Abstract
Ongoing therapy for human parasite infections has a few known drugs but with serious side effects and the problem of drug resistance, impelling us to discover novel drug candidates with newer mechanisms of action. Universally, this has boosted the research in the design and development of novel medicinal agents as antiparasitic drugs with a novel mode of action. Histone deacetylase inhibitors (HDACis) are used in a vast variety of diseases due to their anti-inflammatory properties. Drug repurposing strategies have already approved HDACis as cancer therapeutics and are now under investigation for many parasitic infections. Along with the expression of the gene, histone deacetylase (HDAC) enzymes also act as a slice of great multi-subunit complexes, targeting many non-histones, changing systemic and cellular levels signaling, and producing different cell-based specified effects. Zinc (Zn2+)- and nicotinamide adenine dinucleotide (NAD+)-dependent HDACs of parasites play pivotal roles in the alteration of gene expression of parasites. Some of them are already known to be responsible for the survival of several parasites under odd circumstances; thus, targeting them for therapeutic interventions will be novel for potential antiparasitic targets. This point of view outlines the knowledge of both class-I and class-II HDACis and sirtuin inhibitors that emerged to be the key players in the treatment of human parasitic disorders like Leishmaniasis, Schistosomiasis, Malaria, Trypanosomiasis, and Toxoplasmosis. This review also focuses on repurposing opportunities and challenges in HDAC inhibitors that are preceded by their clinical development as potent new antiparasitic drugs.
1. Introduction
Parasitic disease globally affects a large number of humans and makes up one of the most concerning health problems, with notable morbidity and mortality. It is more prevalent in underdeveloped regions and developing countries. In 2020, 241 million cases and 627,000 deaths were reported worldwide due to malaria [1]. Similarly, in 2015, there were about 214 million clinical patients of malaria, with a mortality of 438,000 [2]. Apart from this disease, other parasitic diseases such as Leishmaniasis, Schistosomiasis, Lymphatic filariasis, and Trypanosomiasis cause notable health burdens. About 250 million patients have been infected with Leishmania and Schistosoma parasite infection [1,3,4]. Every year, the death toll resulting from parasitic diseases is increasing. Some research studies have suggested that these parasites may impair efficient immune responses, increasing the morbidity and mortality of COVID-19 patients. Alternatively, it has also been found that no discernible variations in disease severity were observed between co-infected individuals and naïve COVID-19 cases [5]. However, a Brazilian study indicated that people co-infected with parasite diseases and COVID-19 were less likely to develop severe COVID-19 [6].
RTS,S/AS01 (Mosquirix) and R21/Matrix-M are some of the recently approved vaccines for malaria by the WHO. However, the unavailability of vaccines for various human parasitic diseases makes the problem more urgent. Further, prevention and treatment are also sometimes difficult because of the side effects and developed drug resistance against the currently available therapeutics [7,8,9,10,11]. Thus, identifying novel druggable targets and chemotherapies with newer modes of action is of dire need. Small molecules that react with histones are effective for post-translational modifications of these epigenetic regulatory proteins and are gaining interest as a chemical arsenal as well as probable new leads that could suppress the parasite growth mechanisms [12,13,14].
Clinically, repurposing of the approved drugs is intriguing due to its efficiency in identifying novel applications. This approach expedites market availability and reduces economic costs compared to de novo drug discovery for parasitic diseases in tropical regions [15]. Based upon this approach, drugs approved as histone deacetylase inhibitors (HDACis) for the treatment of cancer are now being repurposed to target various parasitic diseases. However, the repurposing, especially with HDACis, faces hurdles. Variations in HDACis pharmacological profiles between diseases complicate the translation of efficacy from one condition to another. Regulatory complexities hinder their smooth transition, demanding robust evidence for safety and efficacy in the new therapeutic context. Ensuring safety necessitates understanding long-term effects, potential side effects, and interactions with existing treatments, which is also challenging. The absence of standardized drug testing methods adds complexity to evaluating HDAC inhibitors’ effectiveness in novel indications.
Histone and many non-histone proteins are being processed by HDACs that are occasionally called lysine deacetylases (KDACs). Histone/lysine deacetylases remove the acetyl group from histones and other associated proteins, while acetyltransferases append acetyl groups to such proteins. Similarly, the corresponding demethylases and methyltransferases eliminate and attach methyl groups to lysine side chains of proteins, respectively [16]. These post-translational alterations in the eukaryotes synchronize the regulation of transcription, cell cycle, and apoptosis [17,18,19,20]. Some human disorders, like cancers, express an altered expression of epigenetic regulatory proteins/enzymes and could be druggable targets [16,21,22]. HDACis are being investigated for their potential as antiparasitic drugs based on their mechanism of action in cancer, which include cell differentiation, cell cycle arrest, apoptosis induction, and immune system regulation. Studies have demonstrated that HDACis can cause parasites to undergo apoptosis, halt their growth, and alter the host immune system’s reaction to parasitic diseases. Furthermore, it has been discovered that HDACis promote apoptosis and other cell-damaging processes, which may be pertinent in the context of parasite infections, in order to increase tumor cell death [23]. Similarly, several epigenetic regulatory proteins exhibited a vital role during the evolution and life cycle of parasitic pathogens, with little resemblance or remarkable dissimilarity in principle catalytic domains to human proteins, making them compelling antiparasitic drug targets [12,24,25,26,27,28]. Homologs of HDAC have been identified in most parasitic pathogens of humans. The non-identical category of HDACishave exhibited their action in some of these resistance parasites, such as Plasmodium, Leishmania, and Schistosoma species [12,26,27]. Several HDACis have already received approval for therapeutic applications in various cancer types, making them potential candidates for addressing parasitic diseases. Noteworthy among the approved HDACis used in the management of peripheral or cutaneous T-cell lymphoma and applied in combination therapy for multiple myeloma are Belinostat (1) (PXD101), Panobinostat (2) (LBH-589), Vorinostat (3) (SAHA), and Romidepsin (4) (FK228) [29,30,31,32] (Figure 1).
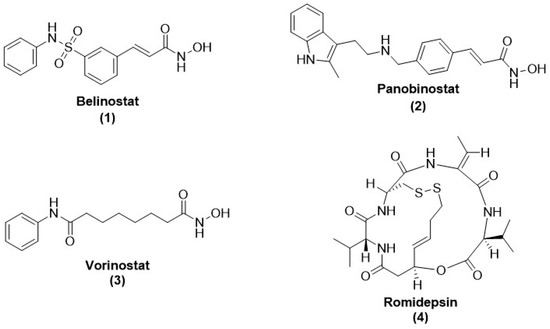
Figure 1.
Chemical structure of FDA-approved drugs acting as HDAC inhibitors. These HDACis have anti-cancer properties.
However, the discovery of antiparasitic drugs is challenging due to partial underlying molecular mechanisms for parasite resistance and a slow pace of identification and validation of novel drug targets. The complex life cycles of parasites with more than one host, morphological form, mode of reproduction (asexual and sexual replication), parasites’ genetic separation, and lack of easy genetic manipulation and biochemical characterization of many species have also sometimes hampered the process of drug discovery. The review is essential to address the urgent global health concern posed by parasitic diseases, which significantly impact human morbidity and mortality, particularly in underdeveloped and developing regions. Despite recent advancements in malaria vaccine development, many parasitic diseases lack effective vaccines, and the current treatments face challenges such as side effects and drug resistance. The review aims to fill a crucial knowledge gap by exploring the potential of HDACis in repurposing parasitic diseases.
2. Human Parasitic Illness
Several parasitic pathogens are involved in various diseases in humans, and the gravity of the disease is directly connected to the host’s immune status. Malaria is a mosquito-borne disease infecting 40% of the population of the African, Southeast Asian, and Western Pacific regions [33]. Six species of Plasmodium cause mortality. As per the WHO 2020, six million deaths were reported due to malaria, with more than two hundred million clinical cases of infection each year [34]. Children below the age of 5 years in indigenous regions affected by malaria have underdeveloped immune systems; thus, they are more susceptible to the threat of serious malarial disease and associated mortality [1]. Recently, the WHO approved Mosquirix (also known as RTS and S/AS01), an antimalarial vaccine, which acts through the active immunization process for extremely affected children [35]. Women having first pregnancies are more vulnerable to a serious form of malaria compared to non-pregnant women [36]. Further, immune-compromised native people with HIV/AIDS and travelers who have visited malaria-endemic regions are also at risk of severe malaria. A frightening signal of the development of resistance leading to treatment failure is also being detected. Plasmodium falciparum has become progressively resistant against chloroquine, and there is a progressive decline in the efficacy of artemisinin and its derivatives, the last line of defense, which leads to a terrifying condition [9,37,38]. The discovery and advancement of novel antimalarial drugs furnished with a novel mode of action is the rationale for the successful treatment of parasites.
Similarly, the causative agent for toxoplasmosis is Toxoplasma gondii, an obligate intracellular protozoan. In healthy persons, toxoplasmosis indications are self-limiting, but it is a more significant threat or is lethal towards women’s fetuses during pregnancy. These parasites are also responsible for serious opportunistic infections in immune-compromised people who received chemotherapy or organ transplants and people with AIDS [39]. The restricted ongoing treatment for toxoplasmosis is the combination therapy of pyrimethamine with a sulfonamide, but it suffers from efficacy problems along with serious adverse events [15,39,40].
One of the crucial neglected parasitic diseases across the globe is Schistosomiasis, which creates severe public health problems, mostly in the Middle East, Asia, South America, and Africa [41,42]. Among six species of Schistosoma, S. mansoni, S. trematodes, and S. haematobium are the leading causes of the disease in humans [43,44]. Globally, the infection of Schistosoma trematodes influences nearly 259 million people, with an annual death rate of 280,000. Further, chronic schistosomiasis develops long-term morbidity in millions of people [45,46,47,48]. Praziquantel is the premier drug to treat schistosomiasis [48,49]. It is an amazingly low-cost antischistosomal agent that is effective against all species of Schistosoma, although its mode of action is still not properly acknowledged [50,51]. It is administered in a single dose through the per oral (P.O.) route with negligible side effects [52]. Due to the emergence of drug resistance, the extended use of praziquantel has produced intense concerns over its potential [50,51,53,54,55]. Several reports have shown its reduced efficacy against some Schistosoma species and the development of drug resistance in some laboratory strains [56,57,58,59,60,61]. Based upon this lacuna, there is a need to develop novel antischistosomal agents.
Chagas disease is caused by Trypanosoma cruzi and has emerged as endemic in Latin America. This is a chronic infectious sickness that affects 6–7 million people worldwide [62]. The acute phase of Chagas disease is treated through benznidazole and nifurtimox [62,63]. Drug resistance is one of the largest constraints, including cytotoxicity, which causes severe side effects upon exposure to these available drugs [62,63,64,65]. Thus, for continuous management and attempts at disease eradication, the development of novel curative molecules with improved modes of action, ensuring increased efficacy and safety, is pivotal.
Another common tropical parasitic disease is Human African Trypanosomiasis (HAT), caused by Trypanosoma brucei. This epidemic developed terrible public health problems in sub-Saharan Africa but is now under check, with 10,000 or fewer cases annually reported [63]. The parasite enters the human body through the Tsetse fly, causing African sleeping sickness [66]. In humans, the parasite thrives freely in the bloodstream and can escalate the host’s immune reaction via antigenic drift [67]. If the parasite remains unchecked, then it leads to central nervous system infection and might be fatal [68]. Presently, the treatment strategy for this disease is based on a restricted number of drugs, such as eflornithine, pentamidine, nifurtimox, melarsoprol, and suramin.
Similarly, Leishmaniasis is a tropical vector-borne infection caused by the genus Leishmania, a protozoan parasite. There are three main manifestations of Leishmaniasis: cutaneous, visceral (kala-azar), and mucocutaneous. Among these, kala-azar is deadly if proper treatment is not undertaken. An estimate suggests 20,000–30,000 annual deaths and 0.9–1.3 million newly reported cases, creating an elevated risk of infection for 350 million people worldwide [69,70]. Leishmaniasis treatment activates a limited number of drug options, like antimony agents, paromomycin, meglumine antimoniate, amphotericin B, sodium stibogluconate, and miltefosine. Nonetheless, these drugs have limited potency and require long-term treatment with parenteral injection, causing severe toxicity [71,72]. The presently available treatments are more problematic in immune-compromised patients who have opportunistic infections like HIV/AIDS [73].
3. Classification and Biological Significance of Human Histone Deacetylases (HDACs)
Biological Significance of Human HDAC Isoforms
Harmonization of cell chromatin structure and gene expression of eukaryotic organisms is completed by pivotal enzymes through histone protein alteration. Histone acetyltransferase enzyme does open chromatin for ease in gene transcription, whereas HDACs oppose the acetylation that leads to a ‘closed’ chromatin state and ultimately suppresses transcription. However, the enhancement of the number of non-histone proteins revealed that HDACs perform several biological functions in eukaryotic cells, such as structural proteins, stability, interactions, and modification of proteins, DNA-binding properties, factors affecting transcription, chaperones, proteins that remodel chromatin, nuclear import proteins, and mediators of signaling [74,75]. Overall, these non-histone proteins play a crucial role in cell invasion, cell cycle regulation, tumor progression, angiogenesis, and apoptosis process [75,76,77]. The influence of HDACs causes many diseases like cancer, immunological disorders, metabolic disorders, and neurodegenerative complications [78]. This ensures HDACs as a remarkable drug target class.
Based upon similarity in sequence and dependence on cofactors, HDACs are classified into four classes in mammals: class-I HDACs (HDAC 1, 2, 3, and 8), class-II HDACs (HDAC 4, 5, 6, 7, 9, and 10), class-III HDACs, and class-IV HDACs (HDAC11). Class-I HDACs are confined in the nucleus and share greater homology to yeast RPD3 [79,80,81]. Class-II HDACs run between the cytoplasm and nucleus specific to tissue and share homology with yeast HDA1 [76,82]. Class-I and class-II HDAC proteins have an indistinguishable catalytic core that utilizes zinc ions as a cofactor but vary in size and structural organization. Class-III (Silent Information Regulator-2 (Sir2)-related protein (sirtuin)) comprise seven nicotinamide adenine dinucleotide (NAD+)-dependent enzymes (SIRT1-7) that share homology with yeast Sir2-related protein. Class-III HDACs have more specificity towards the tissue in the cytoplasm and nucleus and utilize cofactor NAD+. The latest work has spotlighted the role of class-III sirtuins in various functions of the cell, including repression of genes, apoptosis, repair of DNA, and the promotion of durability [53,75,80,83,84,85,86]. The mammalian class-IV HDACs, such as HDAC11, are localized in both the cytoplasm and nucleus and are allied with HDAC6, class-II enzyme, instead of class-multi-protein I HDACs complexes [87]. HDACis consistently target class-I/II HDACs that possess the same catalytic core as a cofactor (Zn2+). Thus, sirtuin activators and inhibitors (SIRTis) are designated as potential therapeutics for multiple diseases, such as cardiovascular diseases, cancer, diabetes, obesity, inflammation, various ageing-related diseases, and metabolic and neuronal diseases [79,81].
