Abstract
Background: Drug development for leishmaniases treatment follows a very selective process in order to propose drug candidates that possess all the qualities that meet the strict specifications of the pharmaceutical industry. Drug resistance is a limiting factor that can impact the lifespan of a marketed drug. It is now essential that the risk of drug resistance be evaluated at the early stage of in vitro studies to discard a lead compound that could quickly generate drug resistance once available on the market. Objectives: This article aims to estimate the risk of drug resistance emergence for a promising drug candidate at the in vitro early stage of drug development. Methods: A sequential method is proposed to study some of the phenotypic characteristics and parameters of resistant parasites such as time to achieve maximal resistance during stepwise drug pressure, resistance amplitude, stability, fitness, and infectivity both in vitro and in vivo. Results: Some examples with drugs in clinical use and former drug candidates are given. Conclusions: This method providing an evaluation of the risk of drug resistance from an in vitro model of Leishmania donovani be extrapolated to other Leishmania species.
1. Introduction
Leishmaniases are tropical and sub-tropical neglected infectious diseases that are caused by the protozoan parasite of the genus Leishmania sp. and transmitted by the bite of a sand fly belonging to the genus Phlebotomus sp. in the Old World and Lutzomyia sp. in the New World [1,2]. About twenty Leishmania species are infectious for humans and are responsible for three different diseases: visceral leishmaniasis, muco-cutaneous leishmaniasis, and cutaneous leishmaniasis. Whereas cutaneous leishmaniasis is often self-healing, visceral leishmaniasis is fatal in the absence of treatment [1].
The epidemiology of leishmaniases is complex, including life cycles based on peridomestic, sylvatic, zoonotic, and anthroponotic transmission as a function of the Leishmania species and the environmental conditions. Many factors such as the climate, the competence of the insect vector, the immune status of the human and/or animal reservoir are involved in the transmission process, and in Southern Europe, visceral leishmaniasis has an important canine reservoir [3]. The different parameters involved in leishmaniases transmission should be studied according to a “One health” approach, combining medical, veterinary, and environmental parameters in the affected areas in order to obtain integrative information for an optimal epidemiological survey. For example, in the North Bihar region of India, the relationship between arsenic in drinking water and selection of antimony resistance demonstrates how environmental stimuli can influence drug resistance in leishmaniasis [4].
Despite the availability of some canine vaccines on the market, there is no efficient vaccine against human leishmaniases. Although the use of insecticide repellent collars efficiently prevents dog contamination and constitutes a first means of prevention. Presently, chemotherapy remains the most efficient method to control the disease [5]. Thus, current antileishmanial chemotherapy includes antimonials, liposomal amphotericin B, and miltefosine as the main drugs in clinical use ([6]; Figure 1). Figure 1 gathers the chemical structures of these antileishmanial drugs and some former drug candidates we worked on.
Figure 1.
Chemical structures of some classical antileishmanial drugs and of some former drugs candidates involved in the present study.
These drugs meet several limitations such as drug resistance phenomenon and toxicity because they are not specific and often expensive. Drug resistance or acquired resistance to therapeutic molecules consists of the reduction in the efficacy of the drug in eliminating the parasite. This parasite phenotype emerges and spreads in contact with the drug and results from adaptation of the parasite to non-lethal drug concentrations. For example, the unresponsiveness to sodium stibogluconate, an antileishmanial drug, was about 43% in the Indian region of Bihar, generating a serious public health problem in Bihar [7].
Many studies are focused on the research of more specific drugs’ capability to not develop drug resistance and reference [8] describes one of them. Drugs for Neglected Diseases initiative (DNDi) boosts drug candidates’ identification, like a novel benzoxaborole preclinical candidate for the treatment of visceral leishmaniasis [9]. The global studies in drug research are now encouraged according to “One Health” approaches with the following requirements: firstly, green chemistry should be prioritized as the main objective of the chemists, with a reduced number of synthesis steps leading to obtaining cheaper drugs, and the elimination of toxic solvents that should be replaced by less toxic ones. Secondly, according to that, the use of chemoinformatic tools applied to computer-assisted drug design would help in reducing the number of compounds synthesis during pharmacomodulations. Thirdly, the “One Health” approach needs to consider toxicological effects through monitoring the behavior of drug candidates and their metabolites in the environment.
One major idealistic point is to select drug candidates for which drug resistance would be absent or very difficult to obtain. There is knowledge of the main causes of drug resistance in Leishmania including the role of ATP-binding cassettes [10]. The first one is based on the intrinsic bioavailability of the drug responsible for accumulation in deep organs. This occurs with antimonials that accumulate within the liver. Thus, during reinfections, the parasites stay in contact with sub-lethal drug concentrations, generating molecular mechanisms of resistance. The second cause is the long half-life of elimination, also putting parasites in contact with sub-lethal drug concentrations during new infections, with miltefosine as an example. The third cause is the drug quality when the patients are treated with counterfeited drugs exhibiting lower concentrations of the active principle [11]. The fourth cause is inherent to the natural variability of drug susceptibility in parasite isolates as a function of geographical areas leading to non-adapted dose regimen. Finally, the last main cause of drug resistance is relative to patient compliance, leading to inadequate drug exposure lower than the therapeutic dose.
As drug-resistant parasites develop within sandflies, they enter the Leishmania life cycle in the field and the efficacy of medical and veterinary treatments could therefore be subsequently affected [12]. Drug development is a long and expensive process. As a direct consequence, it is necessary to evaluate the risk of resistance of a lead compound at an early step of drug development, mainly just after a successful in vivo evaluation. From the results of the evaluation of the resistance risk, the scientists can optimize their selection for obtaining a drug candidate, giving pharmaceutical developers some insight about the life duration of the drug on the market.
As drug resistance compromises the lifespan of a drug, it is thus useful to set up in vitro drug resistance models through the study of different parameters that are able to better predict the behavior of a new antileishmanial drug in the field.
