
XPS Investigation of Sol–Gel Bioactive Glass Synthesized with Geothermal Water



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
- Prata water, collected from a geothermal spring in the Furnas volcanic area (São Miguel Island, Azores), at UTM 649365 E/4182020 N (WGS 84);
- Azeda water, collected from a nearby spring within the same Furnas hydrothermal system, at UTM 649452 E/4182034 N (WGS 84).
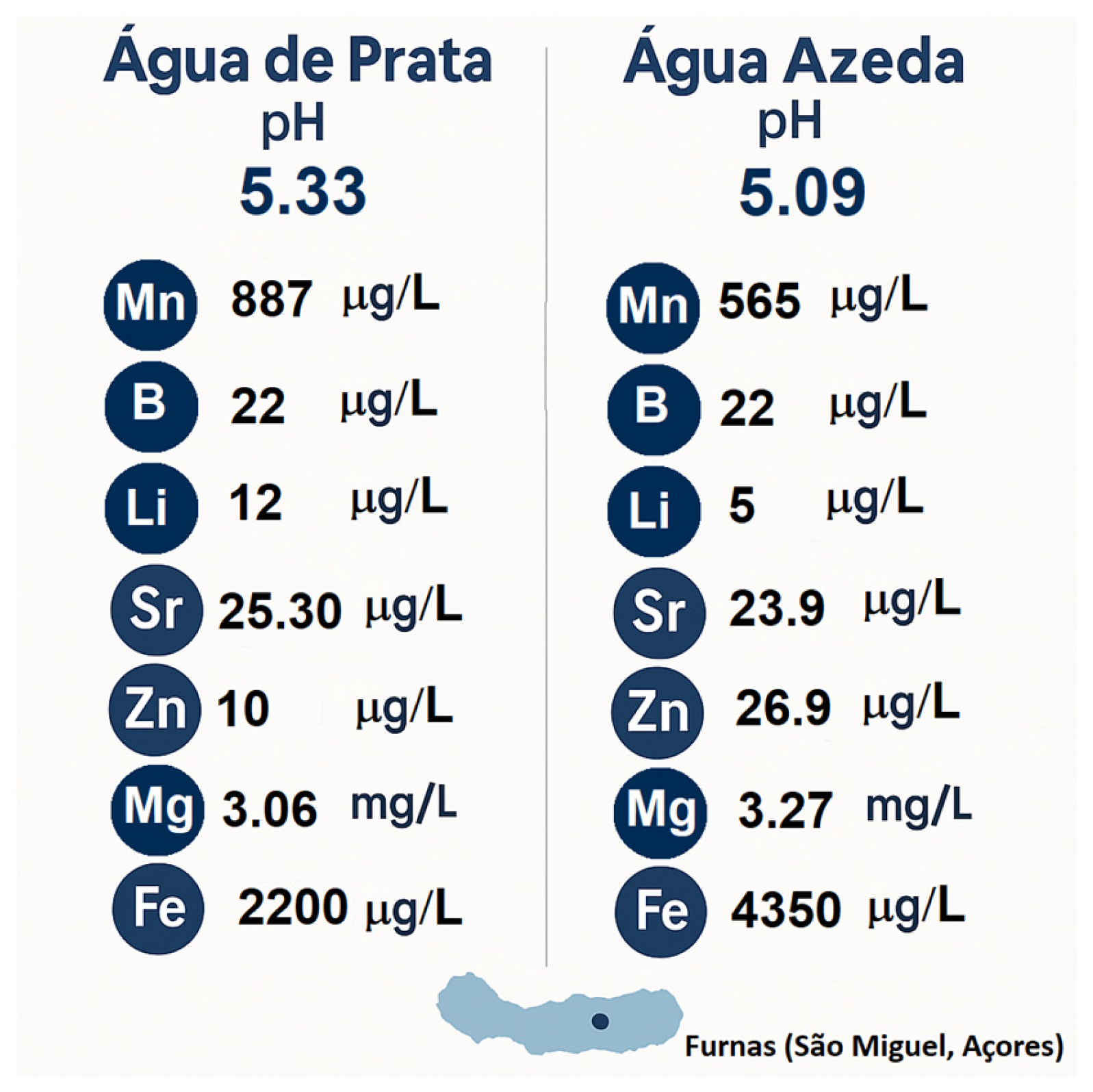
2.2. Geothermal Water Composition and Relevance to Sol–Gel Synthesis
2.2.1. Geochemical Context of Prata Water
2.2.2. Geochemical Context of Azeda Water
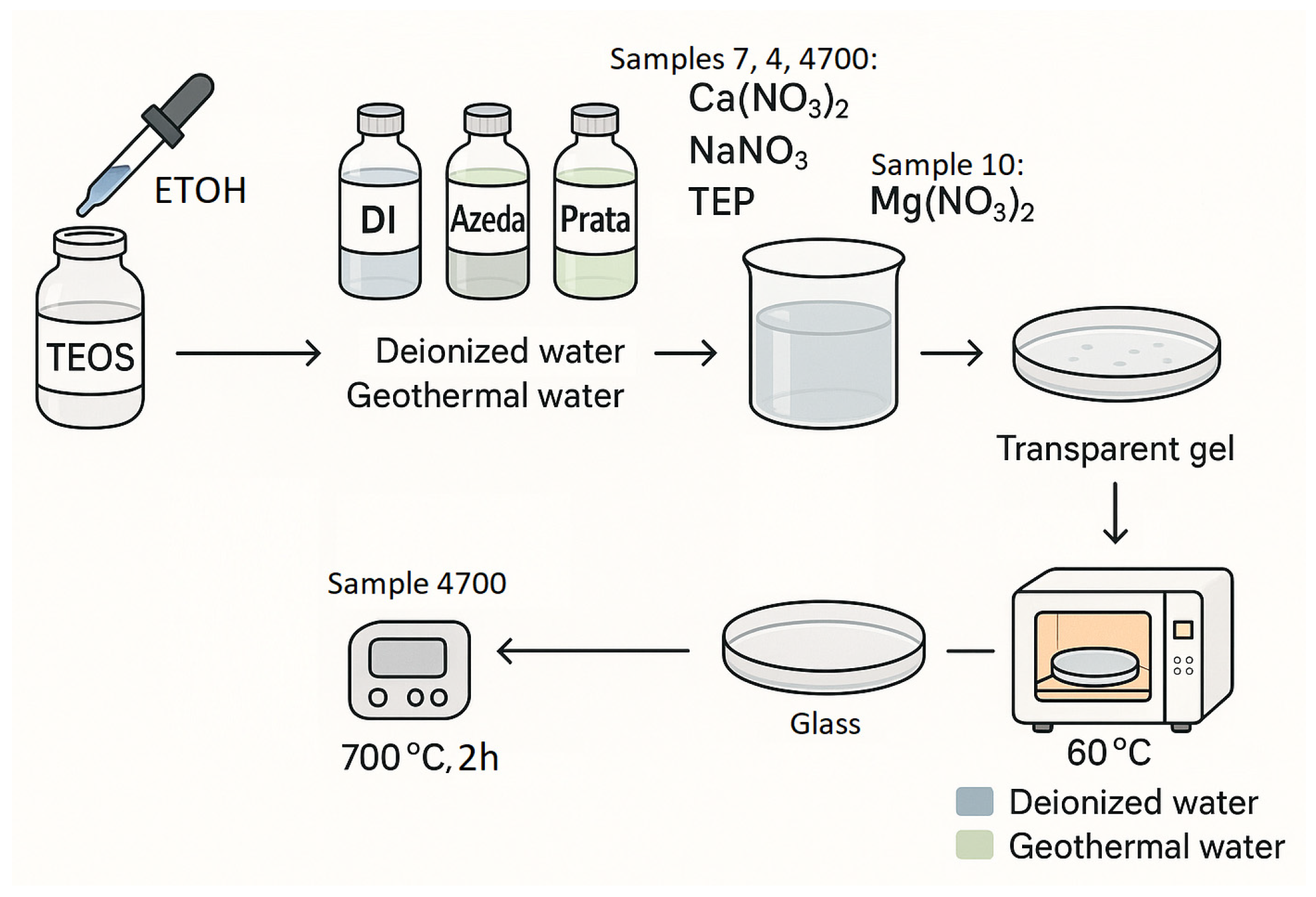
2.3. Synthesis of Bioactive Glasses: 45S5® and 45S5®/MgO
2.3.1. Sample Description
- Sample 7 corresponds to the control bioglass (45S5) synthesized using deionized water acidified with HCl;
- Sample 4 refers to the same 45S5 composition synthesized with Água de Prata, a mineral-rich geothermal water;
- Sample 4700 denotes Sample 4 after thermal treatment at 700 °C:
- Sample 10 represents a modified bioglass in which P2O5 was replaced by 6 wt% MgO, synthesized using Água Azeda.