4. Functions and Roles of Human Parasite Histone Deacetylases (HDACs)
4.1. HDACs of Plasmodium Falciparum
The genome of Plasmodium falciparum comprises five HDAC-encoding genes. Three HDAC genes encode proteins that share mammalian homology with class I or class II (PfHDAC1, PfHDAC2, and PfHDAC3), while two genes are of class-III HDAC homologs (PfSir2A and PfSir2B) [15,25,26,88,89]. PfHDAC1 is confined to the nucleus, which comprises up to 55% amino acid sequences that share similarities to several eukaryotic class-I HDACs. These are transcribed in various stages of the protozoan lifecycle, such as the asexual intraerythrocytic phase, gametocytes, and sporozoites [12,26,88,90,91]. PfHDAC1 plays a key role in post-translational modification of histone and non-histone and gene expression in Plasmodium, which appears crucial for its survival [12,26,88,92,93,94,95]. Homology modelling analysis revealed that PfHDAC1 has an extremely conserved active site tunnel similar to that of human HDACs, but the differences at the entrance to the tunnel of active site differentiate from human HDACs (hHDACs) [92,93]. This dissimilarity may describe the better inhibitory activity of some HDAC inhibitors for P. falciparum as compared to mammalian cells [12,26,88]. Thus, it is also further exploited by new HDACis that could offer selectivity for PfHDAC1 over hHDACs [12,26,88,90,91,92,93]. It is foretold that both PfHDAC2 and PfHDAC3 are proteins with high molecular weight, having less than 14% amino acid residues identical to each other and restricted sequence homology with other class-II HDACs [25,89,90,91]. A knockdown experiment reported that PfHDAC3 inhibits P. falciparum parasite growth and survival in the sexual stage and controls its transcription [25]. Both PfSir2A and PfSir2B have 30% and 38% identical sequences in class-III HDAC (Archaeoglobus fulgidus) and group IV sirtuins, respectively. PfSir2A possesses both histone deacetylase and ADP-ribosyl transferase activity and is also effective for the removal of long-chain and medium fatty acyl groups [96]. The catalytic activity of PfSir2A maintains the length of telomeres, sets up heterochromatin in subtelomeric genomic areas, and modulates a subset of genes of P. falciparum implied in cytoadhesion, antigenic variation, pathogenesis, and virulence [94,97]. PfSir2 is not essential for in vitro growth of P. falciparum, but the regulation of virulence gene expression makes it a powerful target for antidisease therapy [97,98]. Similarly, PfSir2A and PfSir2B function as promoters involved in different telomeric-associated var gene subsets expression that encodes for erythrocyte membrane protein 1 (PfEMP1). It is expressed on the surface of erythrocyte P. falciparum, avoiding immune surveillance of the host at the time of P. falciparum infection [94,97]. PfSir2B also protects the end of the telomere. Knockout studies regarding PfSir2A and PfSir2B suggest non-mortality towards the parasite if any aspect is absent and set up functional dismissal in the parasite. Still, the concurrent knockout of PfSir2A and PfSir2B has not yet been assessed. PfSir2A and PfSir2B not only halt the in vitro growth and development of P. falciparum but are also useful for the regulation of var gene expression, which is in turn vital for the determination of in vivo survivability in the parasite inside a host. It has been suggested as a prospective target for antimalarial therapy. Specifically, P. falciparum’s sirtuins interrupt infected erythrocyte cellular adhesion towards the host cell receptor that conciliates the severity of the disease or obstructs the avoidance of Plasmodium from the innate immune system of the host [94,97]. It has been proposed that the var gene is not identified in other species of Plasmodium. Further, sirtuins (PfSir2A and PfSir2B) can express a subtelomeric gene that can divert the antigen through switching [97].
4.2. HDACs of Toxoplasma
The genome of T. gondii comprises five class-I and class-II HDAC homologs (TgHDAC1-5) besides two homolog subtypes of class-III HDACs, i.e., Sir2, along with the deviation of TgHDAC3 [12]. TgHDAC3 is confined in the nucleus and comprises greater than 60% sequences identical to hHDAC1. The TgHDAC3 sequence is a fragment of a huge multi-protein complex designated as a co-repressor complex of T. gondii, similar to the HDAC3-containing complex of humans. HDACs of T. gondii co-repressor complex encompasses TgHDAC3, T. gondii transducin b-like protein-1 (TgTBL1), two heat-shock protein-70 (HSP70)-like proteins, actin, and T. gondii TCP-1 ring complex (TRiC) subunits [59]. T. gondii HDACs along with deacetylase inhibitory activity additionally correlated with modulation of the gene in distinct forms of this parasite, such as stages of conversion from tachyzoite to bradyzoite [99]. However, the subtelomeric gene family has not been recognized in T. gondii. TgSir2A and TgSir2B proteins are suitably implied in processes other than antigenic variation [97] (Figure 2).
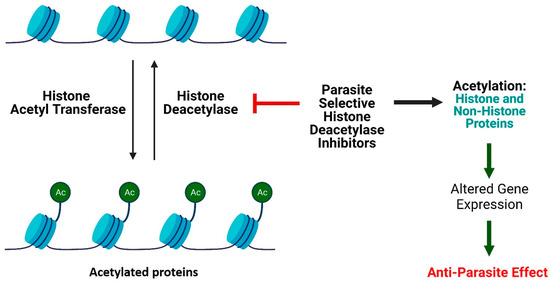
Figure 2.
Mechanism of action of HDAC inhibitors (created with BioRender.com). The dysregulation of dynamics of acetylation and deacetylation is responsible for Antiparasitic activity.
4.3. HDACs of Schistosoma
Schistosoma mansoni comprises three HDACs that have been identified as mammalian class-I HDACs orthologues [100]. Additionally, it also comprises some presumed class-II HDAC orthologues; nevertheless, no specific one has been functionally identified [101]. SmHDAC1 is a class-I homologue that suppresses transcription factor Gal4 or fusion protein Gal4-NF-kβ in mammalian cells through a co-expression or co-transfection system [100]. This suppression was partly reinstituted by the application of HDACicompound 4 (Trichostatin or TSA). Thus, it has been thought that it is HDAC-dependent. Whenever the two histidine residues present in the active site of the SmHDAC1 protein (H176/177A) were altered, simultaneously, Gal4-NF-kβ-dependent transcription was suppressed, specifying the principal role of these residues in the activity of the protein [102]. Still, schistosomes class-III HDACs have not been identified, although expressed sequence tags (ESTs) encoding multiple assumed members are available in the S. mansoni genomic datasets [103].
4.4. HDACs of Trypanosoma
There are four types of class-I/II HDAC orthologs (TbDAC1-4) available in Trypanosoma brucei [12,104,105]. TbDAC1 and TbDAC2 show identical sequences indistinguishable from mammalian class-I HDACs, whereas both TbDAC3 and TbDAC4 are more intimately linked to class-II HDACs [104,105,106,107]. Moreover, TbDAC1 and TbDAC3 are confined in the nucleus and crucial for the survival of parasites, while TbDAC2 and TbDAC4 are chiefly confined to the cytoplasm [106,107]. T. brucei, when present in the bloodstream of the human host, through the periodic switching of single-variant surface glycoproteins (VSGs), evades immunity. These surface glycoproteins are encoded by a massive range of VSG genes, but, at one time, just one VSG expression site is transcriptionally active [106]. Based upon this consideration, TbDAC3 displayed the need for a VSG expression site in the bloodstream and insect-stage cells through the cycling/silencing phenomenon, thus permitting the parasite to avoid the host immune defenses. In contrast, T. brucei carried three distinct homologs of sirtuin (TbSir2rp1-3) [12,108,109]. Nuclear-chromosome-associated protein TbSir2rp1 is not only essential for parasite survival but also for varied antigenicity. Subtelomeric gene suppression occurs in both blood stages and in the insect despite TbSir2rp1 participation in RNA-polymerase-I mediated transcription. Under DNA damaging conditions or during genotoxic stress, TbSir2rp1 is vital for the survival of parasites because, through catalytic reaction, there is an alteration, i.e., deacetylation and ADP-ribosylation, of histone protein, mostly H2A and H2B, responsible for the DNA repair. TbSir2rp1 is overexpressed to provide resistance toward DNA damage in the cell. Based upon this, the selective inhibitors of TbSir2rp1, combined with standard DNA-targeting chemotherapeutics, work to boost antiparasitic efficacy [108,109]. The individual knockouts of TbSir2rp2 and TbSir2rp3 do not affect the differentiation and proliferation forms of the bloodstream as they are mitochondrial proteins [108,109].
TcSir2rp1 genesis cytosolic protein and TcSir2rp3 genesis mitochondrial protein provide code for sirtuins in Trypanosoma cruzi [92,93]. Both sirtuins play vital roles in the multiplication of T. cruzi reproductive forms, host–parasite interactions, and various stages of the lifecycle [110,111]. TcSir2rp1 overexpression diminishes the growth and differentiation of the parasite, whereas TcSir2rp3 overexpression ameliorates the growth and differentiation of the parasite. A recent report showed that the salermide (SIRTi 3) inhibits TcSir2rp3, leading to the inhibition of growth and differentiation of T. cruzi (in vitro and in vivo). This indicates that sirtuin inhibitors are useful for chemotherapy of Chagas disease [111].
4.5. HDACs of Leishmania
The genome of Leishmania is also composed of four genes encoding homologs of class-I/II HDACs, whereas, for homologs of class-III HDACs (sirtuin), only three encoding genes are present [12]. Still, no one class-I/II HDAC species of Leishmania has been practically identified, but only one homolog is upregulated temporarily during differentiation (promastigote–amastigote) conducted without a host model system, signifying a probable role in regulating transcription or in chromatin structure modification [12,112]. Some studies have explored class-III HDACs’ useful roles in Leishmania species. Several research studies must look over the biological responsibility of sirtuins in unusual species of Leishmania specifically, also called Sir2rp1 (Sir2-related protein 1). The existence of this Sir2rp1 protein is observed in the granules of cytoplasm at distinct developmental stages of parasite Leishmania (promastigotes–amastigotes), such as in L. major, L. infantum, and currently in L. amazonensis [12,113,114]. A Leishmania, a major protein with considerable homology to yeast SIR2p Leishmania major Sir2rp1 (Leishmania major SIR2-related protein-1), has been identified, and these protein homologues are demonstrated in different Leishmania species’ cytoplasm and during developmental stages of the parasite (promastigotes and amastigotes) [113,114]. Both L. major Sir2rp1 and L. infantum Sir2rp1 represent ADP-ribosyltransferase and NAD+-dependent deacetylase activities independent of epigenetic silencing, as designated by gene disruption and overexpression studies. This revealed their crucial role in the in vitro and in vivo survivability of two Leishmania species [114,115,116].
L. infantum Sir2rp1, associated with deacetylation of α-tubulin, bears a resemblance to human Sir2 and HDAC6 cytoskeleton network function [115]. Several studies suggested that parasites’ soluble fractions overexpress L. major Sir2rp1 (this HDAC), which co-immunoprecipitates with HSP83, the Leishmania orthologue of mammalian HSP90 [117]. However, intracellular L. infantum Sir2rp1 quantity did not modify the state acetylation of L. infantum HSP83, so it is not so far comprehensible that L. infantum HSP83 is an HDAC substrate [118]. Sirtuins of Leishmania also perform the role of immunomodulation [12,115,118,119]. L. major Sir2rp1 brings out effector B-cell function, production of specific antibodies, as well as energizing differentiation of murine B-cell (in vivo). Recombinant L. major Sir2rp1 purified and expressed from Escherichia coli can activate the function of the B-cell effector, advocating the modulation of the immune system for this HDAC during infection [118]. Likewise, through Toll-like receptor-2 (TLR2)-mediated mode of action, L. infantum Sir2rp1 has the capability to activate splenic B-cells, which leads to upregulation of MHC-II, a major histocompatibility complex, and CD40 and CD86 molecules. It is also responsible for the release of tumor necrosis factor in the working mouse model [119].
The host innate immune system and this activation pathway entail the signaling pathway of Toll-like receptors in the initial identification of Leishmania parasites and may lead to the development of resistance towards infection and provide help for the development of immunity. During monitoring of TLR2-dependent activation of B-cells, after immunization, TLR2-deficient mice could still produce antibodies to L. infantum Sir2rp1, showing that L. infantum Sir2rp1 may also link with other types of cells in the absence of TLR2 [119]. The literature studies on pathogens and signaling of host products through TLR2 and TLR4 remain ambiguous.
The Leishmania Sir2rp1 immunomodulatory role increases the chance that TLR2 and TLR4 protein might function as a probable adjuvant in a multi-component vaccine. Particularly, the sirtuin HDAC proteins are pivotal for the growth of the Leishmania parasite (in vitro and in vivo). Moreover, previously, several researchers have focused on targeting multiple inhibitors with the ambition of recognizing the most effective antileishmanial agents [120,121,122].
5. Opportunities of HDAC Inhibitors as Antiparasitic Agent
5.1. HDAC Inhibitors as Antimalarial Agent
For decades, HDAC inhibitors have been employed as antimalarials. Apicidin (a fungal-derived HDACi) was initially identified as an effective HDACi against Apicomplexan protozoa and Plasmodium parasites [123]. Ever since, many modified HDACi classes have been documented as potent antimalarial agents.