This article aims to propose an in vitro protocol of sequential experiments to gain information about a risk of drug resistance emergence in experimental conditions without immunological considerations, but that are potentially extrapolable to the field. These protocols using Leishmania donovani (MHOM/ET/67/HU3), also called LV9 strain, have been used by our group for the screening of industrial drug compounds of interest and they appeared to be useful for the decision-makers in pharmaceutical companies.
2. Results
The evaluation of the risk that a drug candidate could induce resistance requires an in vitro model that can be used at the early step of drug development, allowing a strict comparison of the resistance characteristics of the drug candidate with those of the drugs that are classically used in clinics. Thus, the following sequential protocol can be proposed in this article to conclude whether a compound is at risk of resistance or not (Figure 2).
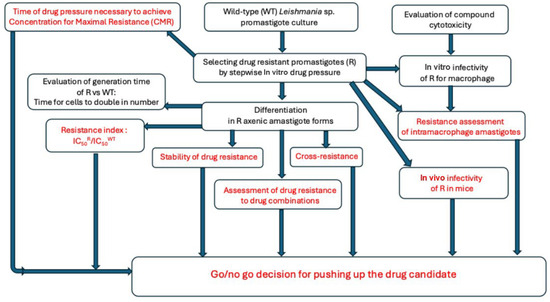
Figure 2.
A flowchart about an early phenotypic evaluation of the risk of drug resistance for an antileishmanial drug candidate.
2.1. Selecting Resistance and Checking Drug Resistance Phenotype
The promastigote forms of Leishmania sp. are easy to maintain in culture with short generation times, facilitating the selection of drug resistance. However, the promastigote parasite model has some predictability limitations as a function of the nature of the drug. Thus, antimonials are not active against the promastigote forms; therefore, axenic and intramacrophage amastigotes have been highly considered in research as they are closer to physiological conditions. This is the reason for which we propose to select drug resistance on promastigote forms first, followed by a differentiation process from promastigotes to amastigotes in an adapted culture medium and a systematic checking of the resistance level on the axenic amastigote model. Drug pressure can then be applied on axenic amastigotes, if necessary, to achieve the maximum level of drug resistance. Thus, in vitro axenic and intramacrophage amastigote models are expected to represent what could really happen in the field despite the fact that many environmental and biological parameters are neglected in such an experimental/artificial model. Comparing the behavior of resistant parasites such as axenic amastigotes and intramacrophage amastigotes is important to better observe whether drug resistance is maintained or not in the intracellular model. Subsequently, the mechanism of drug resistance can be monitored by infecting macrophages with drug-resistant promastigotes and determining the drug IC50 values.
2.1.1. Selecting Drug Resistance by In Vitro Drug Pressure on Promastigote Forms
The principle relies on applying a stepwise in vitro drug pressure at drug concentrations, allowing parasites culture growth. The initial drug pressure can be applied at a drug concentration corresponding to the half of the IC50 values. Several Leishmania parasite subcultures in the presence of such initial drug concentrations are necessary to obtain a growth curve of treated parasites similar to those of non-treated culture (wild-type parasites). By using such sub-lethal drug concentrations, the parasites progressively develop mechanisms of “drug tolerance”. Drug tolerance corresponds to an acquired lower drug susceptibility developed by the parasite on its way to full drug resistance. This is similar to intermediate resistance where the parasites are sub-resistant. Finally, under continuous in vitro drug pressure, the parasites become resistant to the drug. Thus, the next subcultures can be performed with a higher drug concentration, and sequentially, a stepwise drug pressure allows us to reach the maximum level of drug resistance, “CMR”, and is defined as the maximal concentration tolerated by the culture of resistant parasites. Concerning antimonials, we described in the "Materials and Methods" Section that the drug resistance was obtained after in vitro drug pressure of antimony III potassium tartrate (Sb III) because promastigote forms were not susceptible to pentavalent antimonials such as meglumine antimoniate (Sb V). The antimony-resistant axenic amastigotes obtained after differentiation were resistant to meglumine antimoniate with a Resistance Index (RI) of 148 (Table 1 and Table 2).

Table 1.
IC50 values of drugs used in clinics, meglumine antimoniate, miltefosine, and amphotericin B, compared to IC50 of two former drug candidates, sitamaquine and atovaquone. The IC50 values were obtained for both axenic and intramacrophage amastigotes of L. donovani wild-type parasites and their respective drug-resistant lines.
Table 2.
Biological characteristics of the L. donovani-resistant strains obtained by stepwise drug pressure.
Before determining any IC50, it is essential to perform two subcultures without the drug being prevented by this mean, or any error of drug concentration by an effect of drug accumulation. These are long and delicate experiments carried out in triplicate in 24-well flat-bottom sterile plates as parasite cultures can suddenly collapse at any time during the experiment. Table 1 presents the IC50 values of three drugs used in clinics, such as meglumine antimoniate, miltefosine, amphotericin B, and two former drug candidates, sitamaquine and atovaquone, both on axenic and intramacrophage amastigotes of L. donovani wild-type on their respective resistant strain. Table 1 shows that the selection of drug-resistant parasites was successful for all the studied drugs.
2.1.2. Selection of Drug-Resistant Parasites to Classical Antileishmanial Agents
In aiming to define the drug resistance risk of a drug candidate, it is important to compare its resistance characteristics with those of the classical antileishmanial agents and those of former drug candidates. This is the reason for which it is essential to obtain drug-resistant parasites for these specific drugs to establish characteristics comparisons. Several teams have selected drug-resistant Leishmania parasites to antimonials [13], to amphotericin B [14], to miltefosine [15], and also to the former drug candidates sitamaquine [16] and atovaquone [17]. In order to be able to place a drug candidate or a lead compound on a parameter measurement cursor in comparison with the reference products used in the clinic, our team has selected parasites resistant at the CMR to antimonials, amphotericin B, miltefosine, and the abandoned drug candidates, sitamaquine and atovaquone. All the characteristics of drug resistance are presented below in Section 2.2.
