Induced Nephropathy in Obese Spontaneously Hypertensive Rats Via Paradoxical Activation of the Mineralocorticoid Receptor. Hypertension 2007, 50, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Waanders, F.; de Vries, L.V. Aldosterone, from (patho)physiology to treatment in cardiovascular and renal damage. Curr. Vasc. Pharmacol. 2011, 9, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Touyz, R.M. Eplerenone prevents salt-induced vascular remodeling and cardiac fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension 2004, 43, 1252–1257. [Google Scholar] [CrossRef]
- Calo, L.A.; Puato, M. Absence of vascular remodelling in a high angiotensin-II state (Bartter’s and Gitelman’s syndromes): Implications for angiotensin II signalling pathways. Nephrol. Dial Transpl. 2008, 23, 2804–2809. [Google Scholar] [CrossRef][Green Version]
- Susic, D.; Zhou, X.; Frohlich, E.D. Angiotensin blockade prevents salt-induced injury of the renal circulation in spontaneously hypertensive rats. Am. J. Nephrol. 2009, 29, 639–645. [Google Scholar] [CrossRef]
- Varagic, J.; Frohlich, E.D. AT1 receptor antagonism attenuates target organ effects of salt excess in SHRs without affecting pressure. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H853–H858. [Google Scholar] [CrossRef]
- Stier, C.T.; Chander, P. Therapeutic Benefit of Captopril in Salt-Loaded Stroke-Prone Spontaneously Hypertensive Rats Is Independent of Hypotensive Effect. Am. J. Hypertens. 1991, 4, 680–687. [Google Scholar] [CrossRef]
- Cao, W.; Li, A. A Salt-Induced Reno-Cerebral Reflex Activates Renin-Angiotensin Systems and Promotes CKD Progression. J. Am. Soc. Nephrol. 2015, 26, 1619–1633. [Google Scholar] [CrossRef]
- Taylor Norman, E.; Glocka, P. NADPH Oxidase in the Renal Medulla Causes Oxidative Stress and Contributes to Salt-Sensitive Hypertension in Dahl S Rats. Hypertension 2006, 47, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Kitiyakara, C.; Chabrashvili, T. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. 2003, 14, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Shen, Z. Hydrogen Sulfide Inhibits High-Salt Diet-Induced Renal Oxidative Stress and Kidney Injury in Dahl Rats. Oxid. Med. Cell. Longev. 2016, 2016, 15. [Google Scholar] [CrossRef]
- He, F.J.; Marciniak, M. Effect of modest salt reduction on skin capillary rarefaction in white, black, and Asian individuals with mild hypertension. Hypertension 2010, 56, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Smith, F.M.; Morris, L.W. Rapid Microvessel Rarefaction With Elevated Salt Intake and Reduced Renal Mass Hypertension in Rats. Circ. Res. 1996, 79, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Cowley, A.W., Jr. Salt intake and angiotensin II alter microvessel density in the cremaster muscle of normal rats. Am. J. Physiol. 1992, 263, H664–H667. [Google Scholar] [CrossRef]
- Bohle, A.; Mackensen-Haen, S. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res. 1996, 19, 191–195. [Google Scholar] [CrossRef]
- Kang, D.H.; Kanellis, J. Role of the Microvascular Endothelium in Progressive Renal Disease. J. Am. Soc. Nephrol. 2002, 13, 806–816. [Google Scholar] [CrossRef]
- Fellner, R.C.; Cook, A.K. High-salt diet blunts renal autoregulation by a reactive oxygen species-dependent mechanism. Am. J. Physiol. Ren. Physiol. 2014, 307, F33–F40. [Google Scholar] [CrossRef]
- Fellner, R.C.; Guan, Z. Endothelin contributes to blunted renal autoregulation observed with a high-salt diet. Am. J. Physiol. Ren. Physiol. 2015, 309, F687–F696. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Mount, P.F.; Power, D.A. Nitric oxide in the kidney: Functions and regulation of synthesis. Acta Physiol. (Oxf.) 2006, 187, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Manotham, K.; Tanaka, T. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J. Am. Soc. Nephrol. 2004, 15, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Milani, B.; Ansaloni, A. Reduction of cortical oxygenation in chronic kidney disease: Evidence obtained with a new analysis method of blood oxygenation level-dependent magnetic resonance imaging. Nephrol. Dial Transpl. 2017, 32, 2097–2105. [Google Scholar] [CrossRef]
- Prasad, P.V.; Thacker, J. Multi-Parametric Evaluation of Chronic Kidney Disease by MRI: A Preliminary Cross-Sectional Study. PLoS ONE 2015, 10, e0139661. [Google Scholar] [CrossRef]
- Yin, W.J.; Liu, F. Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD-MRI. Eur. J. Radiol. 2012, 81, 1426–1431. [Google Scholar] [CrossRef]
- Khatir, D.S.; Pedersen, M. Evaluation of Renal Blood Flow and Oxygenation in CKD Using Magnetic Resonance Imaging. Am. J. Kidney Dis. 2015, 66, 402–411. [Google Scholar] [CrossRef]
- Pruijm, M.; Hofmann, L. Determinants of renal tissue oxygenation as measured with BOLD-MRI in chronic kidney disease and hypertension in humans. PLoS ONE 2014, 9, e95895. [Google Scholar] [CrossRef]