5.1.1. Cyclictetrapeptide HDAC Inhibitors
Darkin-Ratray et al. observed in vitro potent activity of apicidin (5) against P. falciparum, T. gondii and other protozoan parasites. Hyperacetylation of histones was observed in P. falciparum upon treatment with apicidin. It inhibited the recombinant PfHDAC1 enzymatic activity (IC50 = 1 nM) [95,123]. Notable alterations were observed during the transcriptional phase of the intra-erythrocytic cycle of P. falciparum, with altered expression by other genes [124,125]. These findings revealed a similar effect produced by the compound as in HDACis on higher eukaryotic cells [126,127]. Apicidin caused parasitic histone protein hyperacetylation and genome-wide transcriptional alteration in the protozoan [95,124]. The derivatives of a quinolone, but not derivatives of N-substituted indole, showed their selectivity (up to ~200-fold) for P. falciparum versus mammalian cells at the whole-cell level [128,129,130]. In another study, a comparison was made between apicidin and a series of synthetic analogues for T. cruzi, T. brucei, P. falciparum, and L. donovani [131]. The cyclic tetrapeptide compound FR235222 (6), isolated from Acremonium species in the fermentation broth, showed its potential against tachyzoites of T. gondii and drug-sensitive and drug-resistant P. falciparum-infected erythrocytes with IC50 value of 10 nM [132] (Figure 3).
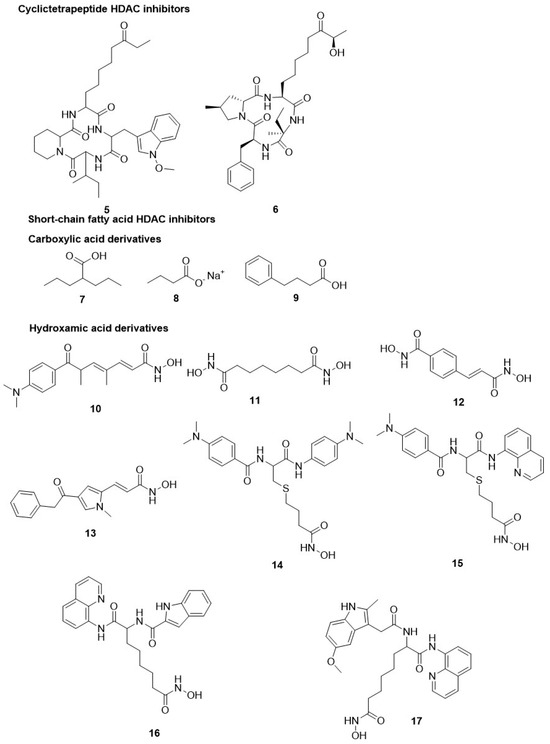
Figure 3.
Chemical structure of cyclictetrapeptide and short-chain fatty acid HDAC inhibitors. Cyclictetrapiptide, carboxylic acid hydroxamic acid derivatives have shown HDAC inhibitory activity.
5.1.2. Short-Chain Fatty Acid HDAC Inhibitors
Several drugs, like valproic acid, sodium butyrate, and its derivatives, are short-chain fatty acid HDACis. These HDAC inhibitors possess weak inhibitory activity in opposition to mammalian HDACs but are also active on additional targets, making their effects tough to ascribe beyond HDAC inhibition. However, some of these compounds have manifested clinical efficacy. Valproic acid (7), an antiepileptic drug, has been extensively used as a mood stabilizer and is now investigated in several clinical trials for HDAC-pertinent diseases [133]. All compounds, valproic acid, sodium butyrate (8), and 4-phenyl butyrate (9), displayed weak selectivity towards the parasite in comparison to HS69 mammalian cells and produced relatively weak activity against tachyzoites of T. gondii, with IC50 values of 1.0, 1.6, and 5.4 mM, respectively [134,135]. Similarly, valproic acid only displayed moderate activity against P. falciparum and S. mansoni. Particularly, with S. mansoni, an ~80% reduction in miracidia development was observed upon treatment with 50 mM valproic acid after 4 h. Nonetheless, there is a loss of viability of schistosomula up to 30% at 5 mM concentration after 7-day continued exposure [104]. Still, valproic acid has so far not been evaluated in animal models for any parasitic disease [104,135] (Figure 3).
5.1.3. Hydroxamate-Based HDAC Inhibitors
Several natural and modified HDACis for class I/II, such as trichostatin A (TSA, 10), vorinostat or suberoylanilide hydroxamic acid (SAHA, 3), suberoyl bis-hydroxamate (SBHA, 11), MW2796 (12), and aroyl-pyrrolyl hydroxyamide (APHAs, 13), have been considered for their antimalarial potential [136]. These compounds displayed inhibitory potential against P. falciparum but exhibited a lack of selectivity affecting the mammalian host cells. However, compound 11 showed effective sensitivity for the parasite compared to cytotoxicity against mammals [12,26,88,95,137,138,139].
TSA hampered PfHDAC1 activity, with an IC50 value of 0.6 nM in parasites. It is more potent than SAHA against P. falciparum, with IC50 values of 0.008–0.120 µM compared to SAHA 0.025–2.2 µM. However, SAHA showed improvement in the selectivity up to 200-fold toward the parasite [12,26,137]. In contrast, compound 11 is less potent with IC50 0.8–2.3 µM but showed higher selectivity towards the parasite than compound 3. Further, this comparative study was conducted in P. berghei-infected BALB/c mice; the administration of compound 11 at an i.p. dose of 200 mg/kg twice a day for 3 days showed significant inhibition of peripheral parasitemia, indicating a cytostatic effect, but the efficacy was not observed in the mice [137]. The early report about compound 11 suggested that hydroxamate-based HDACis would probably be powerful antimalarial agents [12,26,139]. Hence, encouraged by such outcomes, various HDACis displayed in vitro activity with better selectivity against P. falciparum than compounds 3 and 11.
Over the years, the screening of zinc-binding hydroxamic acid compounds revealed varying levels of inhibition of HDACs. These structural patterns resulted in the identification of extensive pharmacophoric groups binding to other important sites for HDACs. The CAP group acts as an enzyme surface identification moiety of protein and binds at the catalytic tunnel entrance. The linker region connects the CAP with the zinc-binding group site. This site is an expandable polar connection unit (CU) that lies between the CAP protein and the linker [12,26,77,88] (Figure 3).
Some L-cysteine-based compounds with thioether linker regions, such as compound 14 and 2-aminobutyric-acid-based compound 15 (2-ASA-9), displayed indistinguishable in vitro antiparasitic activity. Compound 14 displayed an IC50 value of 48 nM against chloroquine-resistant (Dd2), and compound 15 showed IC50 values of 71 and 19 nM against both Dd2 and chloroquine-sensitive (3D7) strains, respectively, of P. falciparum. Compound 15 displayed improved selectivity over the mammalian cells. Among them, compounds 15 and 11 produced a noticeable hyperacetylation of histones in P. falciparum, which led to the inhibition of the growth of P. falciparum in erythrocytes at early and later stages in the parasite life cycle [93]. One reported study revealed that compounds 10, 5, and 15 were explored for their gene expression effects, indicating genome-based alteration of transcription in a range of 2–21% following HDAC inhibition in P. falciparum. However, these three inhibitors overall displayed dissimilar effects on gene expression profile. However, tubulin II was found to be one of the sets of genes that were upregulated after treatment with three HDACis. Hence, they could be recognized as markers of transcription in P. falciparum induced by structurally variable HDACis. Thus, this marker might be used for the development of HDACis identified as antimalarial candidates [140].
Compounds 15, 16, and 3 were also the earliest HDACis tested against the second most crucial human-infecting malaria parasite P. vivax. It causes significant morbidity linked to malaria due to relapses as the parasite remains dormant in the liver [26]. In an ex vivo study, all three hydroxamates inhibited the growth of multidrug-resistant P. falciparum and P. vivax directly isolated from infected patients [141]. Furthermore, a similar activity profile was observed for compounds 3, 15, and 16 against P. falciparum with IC50 values of 310, 533, and 266 nM, respectively, as well as P. vivax with IC50 values of 170, 503, and 278 nM, respectively. HDACis target multiple species of malaria parasites that infect humans; hence, they are highly favorable for further clinical studies. Several series of 2-aminosuberic acid compounds were tested, including compound 17 carrying non-steroidal anti-inflammatory drug (NSAID) components near CAP against P. falciparum, for their inhibitory potential. Compound 17 showed the utmost potential against P. falciparum, with an IC50 value of 0.013 µM, but the selectivity towards the parasite compared to compound 15 did not prevail (Table 1) [142].

Table 1.
In vitro activity of antischistosomal, antimalarial class-I, class-II, class-III, antitrypanosomal class-I/II, antileishmanial class-I/II, and antitoxoplasmal class-I/II/IV HDAC (sirtuin) inhibitors.
Patel et al. performed a high-throughput assay to study the antimalarial efficacy of approximately 2000 HDACis obtained from the compound library. The library was characterized by a moiety of acyl hydrazone as CU and with diverse moieties for CAP protein and zinc-binding group along with the hydrophobic linker length such as 4–6 methylene units [81]. The study revealed that numerous compounds strongly inhibited the growth of P. falciparum and also hampered recombinant PfHDAC1 enzymatic activity. Among them, seventeen derivatives displayed a low range of nanomolar antiparasitic activity for acetylation of histone along with minimal perturbation of human myeloma MM1S cells as an indicator of selectivity [95]. Within this series, the selective inhibition of P. falciparum growth was highly enhanced by the existence of ortho-substituents, such as bromine and hydroxyl, in the aromatic group of CAP protein along with the presence of metal chelator or hydroxamic acid and five methylene units as a linker (compound 18, Table 1) [95]. Another study by Kozikowski et al. on two series of suberoyl amide hydroxamates substituted moieties of triazolyl phenyl and phenyl thiazolyl as CAP protein groups [137,143]. The triazolyl phenyl-based compound 19 showed high efficacy against the multidrug-resistant strains of P. falciparum (C2A and C235), with IC50 values in a range of 0.017–0.035 µM. It was 10-fold more potent compared to its counter congeners such as chloroquine and mefloquine. Compound 19 was 23-fold highly selective for C235 over mammalian cells [143]. In a group of 50 phenyl, thiazolyl hydroxamate-based HDACi, three compounds were highly potent with IC50 values < 3 nM and showed 600-fold selectivity against P. falciparum compared to mammalian cells. Compound 20 (WR301801) was the most favorable HDACi derivative, with IC50 values 0.6–16 nM for several drug-resistant strains, D6, W2, C235, and C2A, of P. falciparum. It displayed notable inhibition of HDAC activity in nuclear extracts of P. falciparum with IC50 ∼10 nM along with a strong in situ parasitic histones hyperacetylation [137]. In another report, compound 20, upon oral administration as monotherapy, at doses of up to 640 mg/kg demonstrated a notable suppression of parasitemia but did not rehabilitate P. berghei-infected mice. However, some mice, but not all mice, were rehabilitated upon treatment with compound 20 (52 mg/kg) when co-administered with a subcurative dose of chloroquine (64 mg/kg) [137]. Similarly, compound 20 on oral administration at a dose of 32 mg/kg/day for 3 days to Aotus monkeys infected with P. falciparum resulted in suppression of the parasite but not complete elimination [137]. Another reported study showed that compound 20 (50 mg/kg/day for 4 days) on i.p. injection with an experimental follow-up period of 6 weeks enhanced survival of infected mice infected with P. berghei and irreversibly downregulated the parasitemia [144]. However, the optimization of the pharmacokinetic properties of compound 20 would show advantageous results as it is quickly hydrolyzed to the corresponding inactive carboxylic acid [137]. These findings suggest the potential of HDACi in mono/combination therapy for malaria treatment [12,26,137,144]. Oyelere et al., reported a series of aryl triazolyl hydroxamate-based HDACis that were screened for their inhibitory activity against L. donovani promastigote stages and asexual blood stage of P. falciparum [145]. Several compounds displayed improved inhibitory activity and selectivity than compound 3 against D6 and W2 strains of P. falciparum. Although several compounds are less active against P. falciparum, they showed a modest potency of inhibition against L. donovani, with 2- to 4-fold better IC50 values than compound 3 and equivalent to the standard drug miltefosine, used to treat visceral Leishmaniasis. Antiparasitic activity depends on polymethylene linker length and the nature of the CAP group. The CAP moiety showed maximum activity against both parasites when 5 and 6 methylene units were present in the spacer region between the CAP and the ZBG. Compounds 21 and 22, with a CAP protein (3′-biphenyltriazolyl moiety) and linker (6 and 5 methylene units), showed the highest selectivity and activity against P. falciparum. Compound 22 also showed its activity against L. donovani with an IC50 of 32 µM [145]. Based upon this concept, Oyelere et al., tested five tricyclic ketolide-based phenyltriazolyl HDACi for antimalarial and antileishmanial activities [146]. During the study, it was noticed that compound 23, with six methylene units as the optimal linker present between the CAP and hydroxamic acid, displayed optimal antimalarial activity. Similarly, compound 24, hydroxamic acid with nine methylene units, displayed the best antileishmanial activity, but the study does not correlate with the PfHDAC1 inhibition [146]. Particularly, compound 23 displayed IC50 in the range of 0.144–0.148 µM against both chloroquine-sensitive (D6) and chloroquine-resistant (W2) P. falciparum strains. However, the compound displayed 7 to 10-fold lesser activity than compound 3 and 10-fold high selectivity against mammalian Vero cells without antileishmanial activity. Further, compound 24 showed anti-L. donovani activity with an IC50 of 5 µM, which was 16-fold stronger than compound 3 with an IC50 of 81 µM [146] (Figure 4).
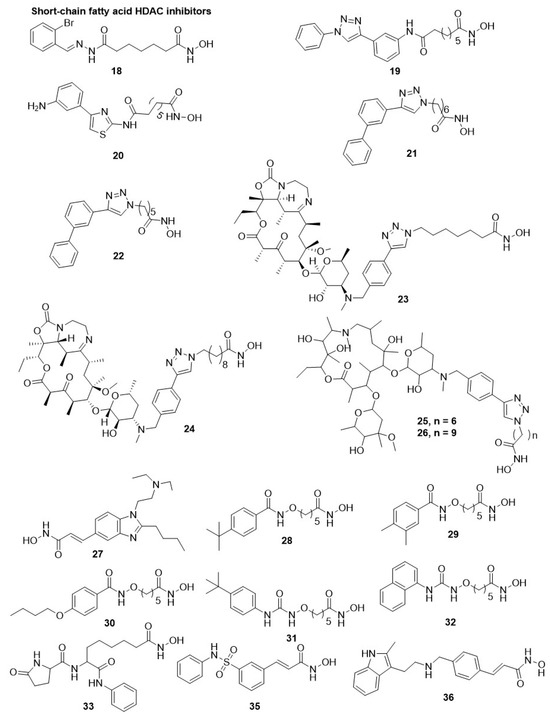
Figure 4.