2.2. The Criteria to Measure the Characteristics of Drug Resistance and the Risk of Resistance Emergence
One major criteria in drug resistance is fitness, defined as the capacity of organisms to survive, reproduce, and be transmitted between hosts in a given environment. The fitness of drug-resistant parasites could be applied not only in vitro but also in vivo through the capacity of drug-resistant parasites to survive and develop through the sand fly vector and mammalian hosts. However, this article is focused on in vitro studies only. Thus, the fitness includes the generation time corresponding to (i) the time necessary for the resistant parasite to double its population versus the wild-type parasite cultures, (ii) the amplitude of drug resistance, and (iii) the time to achieve the maximal drug concentration.
2.2.1. Arbitrary Threshold for Drug Resistance Based on a Significant Difference Between IC50S
The arbitrary and commonly admitted threshold to characterize drug resistance in microbiology requires that the IC50R value of the treated parasites is at minimum 4× times higher than the IC50WT value of the non-treated cultures (wild-type). Thus, the Resistance Index (RI) defined as the ratio RI = IC50R/IC50WT should be higher than 4.
Concerning the drugs evaluated in this article, both regarding axenic and intramacrophage amastigotes, Table 2 shows that the RI values were >4 for all of them, and specifically considering intramacrophage amastigotes, were in a range of 5 for sitamaquine to 45 for atovaquone. Miltefosine and amphotericin B exhibited intermediate RI values of 33 and 20, respectively. Concerning meglumine antimoniate, the resistance level did not allow an evaluation on the intramacrophage amastigotes because of toxicity on the macrophages at the theoretically active concentrations.
In the case that it is not possible to obtain this minimal difference between both IC50 values, meaning the ratio IC50R/IC50WT (Resistance Index) is less than 4, it can be considered that the parasite culture is less susceptible to the drug but not significantly resistant.
In the past, we have obtained such a situation with a 2-substituted-quinoline series (Figure 3) after a long time under drug pressure. In these conditions, we could speculate that the risk of drug resistance is not high on the basis of results obtained using our artificial model. It is notable that the CMR (drug concentration allowing maximal resistance) results exhibit significant variations that are not only species-dependent but also strain-dependent within a defined Leishmania species as expressed in Table 1. However, in the field, many other biological factors linked to the immunological status of the patient, the vector, and the environmental conditions can modulate the response of the parasite to the drugs, and therefore, consequently modulate the level of drug resistance.
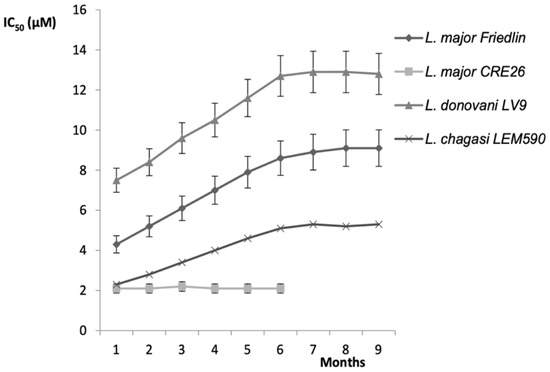
Figure 3.
In vitro susceptibility profiles on four Leishmania sp. parasite models after drug pressure to select resistance to 2-n-propylquinoline (2-PQ) drug [18]. The experiments were carried out in 24-well plastic tissue-culture plates in a final volume of 1 mL and the plates were maintained at 37 °C under an atmosphere of 5% CO2. Axenic amastigotes were initially submitted to a drug pressure corresponding to almost a third of the IC50 value of the soluble formulation of 2-PQ. A stepwise increase in drug concentration was applied as soon as the drug-exposed cultures exhibited a growth rate similar to those of the WT line cultures. This procedure was applied until the maximum concentration allowing parasite growth was reached.
2.2.2. Amplitude of Drug Resistance
The Resistance Index (RI), defined as the ratio IC50R/IC50WT, is a relevant marker of the amplitude of drug resistance. After differentiation of drug-resistant L. donovani LV9 promastigote forms to axenic amastigotes, we compared the RI of the main classical drugs and found that the highest RI value was obtained with antimonials (RI = 148), whereas the lowest value was obtained with a former drug candidate: sitamaquine, with a RI value of 6 (Table 1). Not surprisingly, in vitro amplitude of drug resistance is drug-dependent and probably linked to the primary molecular mechanisms involved, independently of host factors, such as mammalians and insect vectors that constitute environmental conditions able to influence the drug resistance phenomenon.
2.2.3. Time Necessary to Achieve CMR (Concentration to Achieve Maximal Drug Resistance)
The results in Table 2 show the time to achieve CMR from experiments performed in similar conditions in our research team, allowing reliable comparisons between the drugs. The time to achieve CMR is also drug-dependent. Thus, the antimony resistance selection was a relatively rapid process obtained after a 3-month drug pressure, whereas amphotericin B-resistance selection required a 20-month drug pressure. Miltefosine exhibited an intermediate time of 11 months to achieve CMR. Concerning antimonials, it is probable that drug resistance in the field appeared in less than 9 years (Table 2), but the publications were rare at this time. If we consider this hypothesis, we could suggest that there is a positive correlation between the time necessary to achieve CMR in vitro and the time interval from the marketing of the drug and the description of the first cases of drug resistance in the field. This positive correlation also suggests that the in vitro test works as a predictable tool for early detection of the risk of drug resistance emergence in environmental conditions.
2.2.4. Stability of Drug Resistance
To better understand the mechanisms that lead to the emergence of drug-resistant parasites, it is useful for a drug developer to obtain insights about the stability of drug resistance, considering the important polymorphism in the Leishmania world. Thus, a progressive loss of drug resistance in the absence of drug pressure conditions could be good news for therapies applied in the field, allowing alternate drug use without impairing the lifespan of the drug.