2.3.2. Synthesis Procedure
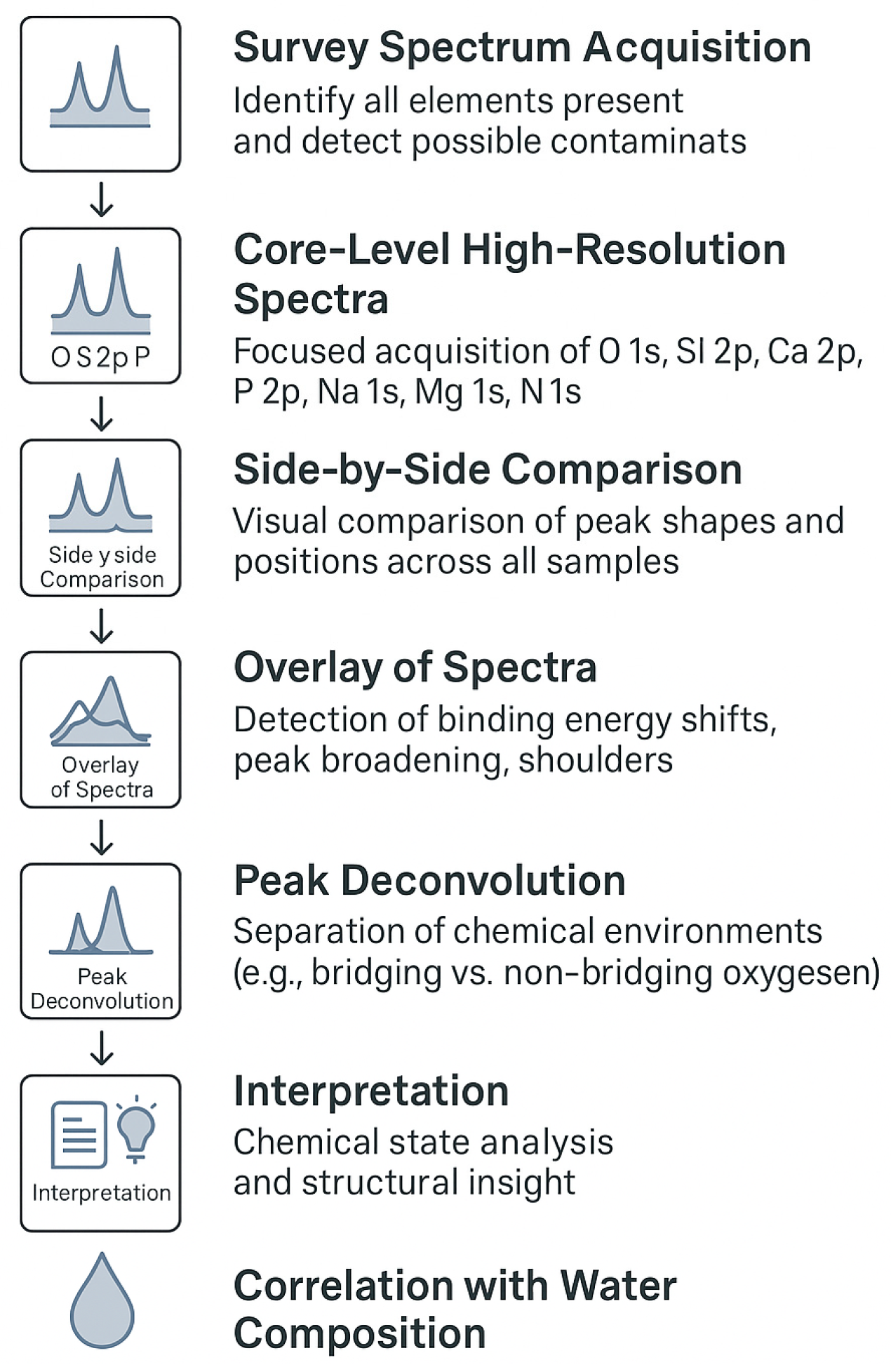
2.4. XPS
3. Results and Discussion
3.1. XPS Analysis
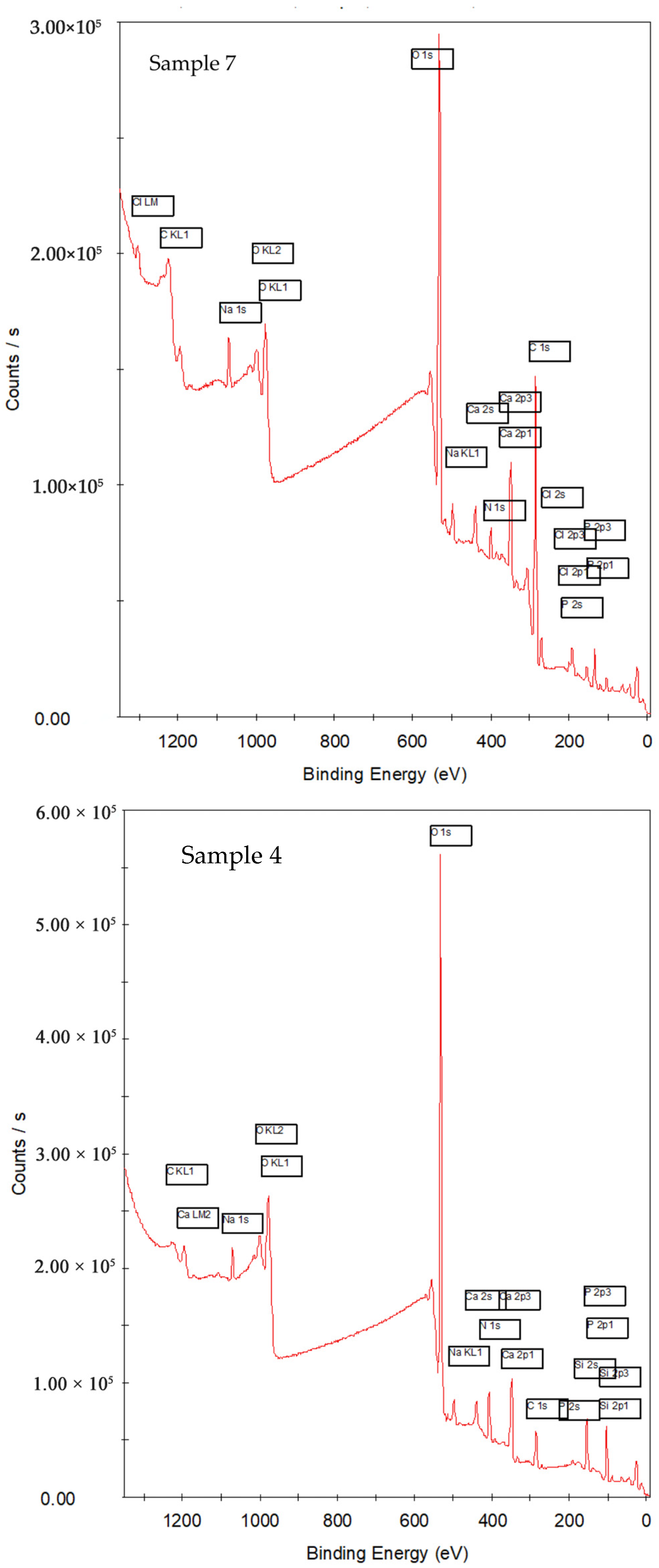
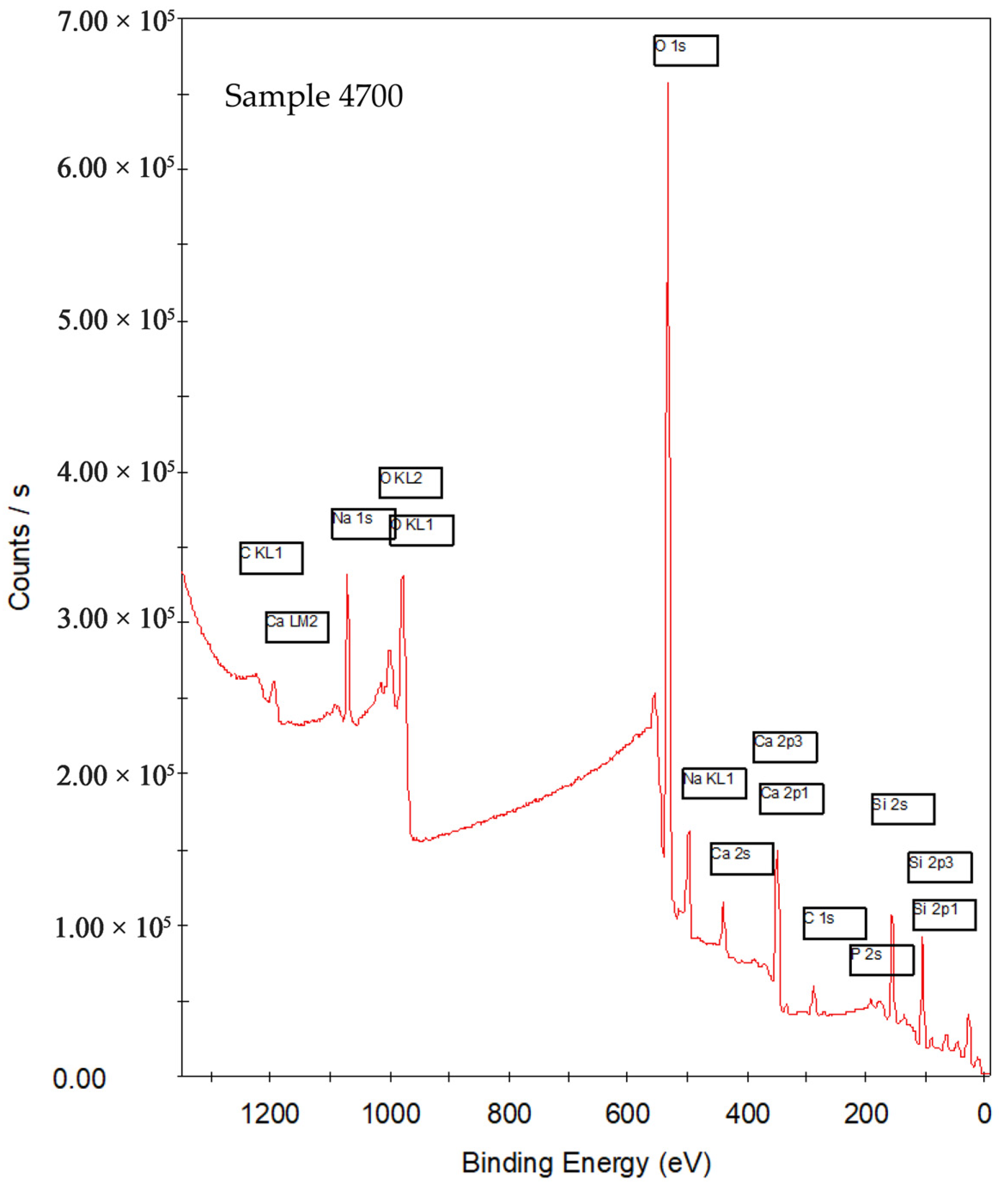
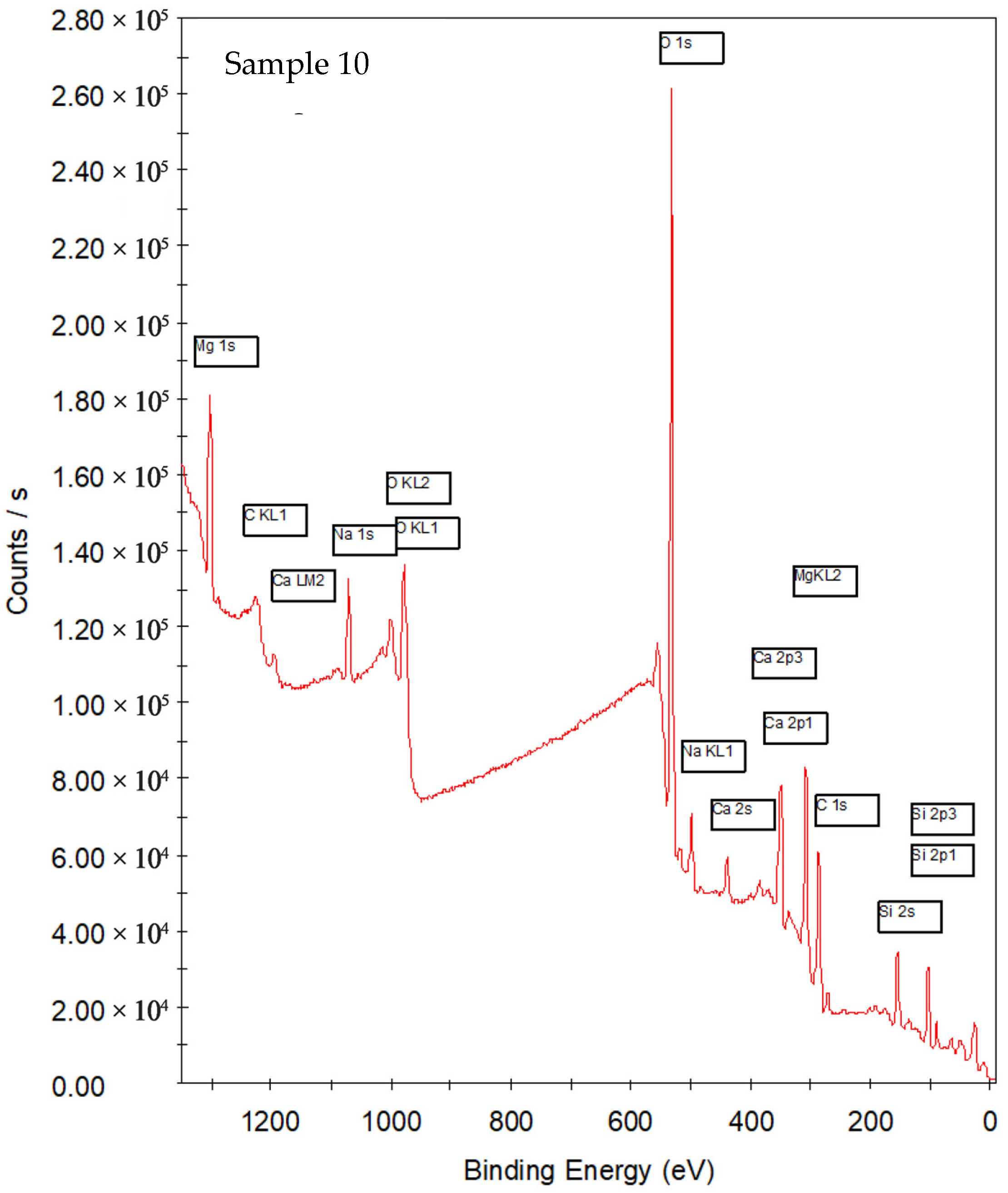
3.1.1. Survey
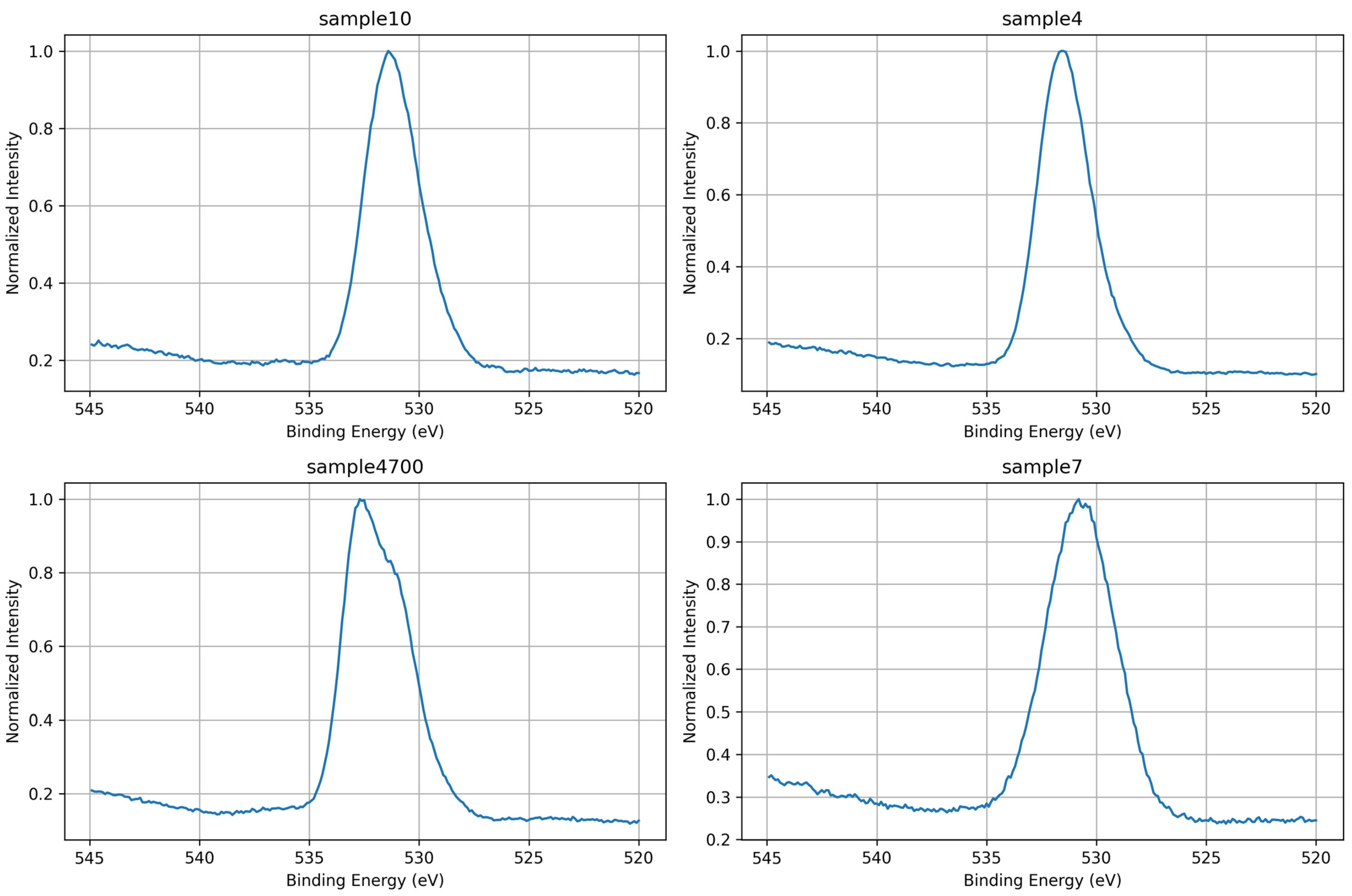
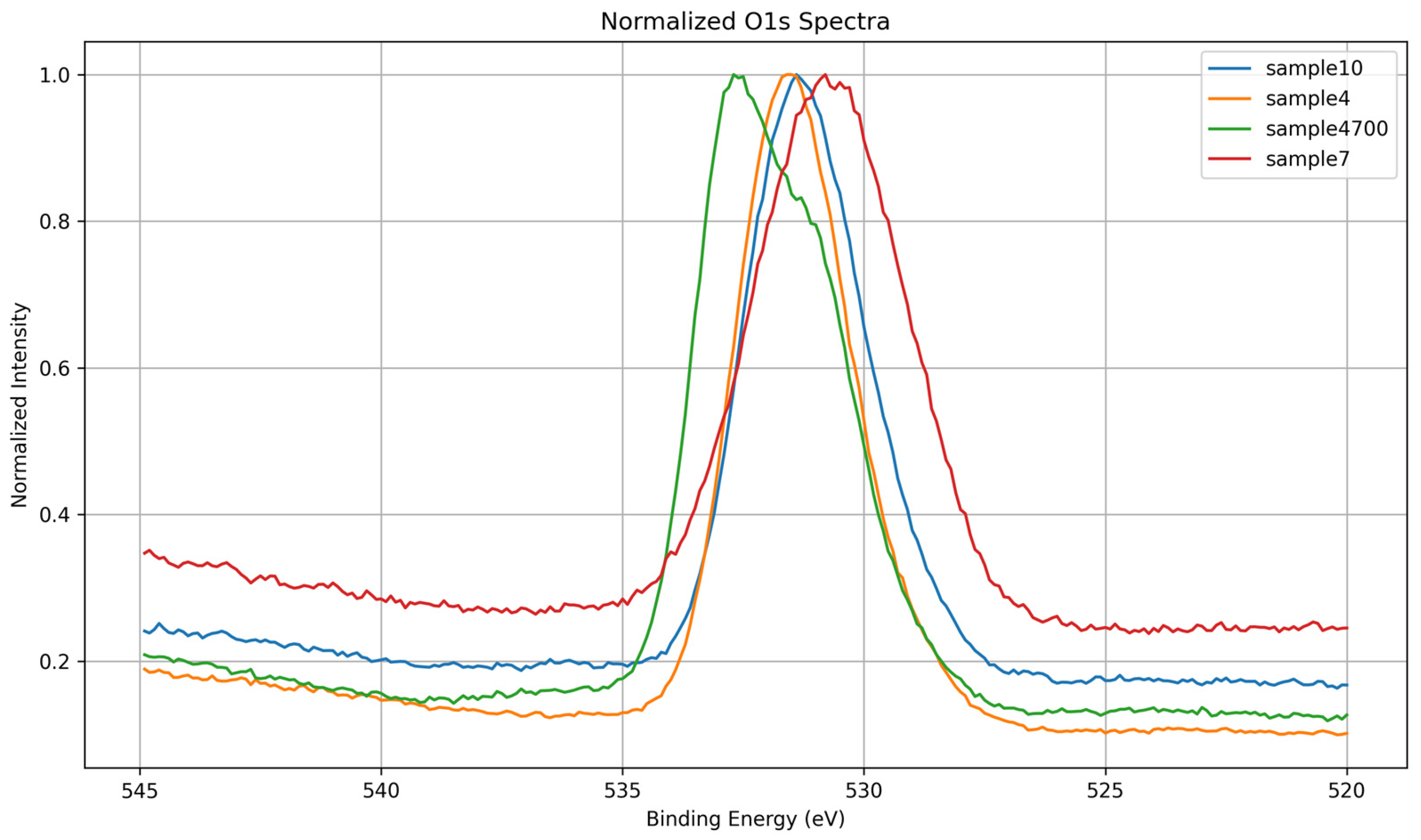
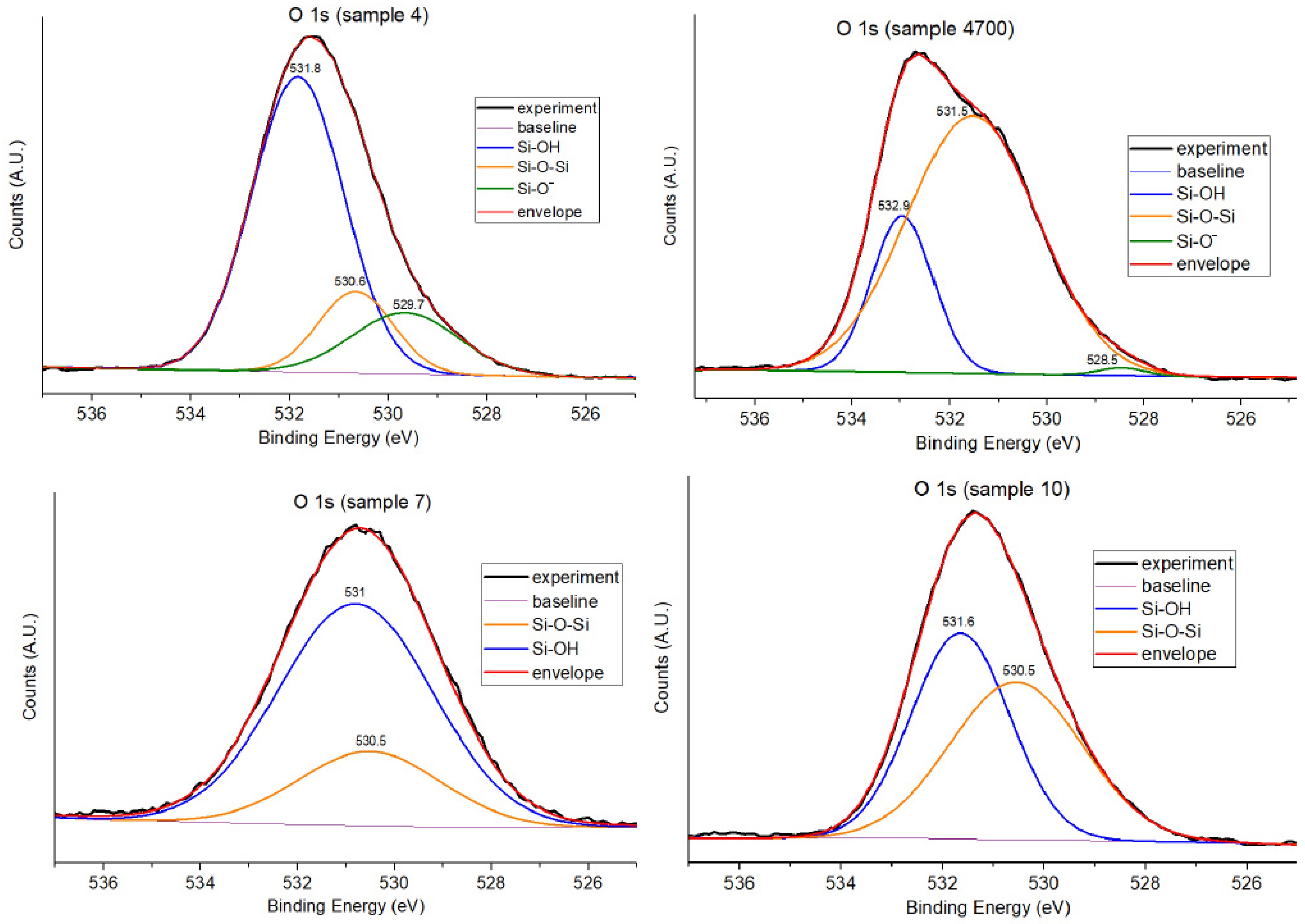
3.1.2. O 1s Core-Level Analysis
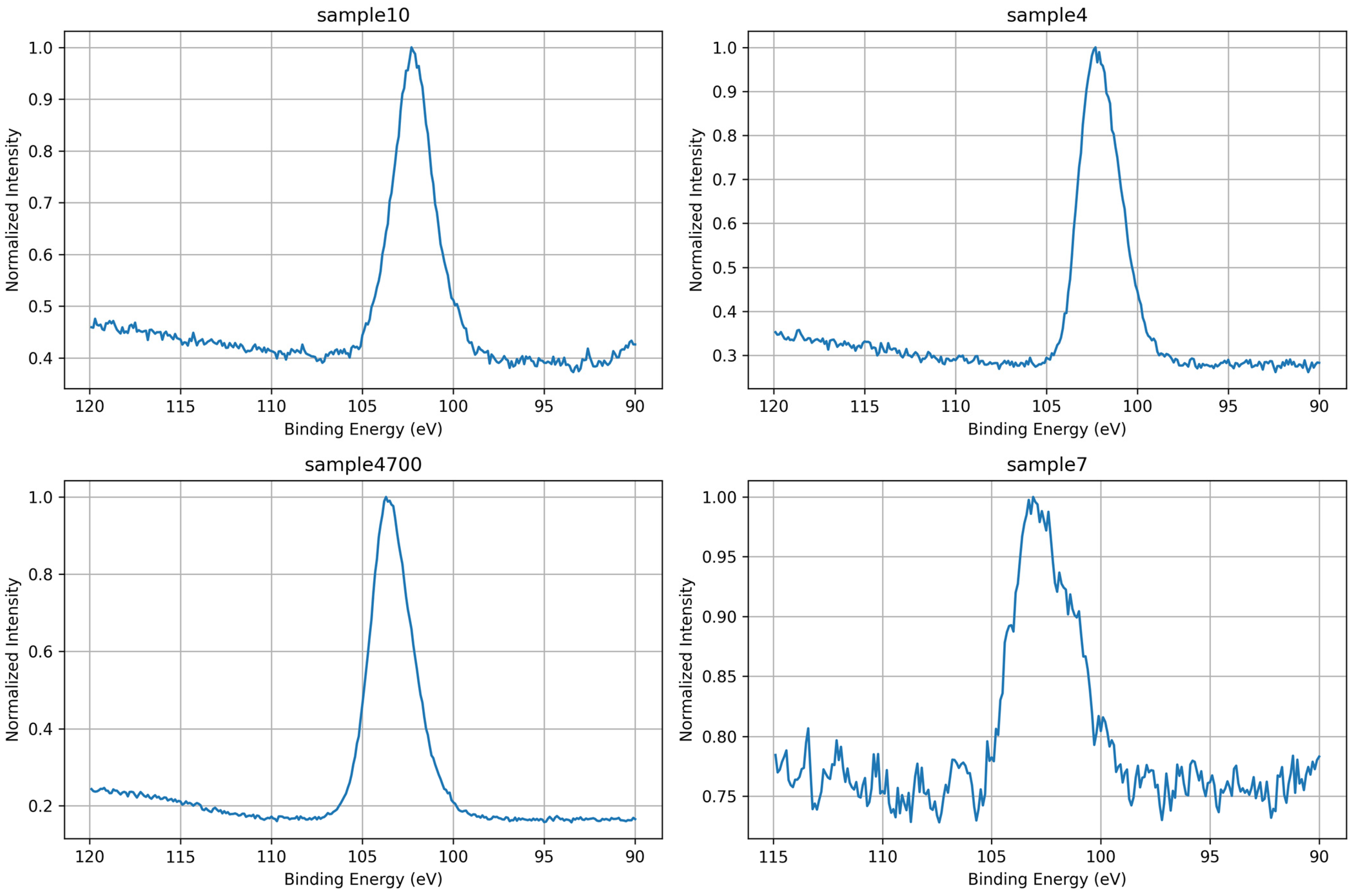
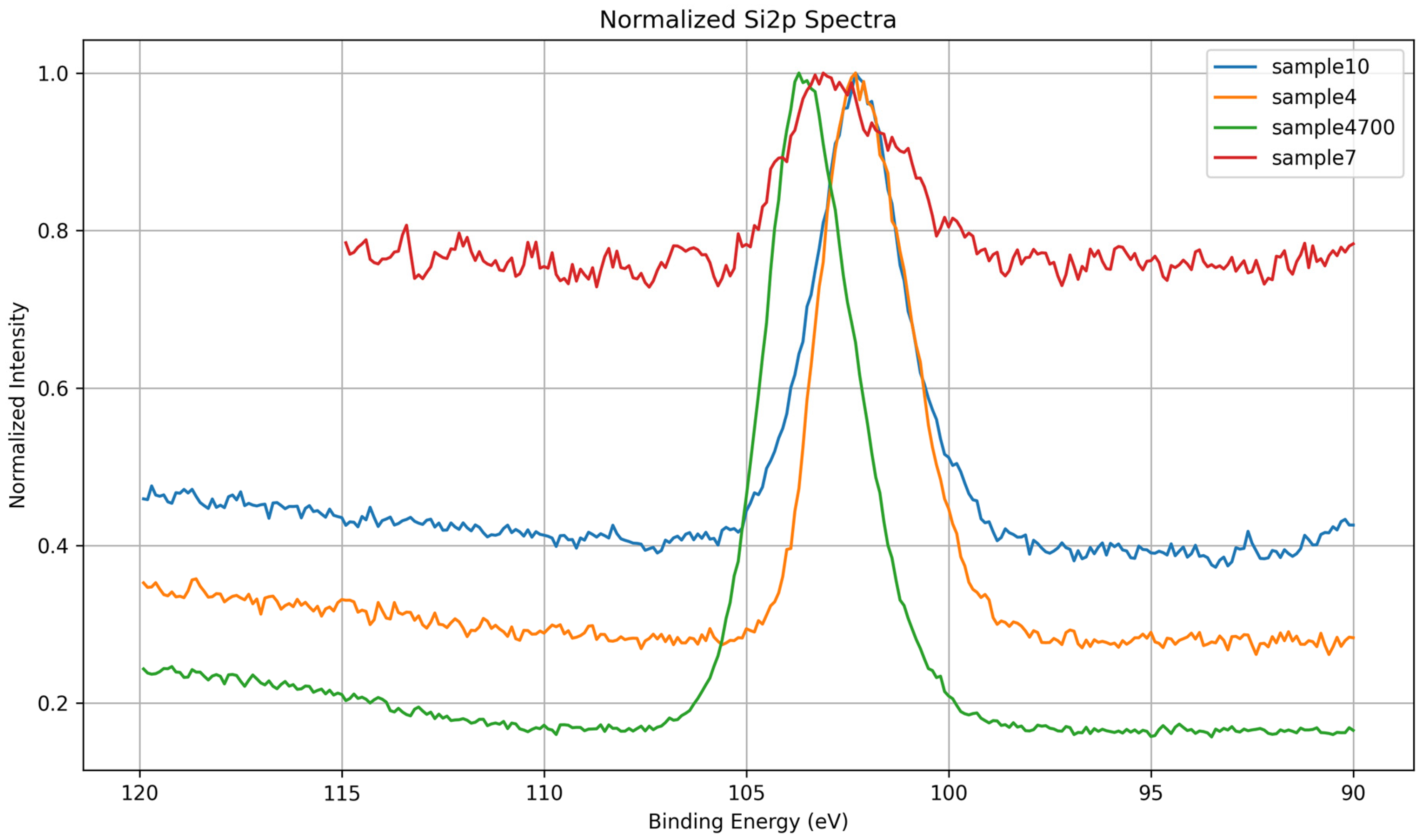
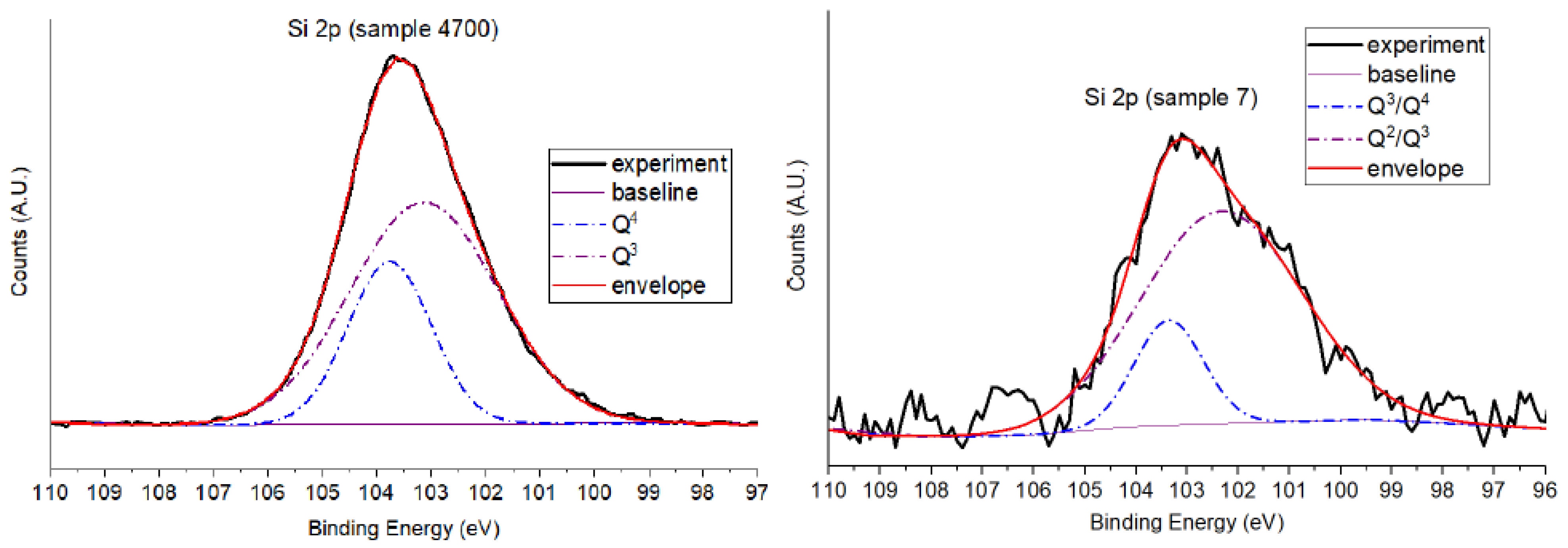
3.1.3. Si 2p Core-Level Analysis
- Q2 corresponds to Si(OSi)2(OH)2;
- Q3 corresponds to Si(OSi)3(OH);
- Q4 corresponds to fully connected Si(OSi)4 species.
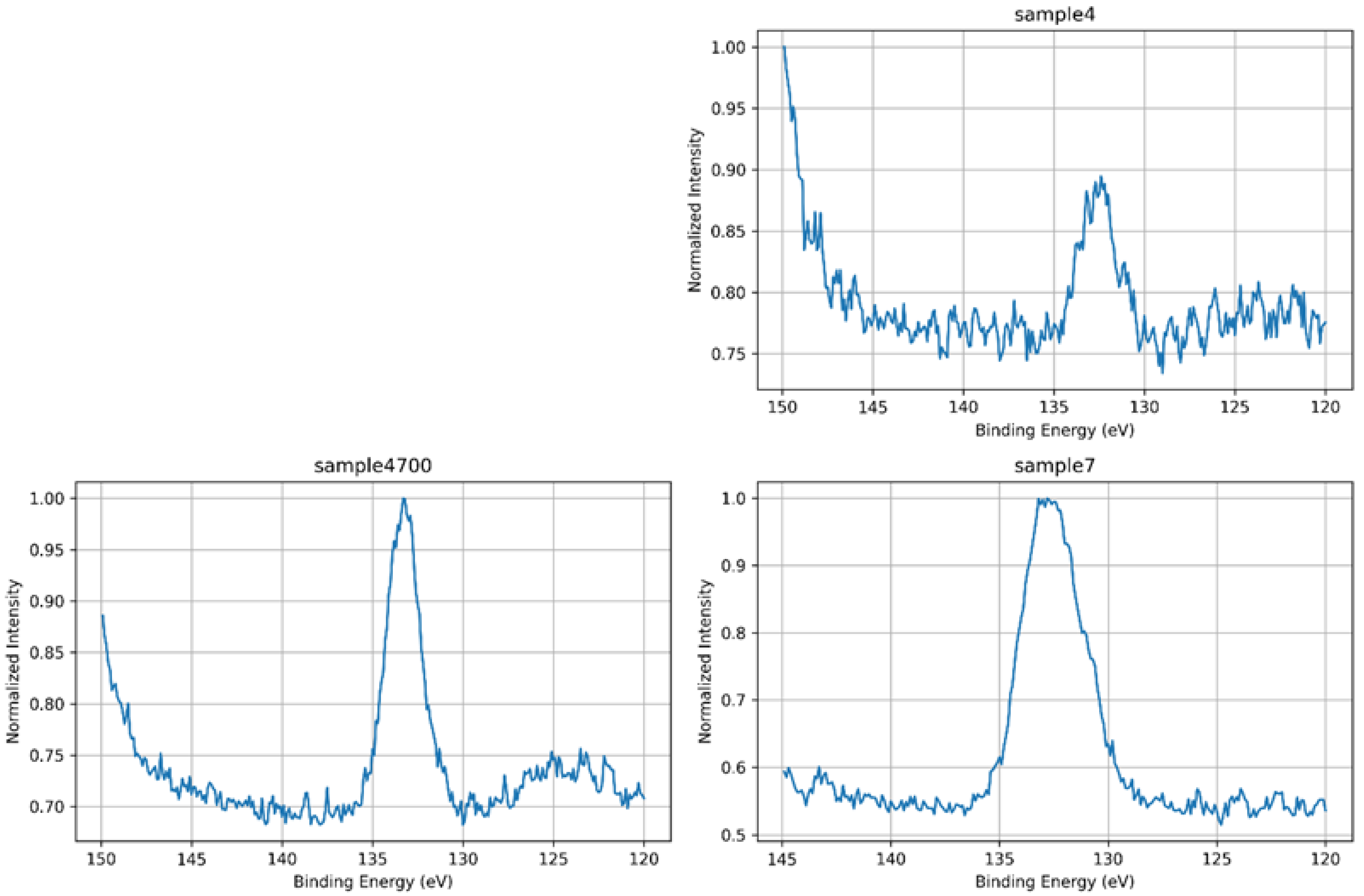
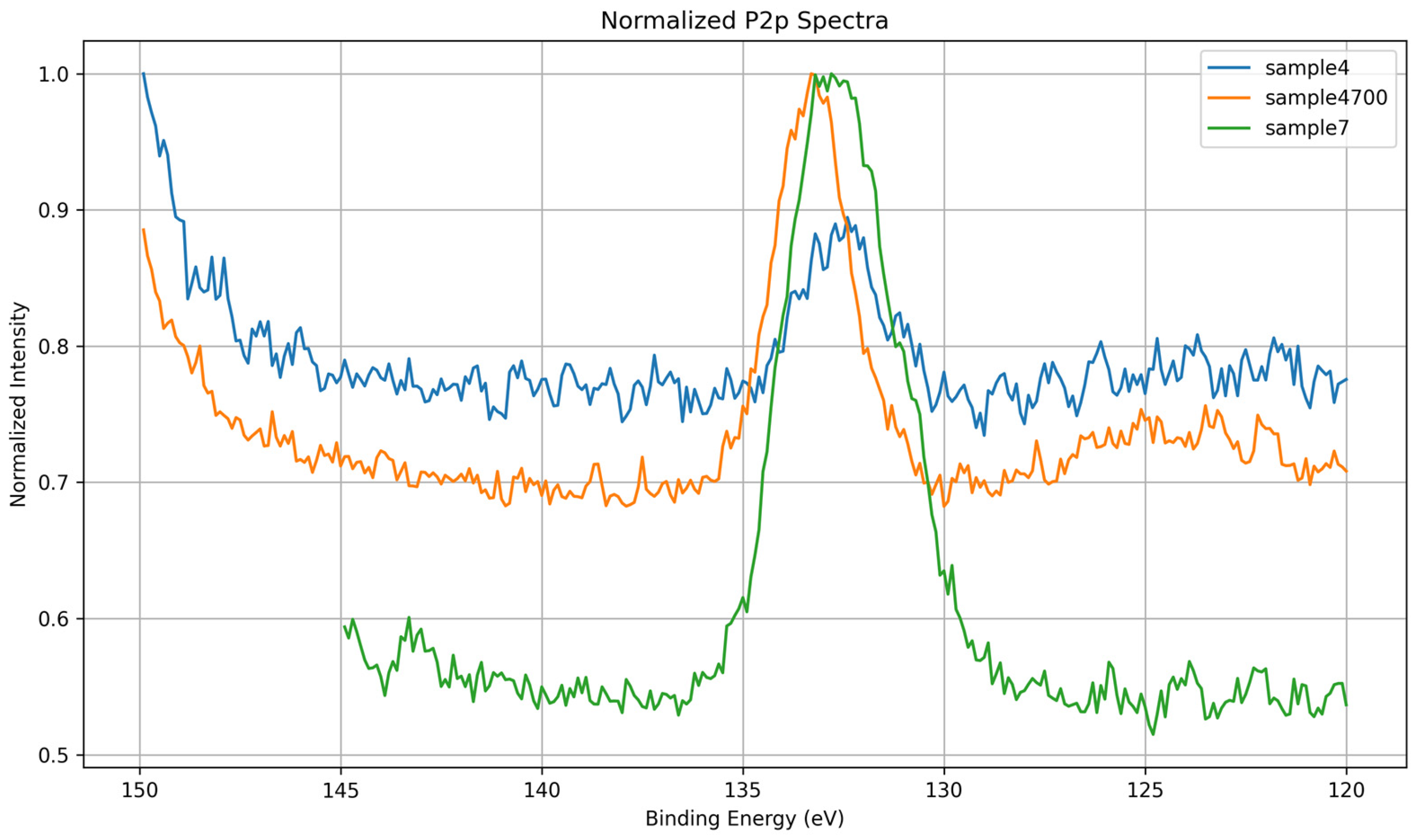
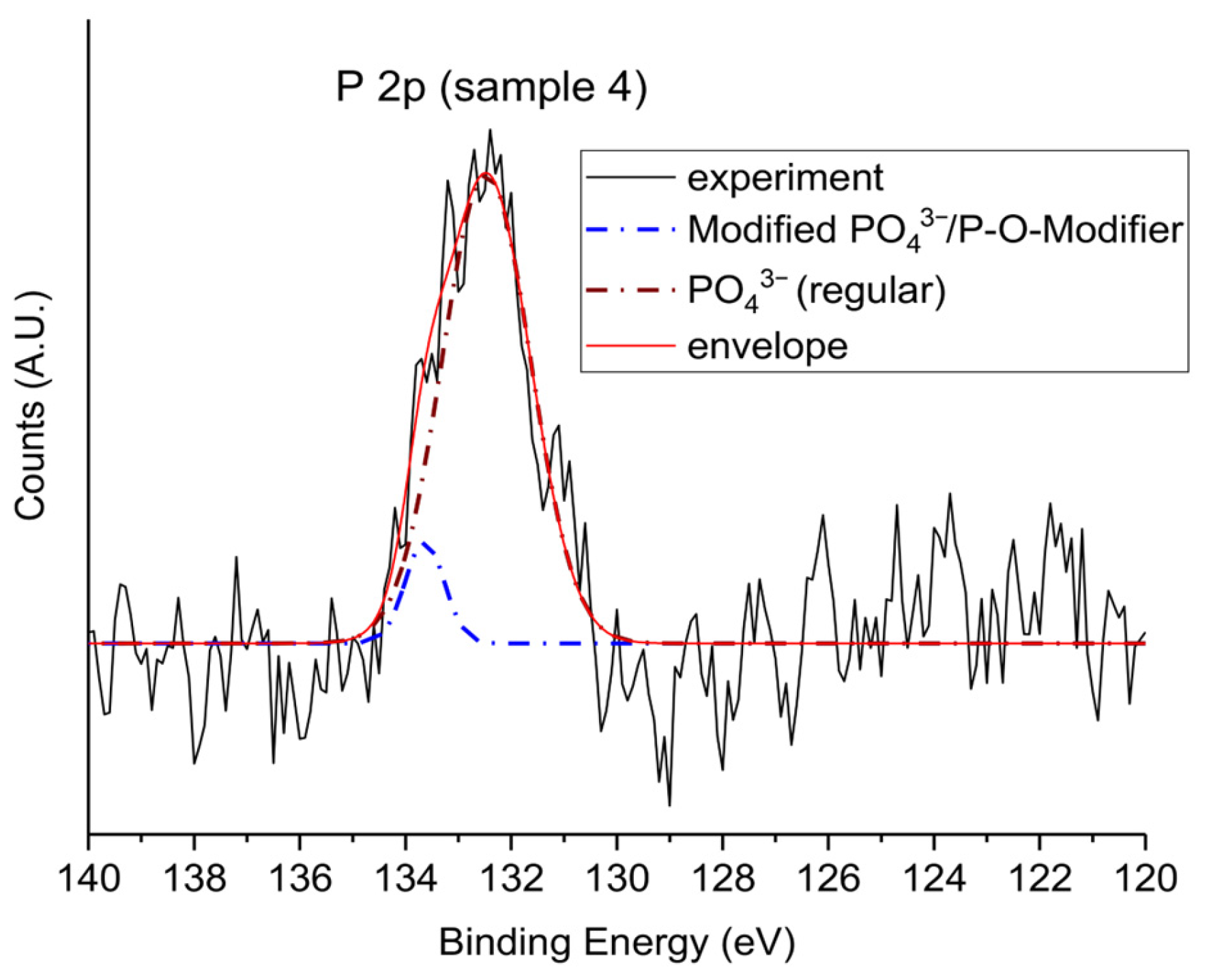
3.1.4. P 2p Core-Level Analysis
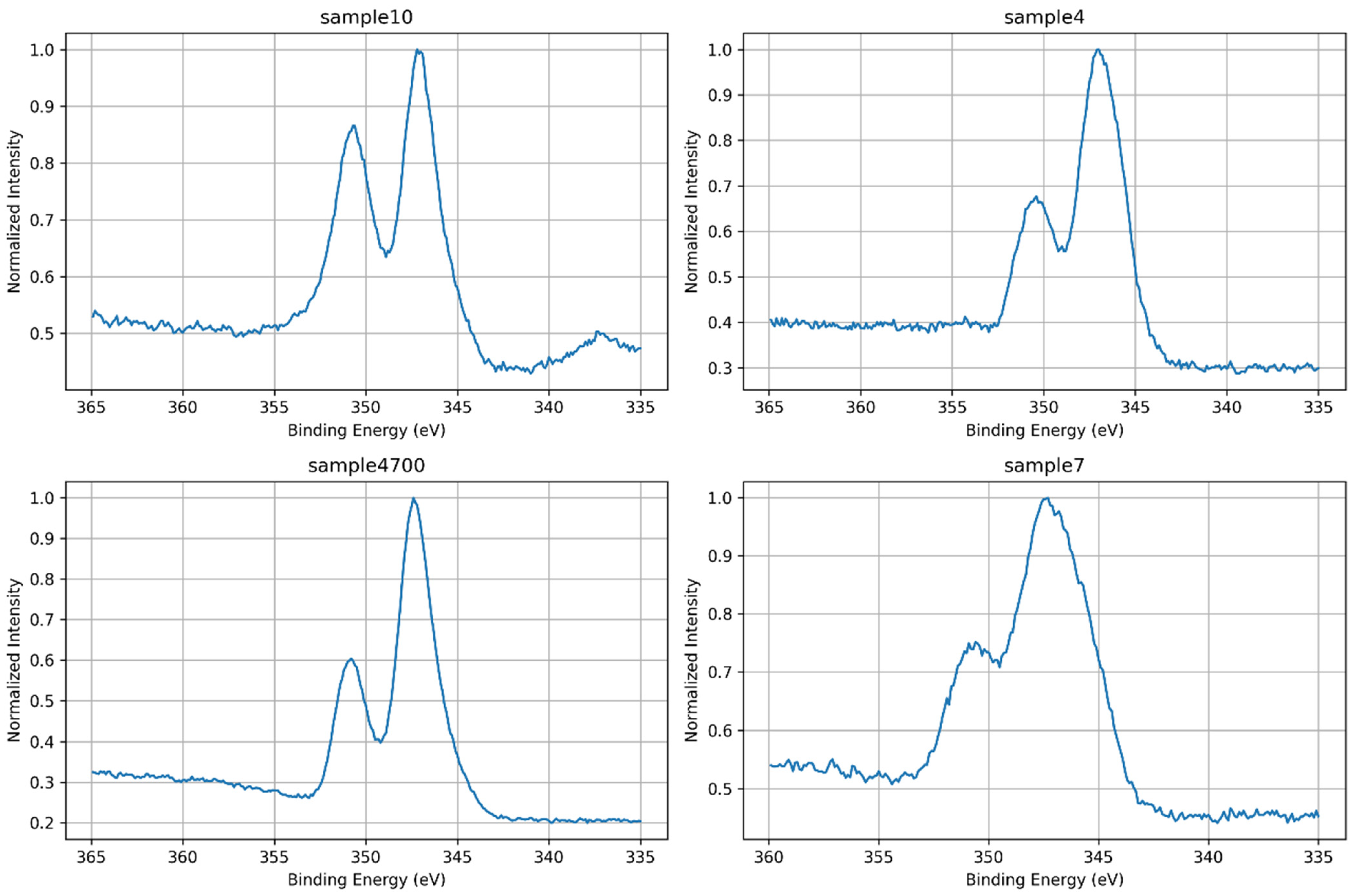