- Pruijm, M.; Milani, B. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Pruijm, M.; Hofmann, L. Effect of sodium loading/depletion on renal oxygenation in young normotensive and hypertensive men. Hypertension 2010, 55, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Riethmuller, C. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Peters, W. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Clausen, P.; Jensen, J.S. Elevated urinary albumin excretion is associated with impaired arterial dilatory capacity in clinically healthy subjects. Circulation 2001, 103, 1869–1874. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Mooij, H.L. Endothelial Glycocalyx Damage Coincides With Microalbuminuria in Type 1 Diabetes. Diabetes 2006, 55, 1127. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Mattson, D.L. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F499–F508. [Google Scholar] [CrossRef]
- Mattson, D.L.; Higgins, D.J. Influence of dietary sodium intake on renal medullary nitric oxide synthase. Hypertension 1996, 27, 688–692. [Google Scholar] [CrossRef]
- De Miguel, C.; Das, S. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1136–R1142. [Google Scholar] [CrossRef]
- Mattson, D.L.; James, L. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 2006, 48, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, R.S.; Shrestha, P. High sodium diet converts renal proteoglycans into pro-inflammatory mediators in rats. PLoS ONE 2017, 12, e0178940. [Google Scholar] [CrossRef] [PubMed]
- Garner, B.R.; Stolarz, A.J. Contribution of the Renal Lymphatic Circulation to the Development of Salt-Sensitive Hypertension. FASEB J. 2019, 33, 678.5. [Google Scholar] [CrossRef]
- Pei, G.; Yao, Y. Lymphangiogenesis in kidney and lymph node mediates renal inflammation and fibrosis. Sci. Adv. 2019, 5, eaaw5075. [Google Scholar] [CrossRef]
- Slagman, M.C.; Kwakernaak, A.J. Vascular endothelial growth factor C levels are modulated by dietary salt intake in proteinuric chronic kidney disease patients and in healthy subjects. Nephrol. Dial Transpl. 2012, 27, 978–982. [Google Scholar] [CrossRef]
- Beaini, S.; Saliba, Y. VEGF-C attenuates renal damage in salt-sensitive hypertension. J. Cell. Physiol. 2019, 234, 9616–9630. [Google Scholar] [CrossRef]
- Mitsides, N.; Alsehli, F.M.S. Salt and Water Retention Is Associated with Microinflammation and Endothelial Injury in Chronic Kidney Disease. Nephron 2019, 12, 1–9. [Google Scholar] [CrossRef]
- Kopp, C.; Linz, P. Elevated tissue sodium deposition in patients with type 2 diabetes on hemodialysis detected by (23)Na magnetic resonance imaging. Kidney Int. 2018, 93, 1191–1197. [Google Scholar] [CrossRef]
- Schneider, M.P.; Raff, U. Skin Sodium Concentration Correlates with Left Ventricular Hypertrophy in CKD. J. Am. Soc. Nephrol. 2017, 28, 1867–1876. [Google Scholar] [CrossRef]
- Wenstedt, E.F.E.; Olde Engberink, R.H.G. Salt-sensitive blood pressure rise in type 1 diabetes patients is accompanied by disturbed skin macrophage influx and lymphatic dilation—A proof-of-concept study. Transl. Res. Provisionally Accepted.
- Selvarajah, V.; Maki-Petaja, K.M. Novel Mechanism for Buffering Dietary Salt in Humans: Effects of Salt Loading on Skin Sodium, Vascular Endothelial Growth Factor C, and Blood Pressure. Hypertension 2017, 70, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Olde Engberink, R.H.G.; Rorije, N.M. Role of the vascular wall in sodium homeostasis and salt sensitivity. J. Am. Soc. Nephrol. 2015, 26, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.; Walter, A. Anionic biopolymers as blood flow sensors. Biosens. Bioelectron. 1996, 11, 281–294. [Google Scholar] [CrossRef]
- Padberg, J.S.; Wiesinger, A. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis 2014, 234, 335–343. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.; Rorije, N.M. The blood pressure lowering potential of sulodexide—A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2015, 80, 1245–1253. [Google Scholar] [CrossRef]
- Packham, D.K.; Wolfe, R. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 123–130. [Google Scholar] [CrossRef]
- Kopp, C.; Linz, P. (23)Na magnetic resonance imaging of tissue sodium. Hypertension 2012, 59, 167–172. [Google Scholar] [CrossRef]
- Olde Engberink, R.H.G.; Selvarajah, V. Clinical impact of tissue sodium storage. Pediatr. Nephrol. 2019. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oppelaar, J.J.; Vogt, L. Body Fluid-Independent Effects of Dietary Salt Consumption in Chronic Kidney Disease. Nutrients 2019, 11, 2779. https://doi.org/10.3390/nu11112779
Oppelaar JJ, Vogt L. Body Fluid-Independent Effects of Dietary Salt Consumption in Chronic Kidney Disease. Nutrients. 2019; 11(11):2779. https://doi.org/10.3390/nu11112779
Chicago/Turabian StyleOppelaar, Jetta J., and Liffert Vogt. 2019. "Body Fluid-Independent Effects of Dietary Salt Consumption in Chronic Kidney Disease" Nutrients 11, no. 11: 2779. https://doi.org/10.3390/nu11112779
APA StyleOppelaar, J. J., & Vogt, L. (2019). Body Fluid-Independent Effects of Dietary Salt Consumption in Chronic Kidney Disease. Nutrients, 11(11), 2779. https://doi.org/10.3390/nu11112779