Thus, meglumine antimoniate, miltefosine, and atovaquone exhibit a slight decrease in drug resistance by 20% after a 6-month period in the absence of drug pressure, whereas amphotericin B and sitamaquine only had a very slight decrease (0 to 10%) of the IC50 values (Table 2).
2.2.5. Cross-Resistance
Cross-resistance can be defined as the ability of a drug-resistant strain to be less susceptible to other drugs. Such therapeutic situations should be anticipated before launching a drug candidate on the market. The determination of IC50 values of the following drugs: meglumine antimoniate, miltefosine, amphotericin B, sitamaquine, and atovaquone on the MA-R, Milt-R, AmB-R, Sita-R, and Atov-R strains has shown that there is no cross-resistance with these drugs as the IC50 values of the drugs are not significantly modified between WT and other drug-resistant strains. For these reasons, obtaining knowledge on the cross-resistance between a drug candidate and a classical antileishmanial drug is highly important to manage the therapeutic schedules to be recommended and applied in the case of Leishmania-resistance emergence.
2.2.6. Drug Combination and Resistance
This approach of drug combination, firstly developed in cancerology, has been extended in parasitology. Thus, drug combination has been proposed as a strategy to escape the phenomenon of drug resistance as it consists of the association of two (or more) active principles having different mechanisms of action. In this way, it is expected to obtain a synergy of drug action, or at least, an additive effect to kill the parasites, eliminating the pathogens quickly enough before it can develop molecular mechanisms of drug resistance. Unfortunately, we now have the proof that in vitro drug pressure using two antileishmanial drugs can result in drug resistance to both of them [19,20]. However, studying drug combination in vitro is very limitative as the interactions of two drugs are certainly completely different in vivo, where many factors should be considered, including drug distribution and metabolism. Hence, we propose to include the evaluation of in vitro drugs combination but not to consider a negative result as redhibitory or discriminant for further evaluation.
2.2.7. In Vitro Infectivity
It is important for a pharmaceutical developer to know the capacity of drug-resistant parasites to be infective for macrophages. Considering the five drug-resistant parasite studies in this article, the capacity to infect macrophages is identical to those of the wild-type strain, considering the percentage of infected macrophages and the mean number of parasites in each infected macrophage.
2.2.8. In Vivo Infectivity on Rodent Models
The capacity of drug-resistant parasites to enter the parasite life cycle in the field mainly depends on two parameters: their ability to infect sand flies and mammalian hosts. The infection of mammal hosts was experimentally performed through parasite injection into laboratory mice and the parasite burden was measured as described in Campos-Vieira et al. [21]. Considering meglumine antimoniate, miltefosine, and atovaquone, we observed that drug-resistant parasite burden in the mice liver was reduced in the range of 40 to 60% in comparison to those of the wild-type parasite burden. Although these results indicate that resistant parasites can infect mammalian hosts, they are not very reflective of what occurs under natural field conditions. Thus, mice were experimentally infected by an intravenous injection of 1.2 × 108 promastigotes whereas the natural infection by a sand fly vector releases several hundred promastigotes within the mice [22]. Concerning amphotericin B and sitamaquine, no parasite can be seen on Giemsa-stained liver smears. An in vitro subculture was necessary to obtain the development of promastigotes 5 to 7 days later, indicating that the parasite is present and viable. Thus, the low parasite burden in the mice could suggest a very low transmission of resistant parasites in the field. The last key-point is the capacity of drug-resistant parasites to infect sand flies from an infected mammalian host. We have not performed this experiment yet; however, we recently published that antimony-resistant Leishmania major Mon-25, responsible for zoonotic cutaneous leishmaniasis, was infective for the sand fly Phlebotomus papatasi [12]. Accordingly, Table 2 shows that the longer it takes to achieve CMR, the lower the parasite’s ability is to infect animal hosts.
2.2.9. In Vivo Infectivity on Competent Insect Vectors
In order to explore the role of drug-resistant parasites within the complete life cycle of Leishmania, it is important to assess the possible transmission of drug-resistant parasites to the sand fly vector, and also from the sand fly to the mammalian host. For example, the results presented in a recent study showed that Phlebotomus papatasi competent vectors could promote circulation of L. major Sb(III)-resistant parasites in the field [12].
3. Discussion
The treatment of leishmaniases has the disposal of few drugs, mainly liposomal amphotericin B, antimonials, miltefosine, and paromomycin, but each of them presents limitations such as (i) parenteral administration (except for miltefosine), (ii) severe adverse effects, (iii) cold chain maintenance, (iv) and often long-term treatments [23]. The chemotherapy of leishmaniases is complex as Leishmania species-dependent variations are observed in drug susceptibility. In addition, the development of drug resistance diminishes the efficacy of these drugs, except for amphotericin B for which only few clinical cases of resistance have been described up to now [24,25]. Thus, the risk of drug resistance is an important aspect to be taken into consideration for a lead compound in drug development.
Accordingly, the methodological sequence proposed in this article should help to decide whether there should be further development of a lead compound by comparing the resistance characteristics of the lead compound versus those of the classical drugs currently used in clinics.
In the past, many studies on antileishmanial drug resistance were performed on the promastigote forms, as they were easy to maintain in culture with short generation times, allowing us to quickly obtain resistant parasites for drug resistance analysis purposes [16,17,19]. Now, the possibility to cultivate axenic amastigotes has adapted to grow and develop outside their host cells in culture medium at acidic pH = 5.5, closer to the physiological conditions, and this has created a long period of debate. Indeed, some scientists consider that axenic amastigote forms are just suffering promastigote parasites. However, the presence of a specific protein “A2” on the membrane of axenic amastigotes was a molecular argument to consider them as real amastigotes, even if there is still a lack of total agreement among the scientific community [26,27]. At any rate, models using host cells currently remain the gold standard in determining compound susceptibility because they reflect the compound’s activity in the natural environment of the parasite in the field. As a consequence, any resistant phenotype observed on promastigote forms, then on axenic amastigotes, should be confirmed on the intramacrophage amastigote model mimicking the in vivo conditions.