Chemical structure of hydroxamic acid HDAC inhibitors. The short chain fatty acid displays diverse HDAC inhibition.
Oyelere et al., further investigated and described the antimalarial and antileishmanial potential of 14 and 15-membered nonpeptide macrocyclics connected to a moiety of phenyltriazolyl, a CAP protein group, as HDACis. All compounds inhibited the proliferation of chloroquine-sensitive (D6) and chloroquine-resistant (W2) strains of P. falciparum [155]. However, the best activity and selectivity against P. falciparum were attained for the six methylene units linker between CAP (triazole ring) and ZBG group (hydroxamate). Among them, compound 25, a skeleton of 15-membered macrolide with an IC50 value of 29 nM against PfHDAC1 enzyme, manifested 11-fold higher antiplasmodial activity with 14-fold enhanced selectivity over mammalian Vero cells compared to compound 3. Surprisingly, the linker group having five to seven methylene units was lacking anti-L. donovani activity on the promastigote stage for both skeletons of a macrolide, whereas the compound having eight or nine methylene units as linker displayed the highest activity similar to ketolide-based HDACis, but distinct from the structure–activity relationship was noticed in aryl triazolyl hydroxamates [145].
Specifically, compound 26 with a skeleton of 14-membered macrolide and methylene units linker was 25-fold more active than compound 3 with an IC50 value of 81 µM, showing the best antileishmanial activity, with IC50 values of 3.2 and 4.7 µM against promastigote and amastigote stages of the parasite, respectively [155]. Andrews et al., identified compound 27 (pracinostat or SB939), an orally active anticancer HDACi, having antiplasmodial activities [156]. Furthermore, compound 27 strongly hampered the proliferation of the asexual-stage of P. falciparum in mammalian erythrocytes, with IC50 values in the range of 0.08–0.15 µM. Compound 27 also caused hyperacetylation on histone and non-histone proteins in the parasite and displayed selectivity over mammalian cells ranging 4 to >1250-fold over tested cell lines [156]. Additionally, an additive effect was first observed for its combination with lopinavir, a protease inhibitor, as antimalarial targeting a liver stage. Compounds 27 strongly inhibit the exo-erythrocytic stage of P. berghei in human HepG2hepatocytes, with an IC50 value of 150 nM [156]. The oral administration of compound 27 up to a dose of 100 mg/kg/day decreased peripheral parasitemia and total parasite load in the ANKA mouse model infected with P. berghei. However, the administration of compound 27 to mice fended off the occurrence of cerebral malaria-like symptoms up to 2–3 weeks after the intervention but did not affect hyperparasitemia in the treated animals [156]. Oyelere et al. also explored a group of 21 HDACi with Pentyloxyamide as a linker and substituted benzene ring as CAP for their effect against different stages of Plasmodium, such as P. falciparum (a sexual blood stage (3D7) cell line), P. berghi (tissue schizontocidal stage), and late stage of P. falciparum gametocyte (IV and V). All compounds exhibited antimalarial activity against P. falciparum asexual form with potency and selectivity enhanced as the bulkiness of alkyl/alkoxy substituents in the phenyl ring at the para position were increased regarding compound 28. Three derivatives, compounds 28, 29, and 30, showed their activity against all three stages of P. falciparum with IC50 values of 0.09, 0.12, and 0.17 µM, respectively. Several compounds of this series displayed better selectivity against the parasite compared to compound 3 for the asexual and exo-erythrocytic life cycle stages of the parasite [157]. Further, Oyelere et al., described the structure–activity relationship and antimalarial activity of a group of HDACi based on alkoxyurea. Some compounds actively inhibited P. falciparum (3D7cell line) and showed gametocytocidal activities at early and late-developmental stages with IC50 values ranging 1.68–6.65 µM [147,148]. Structure–activity relationship study revealed that the hydroxamic acid as a zinc-binding group and 5 methylene units as a linker were important for antiplasmodial activity N-methyl hydroxamic acid, o-hydroxyanilide and o-amino anilide were inactive as a zinc-binding group, while the short-chain analogues with linker less than 5 methylene units displayed lower potency [147]. Furthermore, the 4th position is substituted with bulky alkyl/alkoxy substituent of the phenyl ring of the CAP group. When this group is replaced with bulky aromatic rings leads to the development of more potent and effective compounds against asexual and gametocyte forms of P. falciparum, as denoted by the 4-tert-butyl derivative (31) and 1-naphthyl derivative (32). However, compounds 31 and 32 did not display better potency than compound 3, but, under the test condition, 32 displayed higher selectivity than compound 3 along with gametocidal activity in the asexual and later stages [148].
In 2015, Giannini et al., evaluated twelve analogues for their antiparasitic activity against P. falciparum, T. cruzi, L. donovani, G. lamblia, and T. brucei. These twelve analogues were differentiated at the α position of the anilide (CU) and meta position of the phenyl ring (CAP) and substituted with β-lactam-carboxamides and trifluoro methyl groups, respectively. Additionally, a thiol or hydroxamic acid is present as a zinc-binding group. Compound 33 of this series displayed a significant IC50 of 0.019 µM against P. falciparum [149]. In 2015, Andrews et al., described the potential of four clinically approved HDACi, compounds 34 (romidepsin or FK228), 35 (belinostat), 36 (panobinostat), and 3, for cancer treatment against P. falciparum and T. brucei parasites. All compounds inhibited the growth of P. falciparum parasite, with IC50 values ranging from 0.09–0.13, 0.025–2.2, 0.06–0.13, and 0.01–0.03 µM, respectively. Interestingly only compound 34 was active against the bloodstream form of T. brucei, with an IC50 value of 35 nM, despite lacking mammalian cell selectivity. These four HDACi inhibited the P. falciparum nuclear extract deacetylase activity and recombinant PfHDAC1 due to hyperacetylation of non-histone protein and contrastingly affecting histones (H3 and H4) acetylation. Compounds 3 and 35 did not show selectivity towards malaria parasites over mammalian HEK29 and NFF 3 cells [148] (Figure 5).
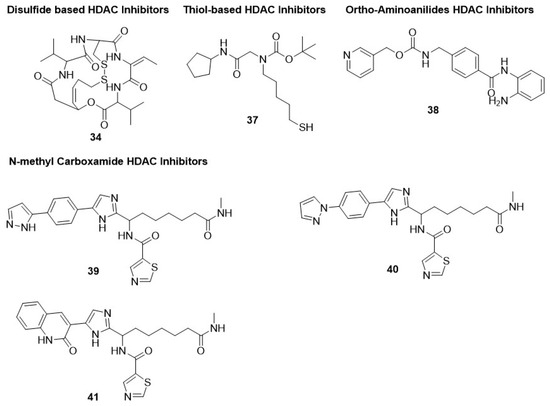
Figure 5.
Chemical structure of disulfide-based, thiol-based, orthoamino-anilide-based, and N-methylcarboxamide-based HDAC inhibitors. These inhibitors displayed anti-malarial activity.
5.1.4. Thiol-Based HDAC Inhibitors
Thiol-based HDAC6-selective inhibitor 37 displayed poor inhibitory potential against chloroquine-sensitive (3D7) and chloroquine-resistant (Dd2) strains of P. falciparum, with IC50 values in the range of 15.2–19.9 µM [150]. Thus, further verification is needed to validate the antimalarial potential of hydroxamate-based pan-HDACi [93] (Figure 5).
5.1.5. Ortho-Aminoanilides HDAC Inhibitors
A similar inference can be made by choosing an ortho-amino anilide group as a zinc-binding group. The precursor of these HDACi in the class-I-HDAC-selective inhibitor is denoted as compound 38 (MS275 or entinostat) [154,155,156]. Multiple trials have been completed on compound 38, revealing its potential as a PfHDAC1 inhibitor and parasitic proliferation inhibitor, with IC50 values of 0.94 and 8 µM, respectively [12,26,88,93,95]. Compounds 39, 40, and 41 are N-methyl carboxamide derivatives that displayed antimalarial potential against P. falciparum [158] (Figure 5).
5.2. Antimalarial Class-III HDAC (Sirtuin) Inhibitors
Fewer Sir2 inhibitors have been tested against P. falciparum-infected erythrocytes for their antiproliferative activity and inhibition of the recombinant PfSir2A protein [12,26]. Generally, the Sir2 inhibitors showed moderate activity against the growth of P. falciparum. A poor homology has been observed between the parasite and other eukaryotic Sir2 proteins [97,98]. Several known natural and synthetic sirtuin inhibitors that have been investigated over the years for growth inhibition of P. falciparum. Compound 42 (sirtinol), 43 (splitomycin), 44 (surfactin), and 45 (hyperforin) displayed IC50 values in the range of 9–13, >10, 9, and 1.5–2 µM, respectively, against P. falciparum [153,154,159,160]. Compound 46 (nicotinamide) is a Sir2-catalysed reaction product and causes non-competitive inhibition of both acetylated peptide and NAD+ but less active at the whole-cell level, with an IC50 value of 9.9 mM for retarding parasite growth [161]. Nicotinic acid, also a product of Sir2-dependent enzymatic pathways, did not show this effect [161].
Compounds 44 and 46 displayed different activity against PfSir2A, with IC50 values of 35 and 51 µM, respectively. Meanwhile, compounds 43 and 44 showed less active activity, with IC50 values of >400 and >50 µM, respectively. Chakrabarty et al., synthesized analogues of lysine-based tripeptide based on a mechanism of competition with the binding pocket of PfSir2 [162]. From the four analogues, three analogues showed equivalent or improved activity, with IC50 values in the range of 23–34 µM against PfSir2A in contrast to compounds 44 and 46 [162]. Compound 47 was highly potent against PfSir2A and also tested against P. falciparum-infected erythrocytes where it displayed parasitic inhibitory potential similar to compounds 34 and 36 with an IC50 value of 9.8 µM [162]. Resveratrol (48) and isonicotinic acid (49) are activators of hSIRT1 that moderately inhibit P. falciparum growth, but during in vitro testing against recombinant PfSir2A, no enzymatic activation or inhibition was reported [79,160]. However, this is fully anticipated as PfSir2A and PfSir2B are not required for the growth and development of the parasite and share a few homological sequences with Sir2 proteins of other eukaryotes [97,98]. In addition, several compounds, 42, 43, 46, and, to a lesser extent, compound 44, displayed lower inhibitory potential against PfSir2A and the in vitro antiproliferative effects due to their multiple unrelated characters at a single gene locus. Thus, the identification and development of inhibitors with a notable enhancement of potency against PfSir2 and the selectivity towards sirtuins of humans could be a functional gadget considering P. falciparum sirtuins’ biology and their pharmacological validation as a target of drugs that either directly or indirectly produce antiparasitic activity through obstructing the parasite avoidance from the host innate immune system [97,98] (Figure 5).
5.3. Antitrypanosomal HDAC Inhibitors
A few compounds were tested against the Trypanosoma species. Compound 10 appeared to prevent the spread of the bloodstream form of T. brucei at a concentration of 7 µM but without affecting a silent variant surface glycoprotein expression site [163]. Another study was conducted where the similar compound 10 hampered recombinant TbDAC3 and TbDAC1 activity by >50% at 5 µM concentration. However, this compound cannot alter the acetylation of the histone protein of the parasite at a concentration of 0.3 µM [146]. Further, four HDACis, compounds 34, 3, 35, and 36, were also tested by Engel et al., for the inhibition of trypanosomal and malaria parasites [164]. The result of the study revealed that all inhibitors effectively inhibit T. brucei but all of them displayed cytotoxicity towards the human NFF and HEK 293 cells at lower concentrations, with a selectivity index less than 1 [136]. However, compound 34 is highly potent against T. brucei with an IC50 value of 35 µM.
In 2012, several clinically approved hydroxymates-based HDACis like long-chain amides, sulphonamides, heterocycle-containing acrylamides, and sulphonylpiperazines were tested against a cultured bloodstream form of T. brucei for their inhibitory effect [165]. The result of the study indicated that long-chain amides with IC50 values > 10 µM displayed a notable inhibitory property on the growth in the range of IC50 values 0.034–1.54 µM. Among them, sulphonylpiperazines with heteroaryl ring attached to the piperazine moiety displayed potent inhibition. Compound 50 displayed an IC50 value of 34 µM and was able to induce parasite death at 2 µg/mL within 4 h after treatment. All four compounds displayed powerful inhibition of the hHDAC activity, with IC50 values in the range of 0.010–0.212 µM [165] (Figure 6).
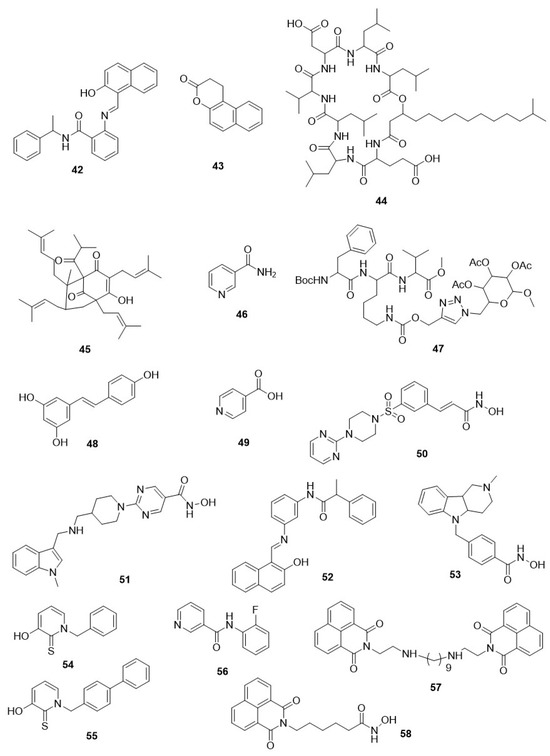
Figure 6.
Chemical structure of other HDAC inhibitors. Diverse chemical structure shows varying range of parasitic HDAC inhibition.