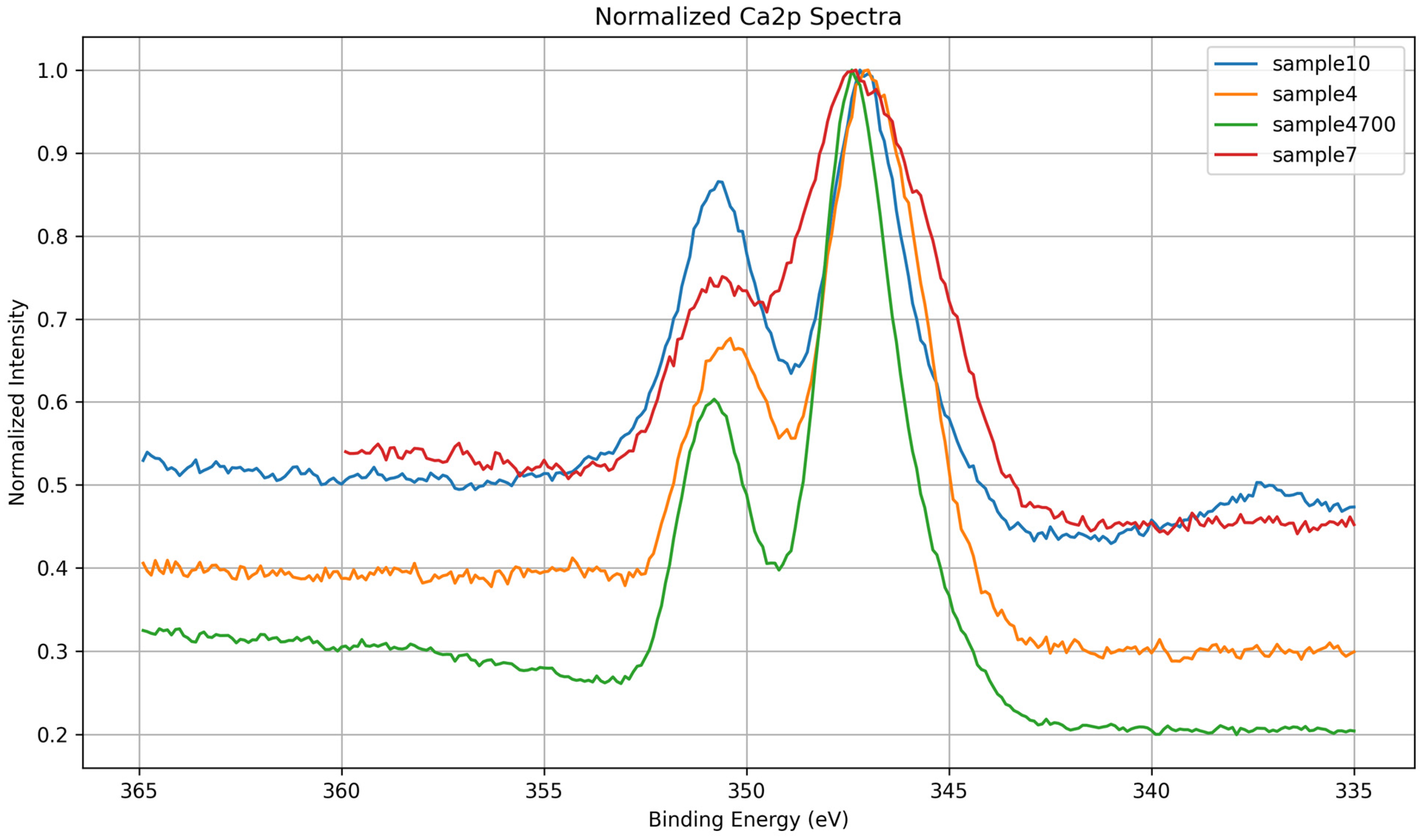
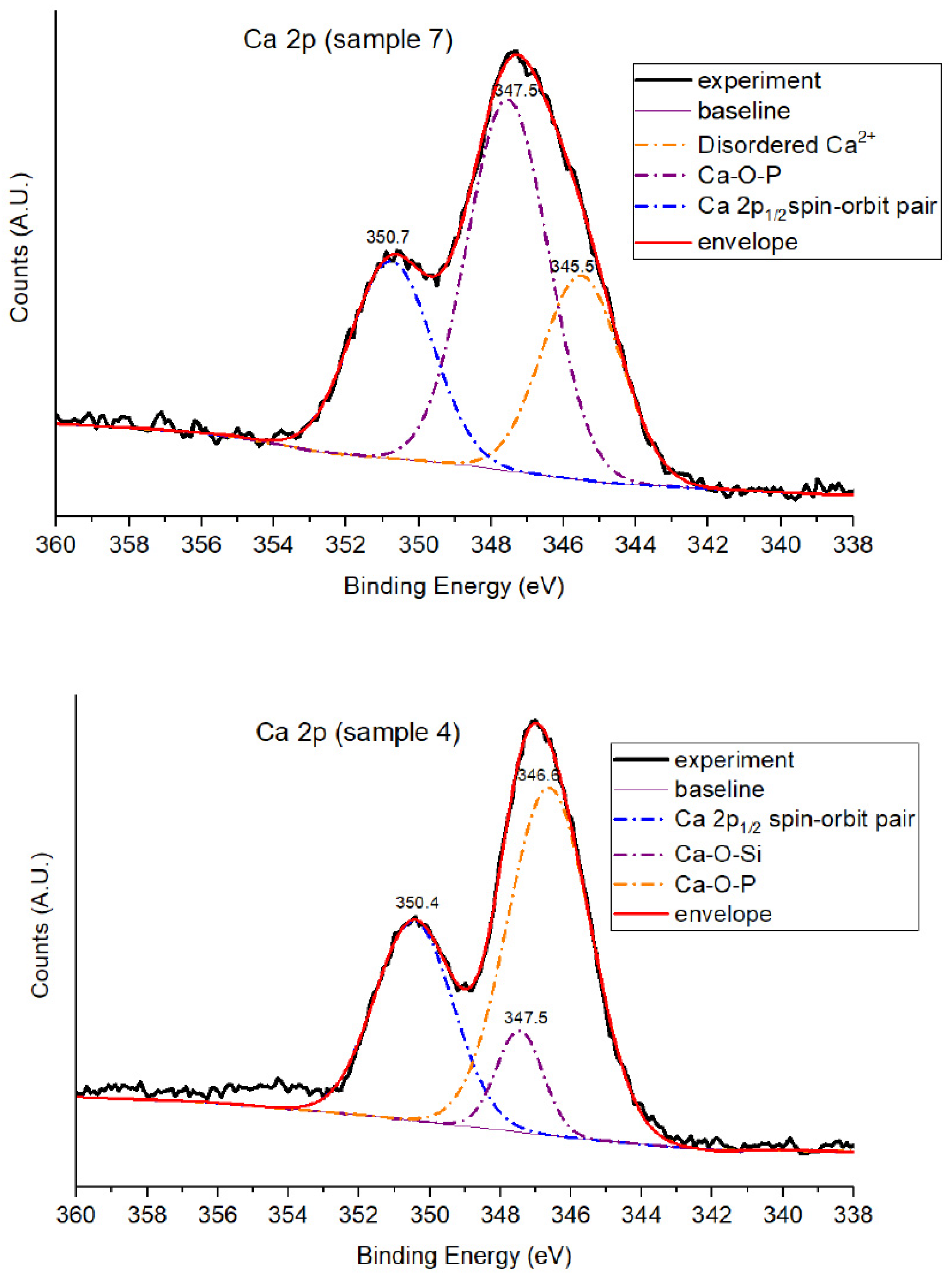
3.1.5. Ca 2p Core-Level Analysis
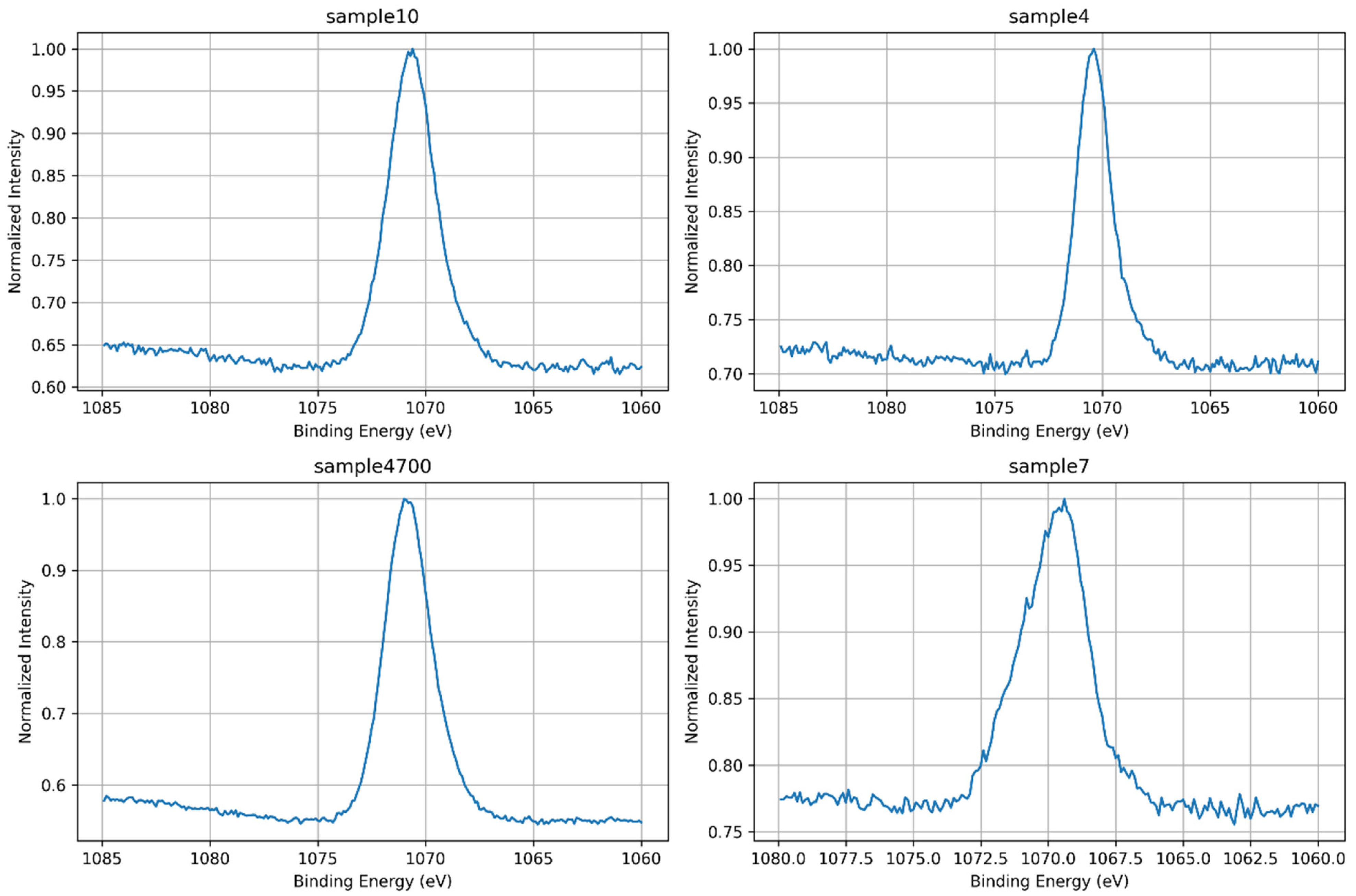
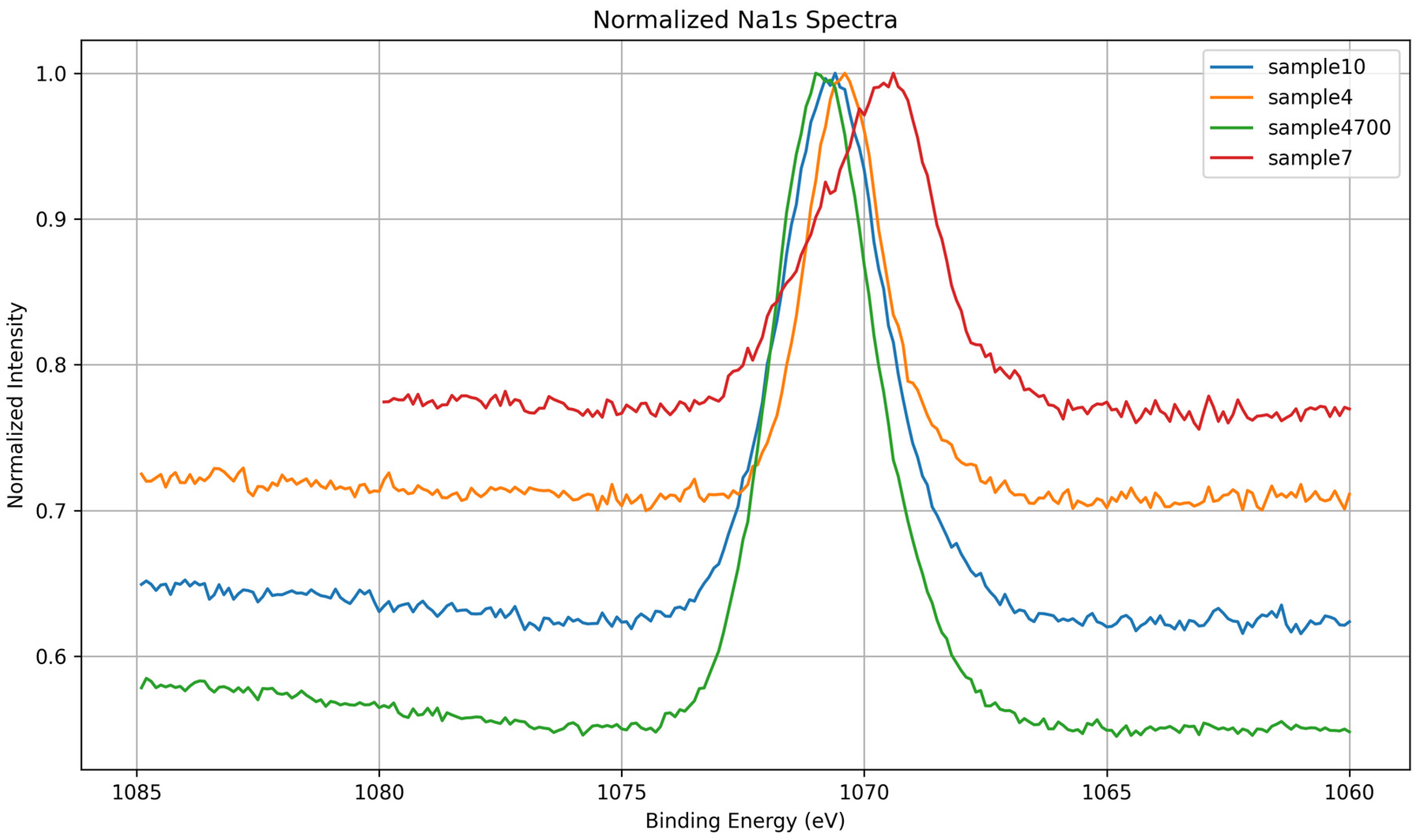
3.1.6. Na 1s Core-Level Analysis
3.1.7. Mg 1s Core-Level Analysis
3.1.8. Summary of Core-Level Trends
4. Conclusions
- The presence of multivalent trace elements in waters such as Água Prata and Água Azeda leads to local rearrangements in the silicate and phosphate networks.
- Thermal treatment enhances network consolidation, reducing hydroxylation and promoting Q4-like domains.
- Modifier ions introduced via mineral waters can replace native Na+ and Ca2+, leading to complex surface chemistries that potentially enhance bioactivity.