In this article, we propose that axenic and intramacrophage amastigote parasites in vitro models are the first tools from which it is possible to obtain relevant information relative to the capacity to achieve the following: (i) resistance for a compound, (ii) the time to achieve the maximum of resistance, (iii) its amplitude, (iv) stability, (v) and the cross-resistance. In addition, in vitro and in vivo infectivity capacity should also be explored to provide to the drug developers suitable information to determine interest in developing the drug in the context of drug resistance. The global work to obtain answers to these questions is estimated at several months and depends on the time to obtain Rmax (the time necessary to achieve maximum of drug resistance).
This methodological sequence is the first way to explore the drug resistance risk through simple in vitro analysis, completed by in vivo experiments on a BALB/c mice animal model. This approach brings insights about the risk of drug resistance for a drug candidate, but it does not claim to reach formal conclusions on the ability of molecules to select resistant parasites, as it does not take into account the immune aspects encountered in the host–parasite interaction. This last point should not be neglected since some drugs such as miltefosine have an immunologic component in their mechanism of action. However, it has been demonstrated that the immunosuppression of Syrian golden hamsters accelerates the occurrence of leishmaniasis relapse without expediting miltefosine-resistance development [28]. Thus, the idealistic expectations for a developer of an antileishmanial drug is an absence of drug resistance or a very low degree of drug resistance.
As the time to achieve maximal resistance (Rmax) is inversely proportional to risk of resistance development, the longer it takes to achieve maximal resistance, the less likely it is for the development of the drug resistance mechanism. So, the time to achieve maximal resistance Rmax in vitro should be very long. In the same way, the lower the Resistance Index (RI) value, the less likely the level of resistance will be. Concerning the resistance reversibility, the higher the reversibility, the longer the likely lifespan of a drug. Considering the in vivo infectivity, the lower the capacity of drug-resistant parasites to infect mammals, the lower the risk of drug extension in the field is. Thus, the evaluation of cross-resistance is highly important to select a substitutive drug in case of cross-resistance.
On the other hand, the relationships between in vitro and in vivo results appear coherent. In fact, considering Amphotericin B, the significant decrease in in vivo infectivity of AmB-R parasites could explain the long time for AmB-resistance emergence in the field. In the case of in vitro resistance, the capacity of in vivo infection is a very important predictive parameter to take into consideration for any drug developer.
In the future, it is essential to include the phlebotomine sand fly insect vector to assess the drug resistance life cycle.
4. Materials and Methods
4.1. Parasite and Cultures
4.1.1. Promastigote Parasites
Promastigote forms of the wild-type Leishmania donovani (MHOM/ET/67/HU3) strain, also called L. donovani LV9, and simply WT in this article, were grown in M199 liquid medium supplemented with 40 mM HEPES, 100 µM adenosine, 0.5 mg/L hemin, 10% heat-inactivated fetal calf serum (FCS), and 50 µg/mL gentamycin at 26 °C in a dark environment. All the experiments were performed with parasites in their logarithmic phase of growth. Drug-resistant lines were obtained for meglumine antimoniate (called MA-R), for miltefosine (Milt-R), for amphotericin B (AmB-R), for sitamaquine (Sita-R), and for atovaquone (Atov-R) according to in vitro continuous stepwise drug pressure as described below. The assays were carried out in duplicate for each drug concentration and the parasites under drug pressure were incubated at 26 °C overnight until the appearance of clusters. The drug-resistant parasites are then stored in liquid nitrogen.
4.1.2. Selection of Drug Pressure on the Promastigote Forms
Leishmania-resistant parasites were selected in vitro in sterile Falcon® flat-bottom 24-well plastic tissue-culture plate, in a final volume of 1 mL, and the plates were maintained at 26 °C in a dark environment. Parasites were initially submitted to an initial drug pressure corresponding to almost third of the IC50 (half maximal inhibitory concentration). The first drug pressure was maintained for a maximum of five passages until the growth of the treated parasite culture was similar to the untreated wild-type parasite culture growth curves. A sequential in vitro stepwise increase in drug concentration was undertaken only when the drug-exposed cultures had a growth rate similar to those of the wild-type cultures. This process was applied until the maximum concentration allowing parasite growth and ran in duplicate for each drug concentration. Concerning antimonials, drug resistance was obtained after in vitro drug pressure of antimony III potassium tartrate (Sb III) because promastigote forms are not susceptible to pentavalent antimonials such as meglumine antimoniate (Sb V).
4.1.3. Axenic Amastigote Forms
The differentiation from promastigotes to axenic amastigotes was carried out as follows: cultures of wild-type and drug-resistant axenic amastigotes of L. donovani were obtained from late log phase growth of promastigotes diluted at 1 × 106/mL in M199 complete medium acidified at pH 5.5 and maintained in culture at 37 °C with under an atmosphere of 5% CO2. The evaluations of activity on axenic forms were adapted from the protocols briefly summarized as follows. Concerning the evaluation on axenic amastigotes, two-fold serial dilutions of the compounds from a maximal concentration of 100 μM or more were performed in 100 μL of complete medium (see above) in 96-well flat-bottomed microplates (Thermo Scientific™, Villebon sur Yvette, France). Axenic amastigotes were then added to each well at a density of 106/mL in a 200 μL final volume. After 72 h of treatment at 37 °C for L. donovani with 5% CO2, 20 μL of resazurin (450 μM) was added to each well and further incubated in the dark for 24 h at 37 °C for L. donovani with 5% CO2 to run the “Alamar blue assay” according to Nociari et al. [29]. In living cells, resazurin is reduced in resorufin that is quantified following the manufacturer’s guidelines. The conversion to resofurin is monitored by measuring the OD570 nm (resorufin) and OD600 nm (resazurin; Lab systems Multiskan MS, Vantaa, Finland) with excitation at 550 nm and emission at 590 nm. Fluorescence intensity was expressed in arbitrary units and the activity of the compounds was expressed as IC50 in µM.