Lately, two recently approved hydroxymate-based HDACis (36, 51) and four hydroxymate-based HDACis that are already available as anticancer agents under clinical trial were investigated to determine their inhibitory capacity on the proliferation of T. brucei in the cultured bloodstream. The result of the study revealed that all compounds moderately inhibited the parasite, with EC50 ranging 0.029 to 11 μM. The most effective compound was 51 (quisinostat) [166]. Compounds 35 and 36, anticancer drugs, were unable to destroy the cultured parasites at the human-tolerated doses after a single dose administration. Furthermore, compound 36, a sustained-acting HDACi when administered with Human African Trypanosomiasis (HAT) drugs, like pentamidine, suramin, melarsoprol, and nifurtimox, did not display a synergistic effect [166]. Accordingly, the application of these two HDACis as single agents or in combination for repurposing in HAT treatment is ruled out. Overall, there is lack of associations between the potency against any isoform of hHDAC and the prevention of T. brucei proliferation. From this idea, the author proposed that HDAC of trypanosome might have a distinctive specificity that could be used for the development of powerful HDACis having selectivity towards the parasite over hHDACs [166].
5.4. Antitrypanosomal Class-III HDAC (Sirtuin) Inhibitors
Compound 46 has been described as a putative inhibitor of T. cruzi sirtuin TcSir2rp1 and a growth inhibitor of the parasite at its developmental stages, including trypomastigote and epimastigote. It is evident by a notable inhibition in the number of amastigotes in the T. cruzi-infected macrophages [167]. However, besides SIRT inhibition, it also possesses a pleiotropic response; i.e., one gene influences many phenotypic expressions [167]. Similarly, Moretti et al., characterized two sirtuins, TcSir2rp1 and TcSir2rp3, of T. cruzi and described that compound 52, an antitumor agent in a different model of cancer, could be a potential SIRTi and inhibit growth and differentiation of T. cruzi [79,111,168,169]. Compound 52 diminished the proliferation of epimastigote, with an EC50 value of 10.6 µM, and prevented its transformation into infective forms. Furthermore, the activity of the recombinant TcSir2rp3 was hampered by compound 52, resulting in an IC50 value of <1 μM. Hence, it could be proposed that the antiparasitic activity of the compound occurred via TcSir2rp3 inhibition.
Experimental evidence holds up this finding that, with the addition of compound 52, the overexpression of TcSir2rp3 and all phenotypic effects were reduced. Appreciably, compound 52 reduced parasitemia in a T. cruzi-infected BALB/c mouse model at a lower dose in comparison to a higher in vivo dose that is used for cancer treatment. Nonetheless, compound 52 was not able to prevent the mortality of mice at this dose. Thus, rationally, it can be used to develop a more potent and selective isoform-based SIRTi of the parasite as a potential agent against T. cruzi infection [111].
Furthermore, a molecular docking study of pan-SIRTi regarding compounds 42, 46, and SIRTi (thiobarbiturate derivatives) of class-III inhibitors revealed that there was strong correlation between human mitochondrial SIRT5 and TcSir2rp3 with respect to strength and binding mode [170]. Although the comparison of TcSir2rp3 catalytic pocket with human protein SIRT5 homologous through in silico surface and structural analysis has permitted the recognition of insignificant but notable structural differences at specific inhibitory catalytic domains, that would probably be used for the development of preferable TcSir2rp3 inhibitors with higher specificity towards T. cruzi [170].
5.5. Antileishmanial HDAC Inhibitors
The response of Leishmania parasite towards HDACi indicates the HDACs’ specificity and importance for survivability [12,120,145,146,155]. Both antimalarial and antileishmanial properties of hydroxamate-based class-I/II HDACis were discussed by Oyelere et al. [155]. Furthermore, they studied the effect of 3-hydroxypyridine-2-thiones (3HPTs) on the L. donovani survivability in amastigote and promastigote forms, which were earlier described as inhibitors of hHDAC6/hHDAC8 [120]. Compound 3, the pan-HDACi, along with compound 53 (tubastatin A), another hydroxamate-based HDACi, and the reference compound PCI-34051, a selective inhibitor of hHDAC6 and hHDAC8, was taken [120]. Compound 53, an hHDAC6-selective inhibitor, produced equal antileishmanial activity at both the developmental stages of L. donovani. This effect was not observed with compound 3 and PCI-34051, a selective hHDAC8 inhibitor. The result also revealed that compounds 54 and 55, 3-HPT-derived HDACi, produced cytotoxicity both at intracellular and extracellular species of L. donovani, with IC50 values ranging from 0.1 to 6.5 µg/mL. Thus, the investigator suggested that the antileishmanial efficacy resulted in L. donovani due to inhibition of HDAC6. This paves a new way for protozoan HDAC6-like activity besides selective isoform inhibitors that might develop a more attentive therapeutic primary plan for Leishmaniasis treatment [120].
5.6. Antileishmanial Class-III HDAC (Sirtuin) Inhibitors
Still, a smaller number of SIRTis furnished with antileishmanial activity have been described [12]. Compound 44 demonstrated developmental-stage-specific antiproliferative properties against L. infantum parasites [171]. The compound led to apoptotic cell death due to DNA double helix breakage and inhibited the multiplication of axenic amastigotes but did not alter the growth of parasite promastigotes with an IC50 value of >60 µM [171]. Overexpression of LmSir2rp1 of parasite results in less susceptibility towards the fragmentation of DNA on the treatment of compound 12 [171].
Similarly, compound 46 can inhibit the growth of L. major; even LmSir2rp1 overexpression did not protect the parasite against this compound [172]. Several in silico and biochemical studies disclosed significant differences between the LmSir2rp1 and catalytic domains of human SIRTs [102]. In another in vitro investigation, four compounds effectively inhibited the axenic amastigote growth of L. infantum, but compound 56, a nicotinamide derivative, inhibited LmSir2rp1 [121]. Later, an investigation revealed that compounds 42 and 46 can also inhibit the recombinant LiSir2rp1, with IC50 values of 194, and 40 μM, respectively, but similar results were also observed against the human SIRT1 enzyme. Thus, selectivity towards the parasite was lacking [122]. Furthermore, Tavares et al., described antileishmanial activity and structure–activity relationship study of twelve compounds belonging to bis-naphthalimidopropyl (BNIP) derivatives that varied in the central alkyl chains length, with 2, 3, or 4 nitrogen atoms, connecting two moieties of BNIP [122]. All derivatives of BNIP could suppress LiSir2rp1, with IC50 values ranging from 7 to 54.7 μM, and displayed selectivity for hSIRT1 in certain cases. The inhibition and selectivity of LiSir2rp1 seem to rely on linker group length and total charge. Diamine BNIP derivatives having linker 4–7 methylene units were less effective than 8–12 methylene linker units. Further, the additions of the positive amino group to the linker do not affect the selectivity or efficacy.
Compound 57 (BNIP9), having nine methylene units in the linker group, displayed IC50 value of 5.7 μM against parasitic LiSir2rp1 with 17-fold enhancement in specificity over hSIRT1 [122]. Furthermore, the derivatives of BNIP could inhibit the intracellular development of L. infantum amastigotes, with IC50 values ranging from 1–10 μM. Still, a linear interaction between LiSir2rp1 inhibitory property and antiproliferative effects in the amastigote stage could not be established. Certain derivatives are committed to have favorable antileishmanial efficacy, as shown in L. infantum-infected BALB/c mouse model [173].
5.7. Antitoxoplasma HDAC Inhibitors
Several HDACis belonging to class I/II with the cyclic tetrapeptides displayed inhibitory activity on the growth of Toxoplasma gondii. Compound 5 with IC50 values ranging from 3 to 15 nM showed its potential but did not possess selectivity over the mammalian cells [123]. The cyclic tetrapeptide compound 6 obtained from the fermentation broth of Acremonium species showed rapid growth inhibitory effects against tachyzoites of T. gondii, with IC50 value of 10 nM and 13-fold specificity over human foreskin fibroblasts [132]. These compounds are effective against drug-sensitive and resistant P. falciparum-infected erythrocytes. Compound 6 caused hyperacetylation of H4 histone of T. gondii through specific inhibition of TgHDAC3 enzyme. The sensitivity for compounds 5 and 6 was reduced due to single-point mutations within TgHDAC3 (T99A and T99I). Hence, these compounds targeted TgHDAC3, providing genetic proof. Intriguingly, compound 6 displayed turning off the enzyme inhibition due to the insertion of two residues (T99 and A98) within the TgHDAC3 catalytic site. This indicated the presence of a simple safeguard in the HDAC3 family of Apicomplexan, which is not available in any other HDACs of eukaryotes [132].
In addition, altered gene expression and stage-specific transformation in T. gondii were completed by TgHDAC3. The treatment with compound 6 converts the replicative tachyzoite to the non-replicative bradyzoite [174]. A successive study narrates that compound 6 was effective in T. gondii cysts treated ex vivo and also affected converted cysts and bradyzoites. Particularly, compound 6 diminished the capacity of bradyzoites for conversion to tachyzoites beyond cyst wall injury; thus, isolated free bradyzoites after cell wall lysis were not able to multiply. Ultimately, the cysts formerly treated with compound 6 injected in mice did not produce infection.
Similar studies were conducted for compound 6 cognate (W363 and W399), which is highly active against tachyzoites and afforded better selectivity of 48–62 fold over human foreskin fibroblast cells. Hence, it could be preferable for in vivo studies in the near future [175]. However, compounds 7, 8, and 9 displayed poor activity against tachyzoites of T. gondii and showed lower selectivity over HS69 mammalian cells [135]. Hydroxamate-based HDACis have also been assessed against T. gondii parasites in the tachyzoite stage [134]. Among them, compounds 10, 58 (scriptaid), and 3 hampered tachyzoites of T. gondii, showing IC50 values of 41, 39, and 83 nM, respectively. However, compound 11 displayed 2–5 times lower potential than others, with IC50 of 213 nM [134]. Likewise, compounds 3 and 58 displayed improved selectivity towards the P. falciparum parasite than compound 10 over mammalian cells [134]. Surprisingly, compounds 10, 3, and 48 at low concentrations of 1–50 nM reduced the infectivity of T. gondii tachyzoite through suppression of proliferation and survival [134]. Very few facts are available on the effectiveness of HDACis belonging to class III against several species of Toxoplasma. A single report suggested that compound 46 is inactive against tachyzoites of T. gondii, with an IC50 value of 50 mM [134].
6. Challenges Regarding HDAC Inhibitors as Antiparasitic Agents
HDACis are a new class of favorable compounds that would function as potential antiparasitic agents. Some of them, such as Vorinostat, Romidepsin, Belinostat, Panobinostat, and many others, show notable in vitro and ex vivo potential [61,176]. A few of them have shown their in vivo potential against particular parasites. Despite the increasing focus on their antiparasitic effects, many censorious points are yet to be explored before taking SIRTi/HDACi towards the clinical phase as antiparasitic candidates [12,26].
The clinical therapy of parasitic infections is quite different compared to other diseases, like cancer. Most morbidities and mortalities are linked with parasitic diseases that happen in underdeveloped areas of the world. Immune-compromised people are at utmost risk. Concomitant infection with dissimilar infective agents is quite a common contributory factor to the development of parasitic disease and needs to be observed because of potential drug–drug interactions. The salient features in prior consideration regarding HDACis for clinical application include: (i) compounds should have high potency with in vivo selectivity towards the parasite compared to normal host cells; (ii) highly active against organisms that are resistant to present drugs; (iii) having an elevated level of safety profile for application in high-risk groups like pregnant women and children; (iv) possess high oral bioavailability and efficacious pharmacokinetic profiles necessitating a single-dose/day; (v) cost-effective to allow potential treatment in hundreds of millions of underprivileged people; and (vi) supportive pharmacokinetics with powerful partner drug to fend off the occurrence of drug resistance.
To take HDACi belonging to class I/II towards clinical trial for parasitic illness, an elevated level of potency and selectivity for parasites against host cells is required. There is an opportunity for optimization of the chemical structure of presently available HDACis belonging to class I/II for therapeutic application against parasites by alteration of the zinc-binding group, CAP protein, and linker. This paves the way for medicinal chemistry programs to specifically target isoforms of parasitic HDAC. Such approaches are still challenging; however, they require cloning and expression of recombinant parasitic HDAC isoforms for enzyme assays and crystallization studies as well as information on the molecular function of these enzymes. This in turn paves the way to specifically pick out HDAC isoforms of a parasite.
The zinc-binding site in HDACs and the presently available tube-shaped pocket between zinc and the HDAC enzyme surface are well-preserved between the enzymes of humans and parasites. The linker moieties for HDACis are not abundant as they are confined by the size and shape of the tunnel to the active site [92,93]. Nonetheless, this region can still be utilized for the development of selective HDACis of the parasite. Heimburg et al., reported selective smHDAC8 inhibitors that displayed a heteroaromatic ring as a linker group [175]. In addition, the linker group length has been initiated to be a major structural decisive factor for both antimalarial and antileishmanial activities of two categories of nonpeptides. Macrocyclic HDACi shows higher antileishmanial activity, with linkers consisting of eight or nine methylene units, while, on the contrary, the foremost antimalarial potency was noticed for spacers with five or six methylene units [146]. The CAP protein group implied in the interplay with the rim at the catalytic tunnel entrance provides a greater extent of dissimilarity and provides room to enhance the activity and selectivity for future antiparasitic HDACis. Furthermore, for increased clinical effectiveness, replacement of the connecting unit could also be launched, such as with Giannini et al., who reported the addition of a γ-lactam carboxamide in α position to the anilide CU of the compound 3 [149]. The zinc-binding hydroxamate, available in several of the most effective HDACis, confers potency and affinity for HDAC enzymes and is not an absolute zinc-binding group. The hydroxamate moiety is also a possible site for metabolic attack and has no selectivity for chelating the zinc ion of HDACs. However, it can tie up to other proteins containing zinc or other metal ions, leading to such probable toxicity issues as those noticed for hydroxamate-carrying inhibitors of matrix metalloprotease [177,178]. However, the alteration of the zinc-binding group was not successful in most cases. Recently, Ontoria et al. were able to use the secondary amide moiety along with the hydroxamate group as a selective HDACi of P. falciparum. This proposed a method for the development of novel HDACis with enhanced metabolic stability and selectivity [158].