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hench, L.L. Bioceramics: From Concept to Clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Ślośarczyk, A.; Paszkiewicza, Z.; Paluszkiewicz, C. FTIR and XRD evaluation of carbonated hydroxyapatite powders synthesized by wet methods. J. Mol. Struct. 2005, 744–747, 657–661. [Google Scholar] [CrossRef]
- Salim, S.A.S.; Mohamad, H.; Noor, S.N.F.M. Influence of MgO on Sol-Gel Derived SiO2-CaO-Na2O-P2O5 Bioglass System. J. Phys. Conf. Ser. 2018, 1082, 012034. [Google Scholar] [CrossRef]
- Vasconcelos, H.C. Glasses as biomaterials. In Overall Aspects of Non-Traditional Glasses: Synthesis, Properties and Applications; Vasconcelos, H.C., Gonçalves, M.C., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2016; pp. 19–38. [Google Scholar] [CrossRef]
- ISO ISO/DIS 23317; Implants for Surgery: In Vitro Evaluation for Apatite Forming Ability of Implant Materials. International Organization for Standardization: Geneva, Switzerland, 2014.
- Gerhardt, L.-C.; Boccaccini, A.R. Bioactive glass and glass-ceramic scaffolds for bone tissue engineering. Materials 2010, 3, 3867–3910. [Google Scholar] [CrossRef]
- Vasconcelos, H.C.; Coutinho, R.; Freire, P.; Farias, A.C.; Gonçalves, M.C.; Silva Pinto, A. Bioglass Prepared via a Mineral Water-assisted Sol–Gel Route; Pérez, M., Mills, J., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2018; pp. 71–104. [Google Scholar]
- Cruz, J.V.; Freire, P.; Costa, A. Mineral waters characterization in the Azores archipelago (Portugal). J. Volcanol. Geotherm. Res. 2010, 190, 353–364. [Google Scholar] [CrossRef]
- Hench, L.L.; West, J.K. The sol-gel process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Cacciotti, I. Cationic and Anionic Substitutions in Hydroxyapatite. In Handbook of Bioceramics and Biocomposites; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–68. [Google Scholar] [CrossRef]