4.1.4. Evaluation of Compounds Cytotoxicity
The cytotoxicity was evaluated on RAW 264.7 murine macrophages. Cells were plated in 96-well flat-bottom microplates at a density of 2 × 104 cells per well. After an incubation of 24 h at 37 °C under an atmosphere of 5% CO2 for RAW264.7 cells, the medium was removed from each well, and 100 μL of DMEM complete liquid medium containing two-fold serial dilutions of the compounds was added to each well. After 48 h of incubation at 37 °C with 5% CO2, 10 μL of resazurin (450 μM) was added to each well, and further incubated in the dark for 4 h at 37 °C with 5% CO2. Cell viability was then monitored as previously described above. The cytotoxicity of the compounds was expressed as CC50 (Cytotoxic Concentration 50%: concentration inhibiting the macrophages growth by 50%).
4.1.5. Intramacrophage Amastigote Forms
The cytotoxicity data allowed us to choose the suitable concentrations of the compounds to evaluate their activity on the intramacrophage amastigotes. Concerning the evaluation on internalized amastigotes into murine cells, RAW 264.7 macrophages (from ATCC) were plated in 96-well flat-bottom microplates at a density of 2 × 104 cells per well and incubated for 24 h at 37 °C with 5% CO2. Axenic amastigotes were differentiated from promastigotes as previously described above, centrifuged at 2000× g for 10 min, resuspended in DMEM complete medium, and then added to each well to reach a 16:1 parasite-to-macrophage ratio. After 24 h of infection in the same conditions, extracellular parasites were removed, and 100 μL of DMEM complete medium containing two-fold serial dilutions of the compounds from a maximal concentration of 100 μM was added to each well. After 48 h of treatment, the medium was removed and replaced by 100 µL of DirectPCR Lysis Reagent (Euromedex, Souffelweyersheim, France) before a series of 3 freeze–thaw cycles at room temperature, with addition of 50 μg/mL proteinase K, and a final incubation at 55 °C overnight to allow cell lysis. Then, 10 μL of each cell extract was then added to 40 μL of DirectPCR Lysis reagent containing SYBR Green I (0.05%; Invitrogen, Waltham, MA, USA). DNA fluorescence was monitored using Mastercycler realplex (Eppendorf, Hamburg, Germany). The activity of the compounds was expressed as IC50 in µM.
4.2. Fitness of Drug-Resistant Leishmania Parasites vs. Wild-Type
4.2.1. Evaluation of Generation Time
The generation time is the time it takes for a population of cells to double in number. It is a marker of the good health of Leishmania parasite cultures. For this study, the generation time is determined by the drug-resistant promastigotes vs. WT by daily cell counting using a Malassez counting chamber slide until the stationary phase of parasite growth was reached. Generation time was expressed in hours.
4.2.2. Amplitude of Drug Resistance and Resistance Index
The Resistance Index (RI), defined as the ratio IC50R/IC50WT corresponds to the measure of the amplitude of drug resistance. RI is an indicator of the possibility of a drug to achieve a high level of resistance.
4.2.3. Time of Drug Pressure Necessary to Achieve CMR Level
The time of drug pressure necessary to achieve the concentration for maximal drug resistance (CMR) is a possible kinetics marker of the capacity of a specific drug to generate resistance in the field. It is expressed as months.
4.2.4. Stability of Drug Resistance
The stability of drug resistance was measured by stopping drug pressure and continuing the subcultures in order to assess the drug susceptibility kinetics. Thus, about 40 subcultures without drug pressure were carried out to assess a possible reversibility of drug resistance mechanism.
4.2.5. Cross-Resistance Phenomenon
Cross-resistance was measured by determining the IC50 values of meglumine animoniate, amphotericin B, miltefosine, sitamaquine, and atovaquone on each drug-resistant strain to observe whether a strain that is resistant to one drug is resistant to other(s).
4.2.6. Drug Combination and Drug Resistance
Resistance to a drug combination is a possibility to be further verified. Thus, the drug pressure of a drug combination can be performed to assess the possibility of resistance to it. Even if no experiment has been performed in this article, this approach can be recommended to better understand this mechanism.
4.2.7. In Vitro Infectivity
The infectivity of drug-resistant promastigotes was assessed using the RAW 264.7 macrophage murine model. Accordingly, cells were plated in 96-well flat-bottom sterile microplates at a density of 2 × 104 cells per well under a 100 µL final volume of DMEM medium. After an incubation of 24 h at 37 °C with 5% CO2, the medium was removed in each well, and a suspension of promastigote forms of WT or drug-resistant parasites at a stationary phase of growth from 7-day-old cultures was used for macrophage infection assays. Thus, promastigotes were centrifuged at 2000× g for 10 min, resuspended in DMEM complete medium, and then added to each well under a 100 µL volume to reach a 20:1 parasite-to-macrophage ratio. After 24 h of infection at 37 °C with 5% CO2, extracellular parasites were removed, and DMEM complete medium (100 μL) was added to each well. Then, after 48 h of incubation, the medium was removed and the slides were fixed with methanol and Giemsa-stained for direct observation. The number of amastigotes in 100 macrophages/well and the mean number of amastigotes/macrophage were determined microscopically.