To restrict the toxicity potential, subsequent studies were carried out to identify compounds with parasite sensitivity for HDACs/SIRTs and their isoform selectivity. The development of certain isoform-specific HDACis has begun to spare hHDACs and is described as the first blueprint of HDACis having selectivity towards the parasite regarding smHDAC8-selective inhibitors. The crystal structures of mHDAC8 form a network with the 3-amido benzohydroxamate group of HDACis. It was established that the two residues, His292 and Lys20, are independently connected by hydrogen bonds to the amide group of the previously mentioned inhibitors. This recommends a scheme for enhancing the activity against smHDAC8 and providing specificity over hHDAC6 and hHDAC1. Nonetheless, the exploration of HDACis with selectivity towards the parasite remains demanding since it requires the cloning and expression of each isoform of recombinant parasite HDAC. To affirm the designing of preferred HDACis, 3D models have been generated for HDACs of parasites and their human homologs through homology modelling [91,92,121,167,170,175,179,180,181]. In silico homology modelling studies of PfHDAC1 have displayed similarity with hHDACs; however, some discrepancies could be utilized for designing novel selective potential inhibitors [92].
7. Discussion
Melesina et al., studied recent homology models of Leishmania, Trypanosoma, and Plasmodium HDACs and differentiated them from S. mansoni and human HDACs. Molecular dynamics and docking studies have identified the existence of various asymmetrical binding cavities and topologies aside from the preserved amino acid residue in the catalytic site that could help in the structure-based design of parasite-specific HDACis [156]. Still, no three-dimensional models have been available for any of the SIRT enzymes of the parasite. However, various three-dimensional models have been developed for TcSir2rp1, LiSir2rp1, TbSir2rp1, and LmSir2rp1 through homology modelling using the crystal structure of hSIRT2 as a template [102,157]. Overall, these studies focused on the nicotinamide adenine dinucleotide (NAD+) binding site of the SIRT active site. The studies revealed notable distinctions regarding compound 46 binding to human and parasite SIRTs and might pave the way for developing selective inhibitors towards the SIRTs of the parasites [121,170,180,182,183]. In addition, the previously discussed selective LiSir2rp1 inhibitor compound 57 binds with the binding area of NAD+ at the LiSir2rp1 in contrast to hSIRT1. Thus, the binding location of NAD+ is utilizable to develop selective SIRT inhibitors for parasites and spare human enzymes [122].
The activity against drug-resistant strains is crucial for any novel antiparasitic agent. The preference for an HDACi for in vivo testing use must be supported by the early observation of prevention of cross-resistance for numerous strains of the parasite. Some antimalarial HDACis have displayed indistinguishable in vitro activities against the strains of P. falciparum that are resistant and sensitive to presently available drugs like mefloquine and chloroquine [93,136,137,138]. Intriguingly, the study of compound 3 and two other 2-aminosuberic acid HDACis showed that they have potential against multidrug-resistant P. vivax and P. falciparum clinical isolates. The vulnerability towards compounds 15 and 16, 2-aminosuberic acid derivative, was linked to the identical traditional antimalarial drug in P. vivax but not in P. falciparum, recommending the probability of variation in the sensitivity profiles of distinct species of Plasmodium [141]. A convenient approach to evaluate the drug resistance potential of novel antiparasitic drugs is in vitro resistant laboratory strains generation or by mutagenesis. While these tolerant or resistant cell lines have not been available for antimalarial HDACi so far, as discussed above, compound 6 and HDACis have been generated that are resistant towards the parasites of T. gondii [132]. Based upon that, the cell line of T. gondii with mutation to the residue of amino acid in the gene of TgHDAC3 showed decreased sensitivity to compound 2 (FR235222). PfHDAC1 homology modelling with bound HDACis displayed that amino acid T76 is prognosticated to be one of the lead binding area residues in PfHDAC1 [92,93]. Additional studies are needed to elucidate the molecular mode of action of HDACi resistance by considering the role of this amino acid on HDAC inhibitory activity along with an in vitro selection of HDACis that are resistant/tolerant towards the parasitic cell lines. However, HDACi resistance mechanisms have recently been initiated to be identified in mammalian cells [184,185]. Particularly, both P-glycoprotein-dependent and -independent mechanisms are connected to the resistance of compound 34 in leukemia, and tolerance to compounds 7 and 3 in cells of adenocarcinoma appears to be self-sufficient by the countenance of multidrug resistance (MDR) [184,185]. This mechanism of resistance developed by two drugs is displayed in numerous parasites [12,26]. Still, now, few studies have been executed to characterize probable companion drugs for HDACis [12,26]. Nonetheless, in rare cases during experiments, favorable additive effects were found, like a combination between compound 20 and chloroquine in mice infected with P. berghei and also in one more combination between compound 27 and lopinavir [156]. In several cases, introductory experimental proofs suggested HDACi potential application in combination therapy of parasitic diseases [12,26,182]. Furthermore, the investigational proof narrates that sirtuin TbSir2rp1 of T. brucei is crucial for the survival of the parasite via DNA repair. In regard to genotoxicity, it has been proposed that TbSir2rp1-selective inhibitors could be used in the form of co-treatment with standard DNA-targeting chemotherapeutic agents to enhance their antiparasitic efficacy [108,111]. Additional studies like considerable in vivo interactivity studies are now needed to assess the potential benefit of combining SIRTi and HDACi therapy. Based upon this phenomenon, the immune-modulatory potentials of SIRTis and HDACis should be considered. Distinct SIRT and HDAC enzymes are displayed to play crucial roles in the antigenic alteration of the parasite, bypassing the host-immune supervision, causing interplay between innate as well as adaptive immunity. HDACis/SIRTis can powerfully affect T-cells, dendritic cells, and macrophages as many cellular components of the immune system [12,76,77,79,80]. Cellular components like dendritic cells, T-cells, and macrophages are key to immunity towards human parasite infections. Further, in vivo therapy with SIRTis and HDACis on host immune reactions and antiparasitic effects should be considered simultaneously. Dendritic cells play a crucial role in the setup of T- and B-cell adaptive responses and can also express co-stimulatory molecules CD40, CD86, and CD80. After HDACi application, CD40, CD86, and CD80 are deceased, followed by enhancement of immunosuppressive molecule indoleamine-2,3-dioxygenase [111]. Several reports have also suggested that HDAC inhibitor treatment increased viral, bacterial, and fungal pathogen susceptibility [110]. HDACis safeguard mice against immune-mediated diseases such as symptoms of septic shock, lupus, and graft-versus-host disease [110,111].
8. Conclusions
The ongoing research is expanding our understanding of the molecular structures and mechanisms of action of HDAC inhibitors in parasitic diseases. The development of selective inhibitors targeting specific parasite HDACs shows promise, although challenges such as drug resistance and variations in sensitivity across parasite species necessitate further investigation. The potential application of HDAC inhibitors in combination therapy and their immune-modulatory effects on host responses highlight the complexity of their roles in antiparasitic strategies. Future studies should focus on elucidating resistance mechanisms, conducting in vivo interactivity studies, and exploring the synergistic effects of HDAC inhibitors with other therapeutic agents to enhance their efficacy against parasitic diseases.
Author Contributions
T.K.M. and R.R.N. contributed equally towards the literature survey and data interpretation, and preparation of the manuscript. A.G. contributed to the preparation of figures and writing of the manuscript. P.T. and D.K. were responsible for the overall supervision, design of the review, critical analysis of the intellectual content, and revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data will be provided upon reasonable request to the corresponding author.
Conflicts of Interest
There are no conflicts of interest among the authors.
References
- World Health Organization. World Malaria Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. World Malaria Report 2014: Summary; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Pigott, D.M.; Bhatt, S.; Golding, N.; Duda, K.A.; Battle, K.E.; Brady, O.J.; Messina, J.P.; Balard, Y.; Bastien, P.; Pratlong, F. Global distribution maps of the leishmaniases. eLife 2014, 3, e02851. [Google Scholar] [PubMed]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Nemati Zargaran, F.; Rostamian, M.; Kooti, S.; Madanchi, H.; Ghadiri, K. Co-infection of COVID-19 and parasitic diseases: A systematic review. Parasite Epidemiol. Control 2023, 21, e00299. [Google Scholar] [CrossRef] [PubMed]
- Gebrecherkos, T.; Gessesse, Z.; Kebede, Y.; Gebreegzabher, A.; Tasew, G.; Abdulkader, M.; Ebuy, H.; Desta, A.; Hailu, A.; Harris, V.; et al. Effect of co-infection with parasites on severity of COVID-19. EClinicalMedicine 2021, 39, 101054. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar]
- Labella, D. Design, Synthesis and Biological Evaluation of Novel Epigenetic Modulators; Sapienza University of Rome: Rome, Italy, 2014. [Google Scholar]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Berg, M.; García-Hernández, R.; Cuypers, B.; Vanaerschot, M.; Manzano, J.I.; Poveda, J.A.; Ferragut, J.A.; Castanys, S.; Dujardin, J.-C.; Gamarro, F. Experimental resistance to drug combinations in Leishmania donovani: Metabolic and phenotypic adaptations. Antimicrob. Agents Chemother. 2015, 59, 2242–2255. [Google Scholar] [CrossRef]
- Takala-Harrison, S.; Jacob, C.G.; Arze, C.; Cummings, M.P.; Silva, J.C.; Dondorp, A.M.; Fukuda, M.M.; Hien, T.T.; Mayxay, M.; Noedl, H. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 2015, 211, 670–679. [Google Scholar] [CrossRef]
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Vert, J.P.; Noble, W.S.; Le Roch, K.G. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. falciparum. Bioessays 2015, 37, 182–194. [Google Scholar]
- Cheeseman, K.; Weitzman, J.B. Host–parasite interactions: An intimate epigenetic relationship. Cell. Microbiol. 2015, 17, 1121–1132. [Google Scholar] [PubMed]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, M.; Sanchez-del-Campo, L.; Fernandez-Perez, M.; Saez-Ayala, M.; Cabezas-Herrera, J.; Rodriguez-Lopez, J. Targeting the epigenetic machinery of cancer cells. Oncogene 2015, 34, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Brien, G.L.; Valerio, D.G.; Armstrong, S.A. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell 2016, 29, 464–476. [Google Scholar] [CrossRef]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer therapy with HDAC inhibitors: Mechanism-based combination strategies and future perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coleman, B.I.; Skillman, K.M.; Jiang, R.H.; Childs, L.M.; Altenhofen, L.M.; Ganter, M.; Leung, Y.; Goldowitz, I.; Kafsack, B.F.; Marti, M. A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe 2014, 16, 177–186. [Google Scholar] [CrossRef] [PubMed]
- T Andrews, K.; N Tran, T.; P Fairlie, D. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr. Pharm. Des. 2012, 18, 3467–3479. [Google Scholar] [CrossRef]
- Marek, M.; Oliveira, G.; Pierce, R.J.; Jung, M.; Sippl, W.; Romier, C. Drugging the schistosome zinc-dependent HDACs: Current progress and future perspectives. Future Med. Chem. 2015, 7, 783–800. [Google Scholar] [PubMed]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef]
- Prince, H.M.; Dickinson, M. Romidepsin for Cutaneous T-cell Lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef]
- Thompson, C.A. Belinostat Approved for Use in Treating Rare Lymphoma; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar]
- Kogan, F.; Kogan, F. Malaria Burden. Remote Sensing for Malaria: Monitoring and Predicting Malaria from Operational Satellites; Springer Nature: Berlin, Germany, 2020; pp. 15–41. [Google Scholar]
- Monroe, A.; Williams, N.A.; Ogoma, S.; Karema, C.; Okumu, F. Reflections on the 2021 World Malaria Report and the Future of Malaria Control. Malar. J. 2022, 21, 154. [Google Scholar] [CrossRef]
- Nadeem, A.Y.; Shehzad, A.; Islam, S.U.; Al-Suhaimi, E.A.; Lee, Y.S. Mosquirix™ RTS, S/AS01 vaccine development, immunogenicity, and efficacy. Vaccines 2022, 10, 713. [Google Scholar]
- Brabin, B.J. An analysis of malaria in pregnancy in Africa. Bull. World Health Organ. 1983, 61, 1005. [Google Scholar]
- Musset, L.; Bouchaud, O.; Matheron, S.; Massias, L.; Le Bras, J. Clinical atovaquone-proguanil resistance of Plasmodium falciparum associated with cytochrome b codon 268 mutations. Microbes Infect. 2006, 8, 2599–2604. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Yeung, S.; White, L.; Nguon, C.; Day, N.P.; Socheat, D.; Von Seidlein, L. Artemisinin resistance: Current status and scenarios for containment. Nat. Rev. Microbiol. 2010, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.B.; Sande, M.A. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N. Engl. J. Med. 1992, 327, 1643–1648. [Google Scholar] [CrossRef]
- Carlier, Y.; Truyens, C.; Deloron, P.; Peyron, F. Congenital parasitic infections: A review. Acta Trop. 2012, 121, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.J.; Ross, A.G.; Li, Y.-S.; McManus, D.P. Diagnosis and management of schistosomiasis. BMJ 2011, 342, d2651. [Google Scholar] [PubMed]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Neglected Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- WHO. Weekly Epidemiological Record Releve Epidemiologique Hebdomadaire; World Health Organization: Geneva, Switzerland, 2015; Volume 316, pp. 25–32. [Google Scholar]
- WHO. Weekly Epidemiological Record Releve Epidemiologique Hebdomadaire; World Health Organization: Geneva, Switzerland, 2016; Volume 91, pp. 53–60. [Google Scholar]
- Dömling, A.; Khoury, K. Praziquantel and schistosomiasis. ChemMedChem 2010, 5, 1420–1434. [Google Scholar] [CrossRef]
- WHO. Expert Committee on the Control of Schistosomiasisx. Bull. World Health Organ. 1993, 71, 657–662. [Google Scholar]
- King, C.H.; Dangerfield-Cha, M. The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 2008, 4, 65–79. [Google Scholar] [CrossRef]
- King, C.H.; Dickman, K.; Tisch, D.J. Reassessment of the cost of chronic helmintic infection: A meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet 2005, 365, 1561–1569. [Google Scholar] [PubMed]
- Chitsulo, L.; Loverde, P.; Engels, D. Schistosomiasis. Nat. Rev. Microbiol. 2004, 2, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L. Praziquantel. Parasitol. Res. 2003, 90 (Supp. 1), S3–S9. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Cioli, D.; Utzinger, J. Praziquantel: Mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, C.R. Chemotherapy of schistosomiasis: Present and future. Curr. Opin. Chem. Biol. 2007, 11, 433–439. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Kusel, J.R.; Coles, G.C.; Cioli, D. Resistance of Schistosoma mansoni to praziquantel: Is there a problem? Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 465–469. [Google Scholar] [CrossRef]
- Doenhoff, M.J.; Pica-Mattoccia, L. Praziquantel for the treatment of schistosomiasis: Its use for control in areas with endemic disease and prospects for drug resistance. Expert Rev. Anti-Infect. Ther. 2006, 4, 199–210. [Google Scholar] [CrossRef]
- Danso-Appiah, A.; De Vlas, S.J. Interpreting low praziquantel cure rates of Schistosoma mansoni infections in Senegal. Trends Parasitol. 2002, 18, 125–129. [Google Scholar] [CrossRef]
- Lawn, S.D.; Lucas, S.B.; Chiodini, P.L. Schistosoma mansoni infection: Failure of standard treatment with praziquantel in a returned traveller: A reply. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 720. [Google Scholar] [CrossRef]
- Bonesso-Sabadini, P.I.P.; Dias, L.C.d.S. Altered response of strain of Schistosoma mansoni to oxamniquine and praziquantel. Memórias Do Inst. Oswaldo Cruz 2002, 97, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Couto, F.F.; Coelho, P.M.Z.; Araújo, N.; Kusel, J.R.; Katz, N.; Jannotti-Passos, L.K.; Mattos, A.C.A. Schistosoma mansoni: A method for inducing resistance to praziquantel using infected Biomphalaria glabrata snails. Mem. Do Inst. Oswaldo Cruz 2011, 106, 153–157. [Google Scholar]
- Fallon, P.G.; Doenhoff, M.J. Drug-resistant schistosomiasis: Resistance to praziquantel and oxamniquine induced in Schistosoma mansoni in mice is drug specific. Am. J. Trop. Med. Hyg. 1994, 51, 83–88. [Google Scholar]
- Vaca, H.R.; Celentano, A.M.; Toscanini, M.A.; Heimburg, T.; Ghazy, E.; Zeyen, P.; Hauser, A.-T.; Oliveira, G.; Elissondo, M.C.; Jung, M. The potential for histone deacetylase (HDAC) inhibitors as cestocidal drugs. PLoS Negl. Trop. Dis. 2021, 15, e0009226. [Google Scholar] [CrossRef] [PubMed]
- Malik, L.H.; Singh, G.D.; Amsterdam, E.A. The epidemiology, clinical manifestations, and management of chagas heart disease. Clin. Cardiol. 2015, 38, 565–569. [Google Scholar] [CrossRef] [PubMed]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). 2016. Available online: http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 10 June 2023).
