

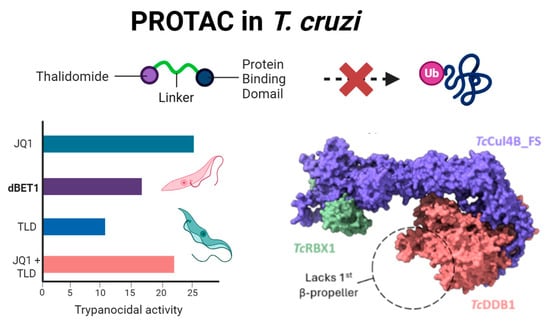
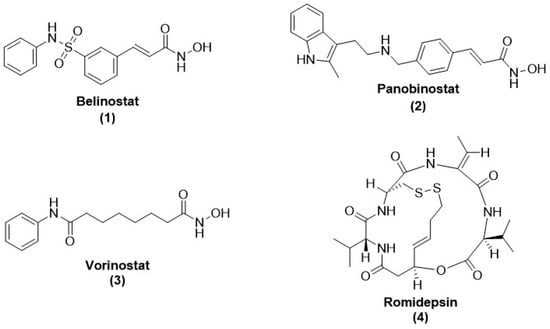
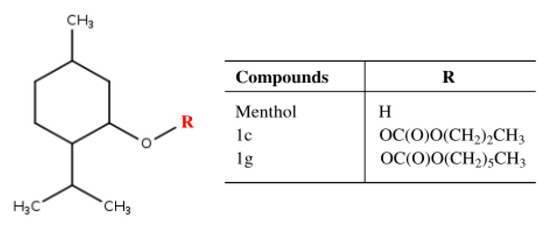
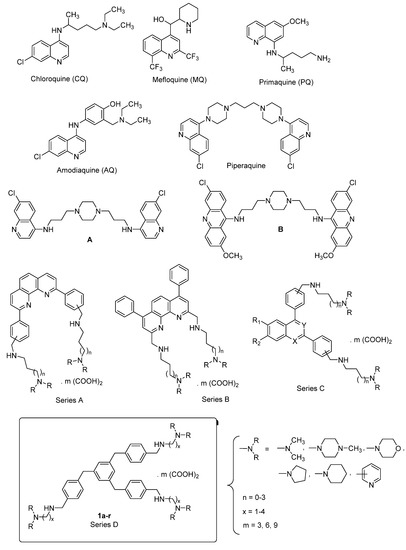
Drugs Drug Candidates 2026, 5(1), 11; https://doi.org/10.3390/ddc5010011 - 31 Jan 2026
Viewed by 755
Abstract
►
Show Figures
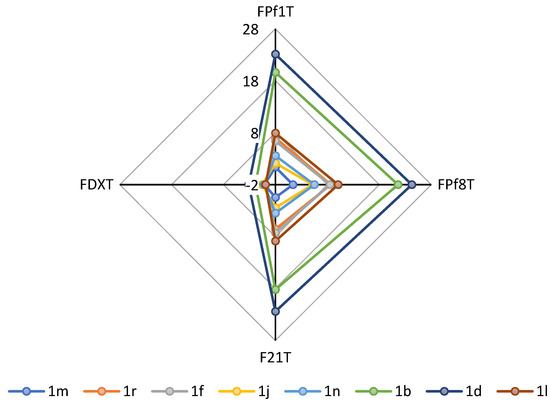
Background/Objectives: The widespread development of anthelmintic resistance in gastrointestinal nematodes constitutes a major production-limiting factor in grazing ruminants. Resistance mechanisms often involve drug efflux transporters like P-glycoprotein (P-gp). This study aimed to evaluate the potential of the phytochemicals cinnamaldehyde (CNM) and thymol (TML)
[...] Read more.
Background/Objectives: The widespread development of anthelmintic resistance in gastrointestinal nematodes constitutes a major production-limiting factor in grazing ruminants. Resistance mechanisms often involve drug efflux transporters like P-glycoprotein (P-gp). This study aimed to evaluate the potential of the phytochemicals cinnamaldehyde (CNM) and thymol (TML) to modulate P-gp activity and enhance the pharmacokinetic profile and efficacy of levamisole (LVM) in lambs. Methods: An ex vivo diffusion assay using sheep ileum was conducted to assess the influence of CNM, TML, and LVM on the transport of the P-gp substrate Rhodamine 123 (Rho123). Subsequently, a clinical trial was performed in lambs naturally infected with resistant nematodes. Animals received LVM (3.75 mg/kg) subcutaneously, either alone or co-administered with CNM or TML (80 mg/kg). Plasma LVM concentrations were analyzed by HPLC, and anthelmintic efficacy was determined via the Fecal Egg Count Reduction (FECR) test. Results: Ex vivo assays demonstrated that CNM, TML and LVM significantly reduced the efflux ratio of Rho123, confirming P-gp inhibition. The pharmacokinetic parameters of LVM did not differ significantly in the co-administered groups. However, the combination of LVM + TML tended to increase the total systemic exposure of LVM. Although all experimental groups showed a significant reduction in EPG between day 0 and day 7 (FECR 50–58%), the magnitude of this reduction did not differ significantly among treatments. Conclusions: While CNM and TML effectively inhibited P-gp activity ex vivo and slightly modified LVM pharmacokinetics, these effects were insufficient to yield clinically meaningful improvements in its efficacy against nematodes under the tested conditions. Future strategies should focus on optimizing delivery systems to maximize phytochemical–drug interactions.
Full article

Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}