4.2.8. In Vivo Infectivity
For each L. donovani drug-resistant strain, 10 BALB/c mice were infected with 1.2 × 108 promastigotes at a stationary phase from 7-day-old cultures by the retro-orbital sinus injection under a volume of 100 µL and to 10 mice with L. donovani WT. Five mice infected with the drug-resistant strain and five mice infected with the WT strain were autopsied at 7, 14, and 21 days post-infection. Animals were kept in the animal facility of Université Paris-Saclay in accordance with institutional guidelines and French legislation for animal protection (APAFIS), which complies with all relevant European Union and international guidelines for experimental animals. The approval code was APAFIS#30494-2021031617294635 v1 and the approval date was 17 May 2024 for a 5-year duration. On the other hand, the spleen and liver were weighed and parasite burdens were determined microscopically as previously described by Balaraman et al. [18]. In addition, a subculture was set up from the liver in M199 complete medium and incubated at 37 °C in an atmosphere of 5% CO2 and the presence of promastigote forms was assessed microscopically 7 days after initiating the subculture.
5. Conclusions
Until a few years ago, the risk of drug resistance for antiparasitic drugs was evaluated once they were released on the market. This paper proposes a methodology as a sequence of simple protocols, allowing a better evaluation of the risk of drug resistance for lead compounds and drug candidates at an early stage of development. Thus, this approach should be initiated as soon as in vivo results suggest that a lead compound can be considered as having a potential as drug candidate.
When observing the resistance characteristics of drugs in clinical use, an idealistic drug candidate will positively reply to the following in vitro criteria to be pushed up in drug development:
- -
- The longest time taken to achieve the concentration for maximal resistance;
- -
- The lowest Resistance Index value IC50R/IC50WT;
- -
- The reversibility of drug resistance;
- -
- An absence of cross-resistance with other antileishmanial drugs;
- -
- A difficult development of drug-resistant parasites in a murine model.
Early in vitro drug resistance studies allow for monitoring of the infective capacity of resistant strains and their fitness, influencing the transmissibility and circulation of drug-resistant strains in the field.
The information collected using this sequence of protocols proposed in this article gives data on the ability of the Leishmania parasite to adapt to the drug pressure applied by a given compound and how fast these adaptations could be acquired. Thus, these data would provide insights about the possible mechanisms of action of the compounds and/or their mechanisms of resistance, identifying a possible cross-resistance phenomenon with combination partners.
However, the polymorphism of Leishmania parasites is responsible for significant differences between parasite species, and also inside one species, between the strains. Therefore, after such a preliminary study on laboratory strains (parasite cultures), it is essential to validate the results obtained by using drug-resistant clinical isolates to confirm these data.
In summary, this methodology aims to become an additional tool for R&D pipelines of antileishmanial compounds, helping the drug designers and researchers by providing a better understanding of phenotypic drug resistance dynamics.
Author Contributions
Conceptualization: P.M.L.; methodology: S.C., N.M., and P.M.L.; validation: P.M.L., N.M., and S.C.; formal analysis: S.C., P.M.L., and N.M.; investigation: S.C., N.M., and P.M.L.; resources: P.M.L., S.C., and N.M.; writing—original draft: P.M.L., N.M., and S.C.; project administration: S.C. and P.M.L.; funding acquisition: S.C. and P.M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by DIM One Health (Domaine d’Intérêt Majeur de la Région Ile de France «One Health», project Leish1Health 2021, and grant number ANR-11-IDEX-0003-02, Région Ile-de-France (DIM1Health).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The COST Action CA21111 “OneHealthDrugs” is acknowledged for its support in the reflection of new strategies to consider environmental aspects in drug development.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Montaner-Angoiti, E.; LLobat, L. Is leishmaniasis the new emerging zoonosis in the world? Vet. Res. Commun. 2023, 47, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Escolar, I.; Morchón, R.; Papadopoulos, E.; Sioutas, G.; Collado-Cuadrado, M.; Infante González-Mohino, E.; Balmori-de La Puente, A. Analysis of the current risk of Leishmania infantum transmission in Greece and its projection. Transbound. Emerg. Dis. 2025, 1087533. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, N.; Mitropoulou, A.; Thom, N. Update on therapy and prevention of canine leishmaniasis. Tierarztl. Prax. Ausg. K Kleintiere Heimtiere 2018, 46, 315–322. [Google Scholar] [CrossRef]
- Majoor, A.; Michel, G.; Marty, P.; Boyer, L.; Pomares, C. Leishmaniases: Strategies in treatment development. Parasite 2025, 32, 18. [Google Scholar] [CrossRef]
- Das, V.N.R.; Ranjan, A.; Bimal, S.; Siddique, N.A.; Pandey, K.; Kumar, N.; Verma, N.; Singh, V.P.; Sinha, P.K.; Bhattacharya, S.K. Magnitude of unresponsiveness to sodium stibogluconate in the treatment of visceral leishmaniasis in Bihar. Natl. Med. J. India 2005, 18, 131–133. [Google Scholar]