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human african trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Matthews, K.R. The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 2005, 118, 283–290. [Google Scholar]
- Maurice, J. New WHO plan targets the demise of sleeping sickness. Lancet 2013, 381, 13–14. [Google Scholar]
- WHO. Leishmaniasis. 2016. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 10 June 2023).
- Pace, D. Leishmaniasis. J. Infect. 2014, 69 (Suppl. S1), S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. Leishmaniasis: An update of current pharmacotherapy. Expert Opin. Pharmacother. 2013, 14, 53–63. [Google Scholar]
- Mishra, J.; Saxena, A.; Singh, S. Chemotherapy of leishmaniasis: Past, present and future. Curr. Med. Chem. 2007, 14, 1153–1169. [Google Scholar] [CrossRef]
- Okwor, I.; Uzonna, J.E. The immunology of Leishmania/HIV co-infection. Immunol. Res. 2013, 56, 163–171. [Google Scholar] [PubMed]
- Cairns, B.R. Emerging roles for chromatin remodeling in cancer biology. Trends Cell Biol. 2001, 11, S15–S21. [Google Scholar] [CrossRef] [PubMed]
- H Beumer, J.; Tawbi, H. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr. Clin. Pharmacol. 2010, 5, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Rotili, D.; Valente, S.; Kazantsev, A.G. Histone deacetylase inhibitors and neurodegenerative disorders: Holding the promise. Curr. Pharm. Des. 2009, 15, 3940–3957. [Google Scholar] [CrossRef]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Tak, P.P.; Reedquist, K.A. Function of histone deacetylase inhibitors in inflammation. Crit. Rev. Immunol. 2011, 31, 233–263. [Google Scholar]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar]
- Gregoretti, I.; Lee, Y.-M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [PubMed]
- Alcaín, F.J.; Villalba, J.M. Sirtuin inhibitors. Expert Opin. Ther. Pat. 2009, 19, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef]
- Pasco, M.Y.; Rotili, D.; Altucci, L.; Farina, F.; Rouleau, G.A.; Mai, A.; Néri, C. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J. Med. Chem. 2010, 53, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M.; Cockell, M.M. The molecular biology of the SIR proteins. Gene 2001, 279, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Rotili, D.; Simonetti, G.; Savarino, A.; Palamara, A.T.; Migliaccio, A.R.; Mai, A. Non-Cancer Uses of Histone Deacetylase Inhibitors: Effects on Infectious Diseases and β-Hemoglobinopathies. Curr. Top. Med. Chem. 2009, 9, 272–291. [Google Scholar]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar]
- Joshi, M.B.; Lin, D.T.; Chiang, P.H.; Goldman, N.D.; Fujioka, H.; Aikawa, M.; Syin, C. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 1999, 99, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Florens, L.; Washburn, M.P.; Raine, J.D.; Anthony, R.M.; Grainger, M.; Haynes, J.D.; Moch, J.K.; Muster, N.; Sacci, J.B.; Tabb, D.L. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002, 419, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Pradhan, A.; Shah, F.; Tekwani, B.L.; Avery, M.A. Structural insights into the Plasmodium falciparum histone deacetylase 1 (PfHDAC-1): A novel target for the development of antimalarial therapy. Bioorganic Med. Chem. 2008, 16, 5254–5265. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.; Tran, T.; Lucke, A.; Kahnberg, P.; Le, G.; Boyle, G.; Gardiner, D.; Skinner-Adams, T.; Fairlie, D. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Hernandez-Rivas, R.; Ralph, S.A.; Montiel-Condado, D.; Ruvalcaba-Salazar, O.K.; Rojas-Meza, A.P.; Mâncio-Silva, L.; Leal-Silvestre, R.J.; Gontijo, A.M.; Shorte, S. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Mazitschek, R.; Coleman, B.; Nguyen, C.; Urgaonkar, S.; Cortese, J.; Barker, R.H., Jr.; Greenberg, E.; Tang, W.; Bradner, J.E. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187. [Google Scholar] [CrossRef]
- Zhu, A.Y.; Zhou, Y.; Khan, S.; Deitsch, K.W.; Hao, Q.; Lin, H. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem. Biol. 2012, 7, 155–159. [Google Scholar] [CrossRef]
- Tonkin, C.J.; Carret, C.K.; Duraisingh, M.T.; Voss, T.S.; Ralph, S.A.; Hommel, M.; Duffy, M.F.; Silva, L.M.d.; Scherf, A.; Ivens, A. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009, 7, e1000084. [Google Scholar] [CrossRef]
- Duraisingh, M.T.; Voss, T.S.; Marty, A.J.; Duffy, M.F.; Good, R.T.; Thompson, J.K.; Freitas-Junior, L.H.; Scherf, A.; Crabb, B.S.; Cowman, A.F. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 2005, 121, 13–24. [Google Scholar] [CrossRef]
- Saksouk, N.; Bhatti, M.M.; Kieffer, S.; Smith, A.T.; Musset, K.; Garin, J.; Sullivan, W.J., Jr.; Cesbron-Delauw, M.-F.; Hakimi, M.-A. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol. 2005, 25, 10301–10314. [Google Scholar] [CrossRef]
- Oger, F.; Dubois, F.; Caby, S.; Noël, C.; Cornette, J.; Bertin, B.; Capron, M.; Pierce, R.J. The class I histone deacetylases of the platyhelminth parasite Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2008, 377, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Theurkauf, W.E.; Baum, H.; Bo, J.; Wensink, P.C. Tissue-specific and constitutive alpha-tubulin genes of Drosophila melanogaster code for structurally distinct proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 8477–8481. [Google Scholar] [PubMed]
- Berriman, M.; Haas, B.J.; LoVerde, P.T.; Wilson, R.A.; Dillon, G.P.; Cerqueira, G.C.; Mashiyama, S.T.; Al-Lazikani, B.; Andrade, L.F.; Ashton, P.D. The genome of the blood fluke Schistosoma mansoni. Nature 2009, 460, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Ingram, A.K.; Horn, D. Histone deacetylases in Trypanosoma brucei: Two are essential and another is required for normal cell cycle progression. Mol. Microbiol. 2002, 45, 89–97. [Google Scholar] [CrossRef]
- Mandava, V.; Fernandez, J.P.; Deng, H.; Janzen, C.J.; Hake, S.B.; Cross, G.A. Histone modifications in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 156, 41–50. [Google Scholar]
- Hertz-Fowler, C.; Figueiredo, L.M.; Quail, M.A.; Becker, M.; Jackson, A.; Bason, N.; Brooks, K.; Churcher, C.; Fahkro, S.; Goodhead, I. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 2008, 3, e3527. [Google Scholar]
- Wang, Q.P.; Kawahara, T.; Horn, D. Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. Microbiol. 2010, 77, 1237–1245. [Google Scholar] [CrossRef]
- Alsford, S.; Kawahara, T.; Isamah, C.; Horn, D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. Microbiol. 2007, 63, 724–736. [Google Scholar] [CrossRef]
- García-Salcedo, J.A.; Gijón, P.; Nolan, D.P.; Tebabi, P.; Pays, E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. EMBO J. 2003, 22, 5851–5862. [Google Scholar]
- Ritagliati, C.; Alonso, V.L.; Manarin, R.; Cribb, P.; Serra, E.C. Overexpression of Cytoplasmic Tc SIR2RP1 and Mitochondrial Tc SIR2RP3 Impacts on Trypanosoma cruzi Growth and Cell Invasion. PLoS Neglected Trop. Dis. 2015, 9, e0003725. [Google Scholar] [CrossRef] [PubMed]
- Moretti, N.S.; da Silva Augusto, L.; Clemente, T.M.; Antunes, R.P.P.; Yoshida, N.; Torrecilhas, A.C.; Cano, M.I.N.; Schenkman, S. Characterization of Trypanosoma cruzi sirtuins as possible drug targets for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 4669–4679. [Google Scholar]
- Saxena, A.; Lahav, T.; Holland, N.; Aggarwal, G.; Anupama, A.; Huang, Y.; Volpin, H.; Myler, P.; Zilberstein, D. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol. Biochem. Parasitol. 2007, 152, 53–65. [Google Scholar] [CrossRef]
- Yahiaoui, B.; Taibi, A.; Ouaissi, A. A Leishmania major protein with extensive homology to silent information regulator 2 of Saccharomyces cerevisiae. Gene 1996, 169, 115–118. [Google Scholar] [PubMed]
- Vergnes, B.; Sereno, D.; Madjidian-Sereno, N.; Lemesre, J.-L.; Ouaissi, A. Cytoplasmic SIR2 homologue overexpression promotes survival of Leishmania parasites by preventing programmed cell death. Gene 2002, 296, 139–150. [Google Scholar] [CrossRef]
- Tavares, J.; Ouaissi, A.; Santarém, N.; Sereno, D.; Vergnes, B.; Sampaio, P.; Cordeiro-Da-Silva, A. The Leishmania infantum cytosolic SIR2-related protein 1 (LiSIR2RP1) is an NAD+-dependent deacetylase and ADP-ribosyltransferase. Biochem. J. 2008, 415, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, B.; Sereno, D.; Tavares, J.; Cordeiro-da-Silva, A.; Vanhille, L.; Madjidian-Sereno, N.; Depoix, D.; Monte-Alegre, A.; Ouaissi, A. Targeted disruption of cytosolic SIR2 deacetylase discloses its essential role in Leishmania survival and proliferation. Gene 2005, 363, 85–96. [Google Scholar]
- Adriano, M.-A.; Vergnes, B.; Poncet, J.; Mathieu-Daude, F.; da Silva, A.C.; Ouaissi, A.; Sereno, D. Proof of interaction between Leishmania SIR2RP1 deacetylase and chaperone HSP83. Parasitol. Res. 2007, 100, 811–818. [Google Scholar] [CrossRef]
- Silvestre, R.; Cordeiro-da-Silva, A.; Tavares, J.; Sereno, D.; Ouaissi, A. Leishmania cytosolic silent information regulatory protein 2 deacetylase induces murine B-cell differentiation and in vivo production of specific antibodies. Immunology 2006, 119, 529–540. [Google Scholar] [CrossRef]
- Silvestre, R.; Silva, A.M.; Cordeiro-da-Silva, A.; Ouaissi, A. The contribution of Toll-like receptor 2 to the innate recognition of a Leishmania infantum silent information regulator 2 protein. Immunology 2009, 128, 484–499. [Google Scholar] [CrossRef]
- Sodji, Q.; Patil, V.; Jain, S.; Kornacki, J.R.; Mrksich, M.; Tekwani, B.L.; Oyelere, A.K. The antileishmanial activity of isoforms 6-and 8-selective histone deacetylase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 4826–4830. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kadam, R.U.; Tavares, J.; Cordeiro, A.; Ouaissi, A.; Roy, N. Structure function analysis of Leishmania sirtuin: An ensemble of in silico and biochemical studies. Chem. Biol. Drug Des. 2008, 71, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.; Ouaissi, A.; Kong Thoo Lin, P.; Loureiro, I.; Kaur, S.; Roy, N.; Cordeiro-da-Silva, A. Bisnaphthalimidopropyl derivatives as inhibitors of Leishmania SIR2 related protein 1. ChemMedChem Chem. Enabling Drug Discov. 2010, 5, 140–147. [Google Scholar] [CrossRef]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cabrera, A.; Kono, M.; Mok, S.; Chaal, B.K.; Haase, S.; Engelberg, K.; Cheemadan, S.; Spielmann, T.; Preiser, P.R. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010, 28, 91–98. [Google Scholar]
- Chaal, B.K.; Gupta, A.P.; Wastuwidyaningtyas, B.D.; Luah, Y.-H.; Bozdech, Z. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 2010, 6, e1000737. [Google Scholar]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar]
- Peart, M.J.; Smyth, G.K.; Van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Meinke, P.T.; Colletti, S.L.; Doss, G.; Myers, R.W.; Gurnett, A.M.; Dulski, P.M.; Darkin-Rattray, S.J.; Allocco, J.J.; Galuska, S.; Schmatz, D.M. Synthesis of apicidin-derived quinolone derivatives: Parasite-selective histone deacetylase inhibitors and antiproliferative agents. J. Med. Chem. 2000, 43, 4919–4922. [Google Scholar]
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 2. Bioorganic Med. Chem. Lett. 2001, 11, 113–117. [Google Scholar]