- Pomel, S.; Cojean, S.; Pons, V.; Cintrat, J.C.; Nguyen, L.; Vacus, J.; Pruvost, A.; Barbier, J.; Gillet, D.; Loiseau, P.M. An adamantamine derivative as a drug candidate for the treatment of visceral leishmaniasis. J. Antimicrob. Chemother. 2021, 76, 2640–2650. [Google Scholar] [CrossRef]
- Mowbray, C.E.; Braillard, S.; Glossop, P.A.; Whitlock, G.A.; Jacobs, R.T.; Speake, J.; Bharathi, P.; Nare, B.; Maes, L.; Yardley, V.; et al. DNDI-6148: A novel benzoxaborole preclinical candidate for the treatment of visceral leishmaniasis. J. Med. Chem. 2021, 64, 16159–16176. [Google Scholar] [CrossRef]
- de Santana, M.B.R.; Miranda, G.O.; Carvalho, L.P. ATP-binding cassette transporters and drug resistance in cutaneous leishmaniasis. Int. J. Infect. Dis. 2025, 151, 107315. [Google Scholar] [CrossRef]
- Kumari, A.; Siddiqui, N.A.; Kumari, S.; Murti, K.; Kumar, R.; Pandey, K.; Padmakar, S.; Pal, B. Combination Therapy for Post-Kala-Azar Dermal Leishmaniasis: A literature review of current evidence. Indian J. Dermatol. 2024, 69, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Mekarnia, N.; Benellal, K.E.; Sadlova, J.; Vojtkova, B.; Mauras, A.; Imbert, N.; Longhitano, M.; Harrat, Z.; Volf, P.; Loiseau, P.M. Effect of Phlebotomus papatasi on the fitness, infectivity and antimony-resistance phenotype of antimony-resistant Leishmania major Mon-25. Int. J. Parasitol. Drugs Drug. Resist. 2024, 25, 100554. [Google Scholar] [CrossRef] [PubMed]
- Arana, F.E.; Perez-Victoria, J.M.; Repetto, Y.; Morello, A.; Castanys, S.; Gamarro, F. Involvement of thiol metabolism in resistance to glucantime in Leishmania tropica. Biochem. Pharmacol. 1998, 56, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Mbongo, N.; Loiseau, P.M.; Billion, M.A.; Robert-Gero, M. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 1998, 42, 352–357. [Google Scholar] [CrossRef]
- Seifert, K.; Matu, S.; Javier Perez-Victoria, F.; Castanys, S.; Gamarro, F.; Croft, S.L. Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine). Int. J. Antimicrobial. Agents 2003, 22, 380–387. [Google Scholar] [CrossRef]
- Bories, C.; Cojean, S.; Huteau, F.; Loiseau, P.M. Selection and phenotype characterisation of sitamaquine-resistant promastigotes of Leishmania donovani. Biomed. Pharmacother. 2008, 62, 164–167. [Google Scholar] [CrossRef]
- Cauchetier, E.; Loiseau, P.M.; Lehman, J.; Rivollet, D.; Fleury, J.; Astier, A.; Deniau, M.; Paul, M. Characterisation of atovaquone resistance in Leishmania infantum promastigotes. Int. J. Parasitol. 2002, 32, 1043–1051. [Google Scholar] [CrossRef]
- Balaraman, K.; Campos Vieira, N.; Moussa, F.; Vacus, J.; Cojean, S.; Pomel, S.; Bories, C.; Figadère, B.; Kesavan, V.; Loiseau, P.M. In vitro and in vivo antileishmanial properties of a 2-n-propylquinoline hydroxypropyl β-cyclodextrin formulation and pharmacokinetics via intravenous route. Biomed. Pharmacother. 2015, 76, 127–133. [Google Scholar] [CrossRef]
- Garcia-Hernandez, R.; Manzano, J.I.; Castanys, S.; Gamarro, F. Leishmania donovani develops resistance to drug combinations. PLoS Negl. Trop. Dis. 2012, 6, e1974. [Google Scholar] [CrossRef]
- Berg, M.; Garcia-Hernandez, R.; Cuypers, B.; Vanaerschot, M.; Manzano, J.; Poveda, J.A.; Ferragut, J.A.; Catsanys, S.; Dujardin, J.A.; Gamarro, F. Experimental resistance to drug combinations in Leishmania donovani: Metabolic and phenotypic adaptations. Antimicrob. Agents Chemother. 2015, 59, 2242–2255. [Google Scholar] [CrossRef]
- Campos-Vieira, N.; Vacus, J.; Fournet, A.; Baudouin, R.; Bories, C.; Séon-Méniel, B.; Figadère, B.; Loiseau, P.M. Antileishmanial activity of a formulation of 2-n-propylquinoline by oral route in mice model. Parasite 2011, 18, 333–336. [Google Scholar] [CrossRef]
- Kimblin, N.; Peters, N.; Debrabant, A.; Secundino, N.; Egen, J.; Lawyer, P.; Fay, M.P.; Kamhavi, S.; Sacks, D. Quantification of the infectious dose of Leishmania major transmitted to the skin by single sand flies. Proc. Natl. Acad. Sci. USA 2008, 105, 10125–10130. [Google Scholar] [CrossRef]
- Santos, S.S.; de Araujo, R.V.; Giarolla, J.; Seoud, O.E.; Ferreira, E.I. Searching for drugs for Chagas disease, leishmaniasis and schistosomiasis: A review. Int. J. Antimicrob. Agents 2020, 55, 105906. [Google Scholar] [CrossRef] [PubMed]
- Purkait, B.; Kumar, A.; Nandi, N.; Sardar, A.H.; Das, S.; Kumar, S.; Paney, K.; Ravidas, V.; Kumar, M.; De, T. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob. Agents Chemother. 2012, 56, 1031–1041. [Google Scholar] [CrossRef]
- Ferreira, B.A.; Coser, E.M.; de la Roca, S.; Aoki, J.I.; Branco, N.; Soares, G.H.C.; Lima, M.I.S.; Coelho, A.C. Amphotericin B resistance in Leishmania amazonensis: In vitro and in vivo characterization of a Brazilian clinical isolate. PLoS Negl. Trop. Dis. 2024, 18, e0012175. [Google Scholar] [CrossRef]
- Zhang, W.W.; Charest, H.; Ghedin, E.; Matlashewski, G. Identification and overexpression of the A2 amastigote-specific protein in Leishmania donovani. Mol. Biochem. Parasitol. 1996, 78, 79–90. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. Loss of virulence in Leishmania donovani deficient in an amastigote-specific protein, A2. Proc. Natl. Acad. Sci. USA 1997, 94, 8807–8811. [Google Scholar] [CrossRef]
- Hendrickx, S.; Bulté, D.; Van den Kerhof, M.; Cos, P.; Delputte, P.; Maes, L.; Caljon, G. Immunosuppression of Syrian golden hamsters accelerate relapse but not the emergence of resistance in Leishmania infantum following recurrent miltefosine pressure. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.M.; Shalev, A.; Benias, P.; Russo, C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J. Immunol. Methods 1998, 213, 157–167. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).