- Colletti, S.L.; Myers, R.W.; Darkin-Rattray, S.J.; Gurnett, A.M.; Dulski, P.M.; Galuska, S.; Allocco, J.J.; Ayer, M.B.; Li, C.; Lim, J. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: Structure–activity relationships of apicidin. Part 1. Bioorganic Med. Chem. Lett. 2001, 11, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Kranz, M.; Ladlow, M.; Taylor, S.; Berst, F.; Holmes, A.B.; Keavey, K.N.; Jaxa-Chamiec, A.; Seale, P.W.; Stead, P. The synthesis of cyclic tetrapeptoid analogues of the antiprotozoal natural product apicidin. Bioorganic Med. Chem. Lett. 2001, 11, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Bougdour, A.; Maubon, D.; Baldacci, P.; Ortet, P.; Bastien, O.; Bouillon, A.; Barale, J.-C.; Pelloux, H.; Ménard, R.; Hakimi, M.-A. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med. 2009, 206, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef]
- Strobl, J.S.; Cassell, M.; Mitchell, S.M.; Reilly, C.M.; Lindsay, D.S. Scriptaid and suberoylanilide hydroxamic acid are histone deacetylase inhibitors with potent anti–Toxoplasma gondii activity in vitro. J. Parasitol. 2007, 93, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Jones-Brando, L.; Torrey, E.F.; Yolken, R. Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr. Res. 2003, 62, 237–244. [Google Scholar] [PubMed]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef]
- Dow, G.S.; Chen, Y.; Andrews, K.T.; Caridha, D.; Gerena, L.; Gettayacamin, M.; Johnson, J.; Li, Q.; Melendez, V.; Obaldia III, N. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 3467–3477. [Google Scholar] [CrossRef]
- Andrews, K.T.; Walduck, A.; Kelso, M.J.; Fairlie, D.P.; Saul, A.; Parsons, P.G. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int. J. Parasitol. 2000, 30, 761–768. [Google Scholar] [CrossRef]
- Mai, A.; Cerbara, I.; Valente, S.; Massa, S.; Walker, L.A.; Tekwani, B.L. Antimalarial and antileishmanial activities of aroyl-pyrrolyl-hydroxyamides, a new class of histone deacetylase inhibitors. Antimicrob. Agents Chemother. 2004, 48, 1435. [Google Scholar] [CrossRef][Green Version]
- Andrews, K.T.; Gupta, A.P.; Tran, T.N.; Fairlie, D.P.; Gobert, G.N.; Bozdech, Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PLoS ONE 2012, 7, e31847. [Google Scholar] [CrossRef] [PubMed]
- Marfurt, J.; Chalfein, F.; Prayoga, P.; Wabiser, F.; Kenangalem, E.; Piera, K.A.; Fairlie, D.P.; Tjitra, E.; Anstey, N.M.; Andrews, K.T. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 2011, 55, 961–966. [Google Scholar] [PubMed]
- Wheatley, N.C.; Andrews, K.T.; Tran, T.L.; Lucke, A.J.; Reid, R.C.; Fairlie, D.P. Antimalarial histone deacetylase inhibitors containing cinnamate or NSAID components. Bioorganic Med. Chem. Lett. 2010, 20, 7080–7084. [Google Scholar]
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448. [Google Scholar] [CrossRef] [PubMed]
- Agbor-Enoh, S.; Seudieu, C.; Davidson, E.; Dritschilo, A.; Jung, M. Novel inhibitor of Plasmodium histone deacetylase that cures P. berghei-infected mice. Antimicrob. Agents Chemother. 2009, 53, 1727–1734. [Google Scholar] [CrossRef]
- Patil, V.; Guerrant, W.; Chen, P.C.; Gryder, B.; Benicewicz, D.B.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. Antimalarial and antileishmanial activities of histone deacetylase inhibitors with triazole-linked cap group. Bioorganic Med. Chem. 2010, 18, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Mwakwari, S.C.; Guerrant, W.; Patil, V.; Khan, S.I.; Tekwani, B.L.; Gurard-Levin, Z.A.; Mrksich, M.; Oyelere, A.K. Non-peptide macrocyclic histone deacetylase inhibitors derived from tricyclic ketolide skeleton. J. Med. Chem. 2010, 53, 6100–6111. [Google Scholar] [CrossRef]
- Hansen, F.K.; Skinner-Adams, T.S.; Duffy, S.; Marek, L.; Sumanadasa, S.D.; Kuna, K.; Held, J.; Avery, V.M.; Andrews, K.T.; Kurz, T. Synthesis, Antimalarial Properties, and SAR Studies of Alkoxyurea-Based HDAC Inhibitors. ChemMedChem 2014, 9, 665–670. [Google Scholar] [CrossRef]
- Trenholme, K.; Marek, L.; Duffy, S.; Pradel, G.; Fisher, G.; Hansen, F.K.; Skinner-Adams, T.S.; Butterworth, A.; Ngwa, C.J.; Moecking, J. Lysine acetylation in sexual stage malaria parasites is a target for antimalarial small molecules. Antimicrob. Agents Chemother. 2014, 58, 3666–3678. [Google Scholar]
- Giannini, G.; Battistuzzi, G.; Vignola, D. Hydroxamic acid based histone deacetylase inhibitors with confirmed activity against the malaria parasite. Bioorganic Med. Chem. Lett. 2015, 25, 459–461. [Google Scholar] [CrossRef]
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure−selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Altucci, L. Epi-drugs to fight cancer: From chemistry to cancer treatment, the road ahead. Int. J. Biochem. Cell Biol. 2009, 41, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Paonessa, G.; Ponzi, S.; Ferrigno, F.; Nizi, E.; Biancofiore, I.; Malancona, S.; Graziani, R.; Roberts, D.; Willis, P. Discovery of a selective series of inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. [Google Scholar] [CrossRef]
- Verotta, L.; Appendino, G.; Bombardelli, E.; Brun, R. In vitro antimalarial activity of hyperforin, a prenylated acylphloroglucinol. A structure–activity study. Bioorganic Med. Chem. Lett. 2007, 17, 1544–1548. [Google Scholar]
- Prusty, D.; Mehra, P.; Srivastava, S.; Shivange, A.V.; Gupta, A.; Roy, N.; Dhar, S.K. Nicotinamide inhibits Plasmodium falciparum Sir2 activity in vitro and parasite growth. FEMS Microbiol. Lett. 2008, 282, 266–272. [Google Scholar]
- Guerrant, W.; Mwakwari, S.C.; Chen, P.C.; Khan, S.I.; Tekwani, B.L.; Oyelere, A.K. A structure–activity relationship study of the antimalarial and antileishmanial activities of nonpeptide macrocyclic histone deacetylase inhibitors. ChemMedChem 2010, 5, 1232–1235. [Google Scholar] [CrossRef]
- Sumanadasa, S.D.; Goodman, C.D.; Lucke, A.J.; Skinner-Adams, T.; Sahama, I.; Haque, A.; Do, T.A.; McFadden, G.I.; Fairlie, D.P.; Andrews, K.T. Antimalarial activity of the anticancer histone deacetylase inhibitor SB939. Antimicrob. Agents Chemother. 2012, 56, 3849–3856. [Google Scholar] [CrossRef]
- Hansen, F.K.; Sumanadasa, S.D.; Stenzel, K.; Duffy, S.; Meister, S.; Marek, L.; Schmetter, R.; Kuna, K.; Hamacher, A.; Mordmüller, B. Discovery of HDAC inhibitors with potent activity against multiple malaria parasite life cycle stages. Eur. J. Med. Chem. 2014, 82, 204–213. [Google Scholar] [CrossRef]
- Chakrabarty, S.P.; Saikumari, Y.K.; Bopanna, M.P.; Balaram, H. Biochemical characterization of Plasmodium falciparum Sir2, a NAD+-dependent deacetylase. Mol. Biochem. Parasitol. 2008, 158, 139–151. [Google Scholar] [CrossRef]
- Merrick, C.J.; Duraisingh, M.T. Plasmodium falciparum Sir2: An unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot. Cell 2007, 6, 2081–2091. [Google Scholar] [CrossRef]
- Gey, C.; Kyrylenko, S.; Hennig, L.; Nguyen, L.H.D.; Büttner, A.; Pham, H.D.; Giannis, A. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: Inhibitors of human sirtuins SIRT1 and SIRT2. Angew. Chem. Int. Ed. 2007, 46, 5219–5222. [Google Scholar]
- Chakrabarty, S.P.; Ramapanicker, R.; Mishra, R.; Chandrasekaran, S.; Balaram, H. Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity. Bioorganic Med. Chem. 2009, 17, 8060–8072. [Google Scholar]
- Sheader, K.; te Vruchte, D.; Rudenko, G. Bloodstream form-specific up-regulation of silent vsg expression sites and procyclin in Trypanosoma brucei after inhibition of DNA synthesis or DNA damage. J. Biol. Chem. 2004, 279, 13363–13374. [Google Scholar] [PubMed]
- Respuela, P.; Ferella, M.; Rada-Iglesias, A.; Åslund, L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J. Biol. Chem. 2008, 283, 15884–15892. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; Taylor, M.C.; Horn, D.; Loza, E.; Kalvinsh, I.; Björkling, F. Inhibitors of human histone deacetylase with potent activity against the African trypanosome Trypanosoma brucei. Bioorganic Med. Chem. Lett. 2012, 22, 1886–1890. [Google Scholar]
- Carrillo, A.K.; Guiguemde, W.A.; Guy, R.K. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorganic Med. Chem. 2015, 23, 5151–5155. [Google Scholar] [CrossRef]
- Soares, M.B.; Silva, C.V.; Bastos, T.M.; Guimarães, E.T.; Figueira, C.P.; Smirlis, D.; Azevedo, W.F., Jr. Anti-Trypanosoma cruzi activity of nicotinamide. Acta Trop. 2012, 122, 224–229. [Google Scholar] [CrossRef]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Nebbioso, A.; Paolini, C.; Huidobro, C.; Lara, E.; Mellini, P.; Lenoci, A.; Pezzi, R.; Botta, G. Discovery of salermide-related sirtuin inhibitors: Binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J. Med. Chem. 2012, 55, 10937–10947. [Google Scholar]
- Sacconnay, L.; Smirlis, D.; Queiroz, E.F.; Wolfender, J.L.; Soares, M.B.P.; Carrupt, P.-A.; Nurisso, A. Structural insights of SIR2rp3 proteins as promising biotargets to fight against Chagas disease and leishmaniasis. Mol. BioSyst. 2013, 9, 2223–2230. [Google Scholar] [CrossRef][Green Version]
- Vergnes, B.; Vanhille, L.; Ouaissi, A.; Sereno, D. Stage-specific antileishmanial activity of an inhibitor of SIR2 histone deacetylase. Acta Trop. 2005, 94, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Sereno, D.; Alegre, A.M.; Silvestre, R.; Vergnes, B.; Ouaissi, A. In vitro antileishmanial activity of nicotinamide. Antimicrob. Agents Chemother. 2005, 49, 808–812. [Google Scholar] [CrossRef]
- Tavares, J.; Ouaissi, A.; Silva, A.M.; Lin, P.K.T.; Roy, N.; Cordeiro-da-Silva, A. Anti-leishmanial activity of the bisnaphthalimidopropyl derivatives. Parasitol. Int. 2012, 61, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Maubon, D.; Bougdour, A.; Wong, Y.-S.; Brenier-Pinchart, M.-P.; Curt, A.; Hakimi, M.-A.; Pelloux, H. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: Inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob. Agents Chemother. 2010, 54, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.-T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from Schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Langley, B.C. Functional differences in epigenetic modulators superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061. [Google Scholar] [CrossRef]
- Ghazy, E.; Abdelsalam, M.; Robaa, D.; Pierce, R.J.; Sippl, W. Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals 2022, 15, 80. [Google Scholar]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Maolanon, A.R.; Madsen, A.S.; Olsen, C.A. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem. Biol. 2016, 23, 759–768. [Google Scholar] [CrossRef]
- Kaur, S.; Shivange, A.V.; Roy, N. Structural analysis of trypanosomal sirtuin: An insight for selective drug design. Mol. Divers. 2010, 14, 169–178. [Google Scholar] [CrossRef]
- Zheng, W. Sirtuins as emerging anti-parasitic targets. Eur. J. Med. Chem. 2013, 59, 132–140. [Google Scholar] [PubMed]
- Kannan, S.; Melesina, J.; Hauser, A.-T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [PubMed]
- Schiedel, M.; Marek, M.; Lancelot, J.; Karaman, B.; Almlöf, I.; Schultz, J.; Sippl, W.; Pierce, R.J.; Romier, C.; Jung, M. Fluorescence-based screening assays for the NAD+-dependent histone deacetylase smSirt2 from Schistosoma mansoni. J. Biomol. Screen. 2015, 20, 112–121. [Google Scholar] [PubMed]
- Yamada, H.; Arakawa, Y.; Saito, S.; Agawa, M.; Kano, Y.; Horiguchi-Yamada, J. Depsipeptide-resistant KU812 cells show reversible P-glycoprotein expression, hyper-acetylated histones, and modulated gene expression profile. Leuk. Res. 2006, 30, 723–734. [Google Scholar] [CrossRef]
- Fedier, A.; Dedes, K.J.; Imesch, P.; Von Bueren, A.O.; Fink, D. The histone deacetylase inhibitors suberoylanilide hydroxamic (Vorinostat) and valproic acid induce irreversible and MDR1-independent resistance in human colon cancer cells. Int. J. Oncol. 2007, 31, 633–641. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Kutamba, E.; Mugarura, K.; Opoka, R.O. Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev. Anti-Infect. Ther. 2010, 8, 997–1008. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).