- Hung, C.-C.; Chaya, A.; Liu, K.; Verdelis, K.; Sfeir, C. The role of magnesium ions in bone regeneration involves the canonical Wnt signaling pathway. Acta Biomater. 2019, 98, 246–255. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, W.; Tian, P.; Meng, F.; Zhu, H.; Jiang, X.; Liu, X.; Chu, P.K. Stimulation of bone growth following zinc incorporation into biomaterials. Biomaterials 2014, 35, 6882–6897. [Google Scholar] [CrossRef]
- Kołodziejska, B.; Stępień, N.; Kolmas, J. The Influence of Strontium on Bone Tissue Metabolism and Its Application in Osteoporosis Treatment. Int. J. Mol. Sci. 2021, 22, 6564. [Google Scholar] [CrossRef]
- Jacobs, A.; Renaudin, G.; Forestier, C.; Nedelec, J.-M.; Descamps, S. Biological properties of copper-doped biomaterials for orthopedic applications: A review of antibacterial, angiogenic and osteogenic aspects. Acta Biomater. 2020, 117, 21–39. [Google Scholar] [CrossRef]
- Rondanelli, M.; Faliva, M.A.; Peroni, G.; Infantino, V.; Gasparri, C.; Iannello, G.; Perna, S.; Riva, A.; Petrangolini, G.; Tartara, A. Pivotal role of boron supplementation on bone health: A narrative review. J. Trace Elem. Med. Biol. 2020, 62, 126577. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Chin, K.-Y.; Ima-Nirwana, S. The Skeletal-Protecting Action and Mechanisms of Action for Mood-Stabilizing Drug Lithium Chloride: Current Evidence and Future Potential Research Areas. Front. Pharmacol. 2020, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, W.; Wang, M.; Backman, L.J.; Chen, J. Effects of Zinc, Magnesium, and Iron Ions on Bone Tissue Engineering. ACS Biomater. Sci. Eng. 2022, 8, 2321–2335. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.M.; Flores-Sahagun, T.H.S.; Paredes, R.C. A perspective on molybdenum biocompatibility and antimicrobial activity for applications in implants. J. Mater. Sci. 2015, 51, 2806–2816. [Google Scholar] [CrossRef]
- Qi, L.; Zhao, T.; Yan, J.; Ge, W.; Jiang, W.; Wang, J.; Gholipourmalekabadi, M.; Lin, K.; Wang, X.; Zhang, L. Advances in magnesium containing bioceramics for bone repair. Biomater. Transl. 2024, 5, 3–20. [Google Scholar]
- Exley, C. The toxicity of aluminium in humans. Morphologie 2016, 100, 51–55. [Google Scholar] [CrossRef]
- Alzahrani, A.S. A Review of Glass and Crystallizations of Glass-Ceramics. Adv. Mater. Phys. Chem. 2022, 12, 261–288. [Google Scholar] [CrossRef]
- Dziadek, M.; Zagrajczuk, B.; Jelen, P.; Olejniczak, Z.; Cholewa-Kowalska, K. Structural variations of bioactive glasses obtained by different synthesis routes. Ceram. Int. 2016, 42, 14700–14709. [Google Scholar] [CrossRef]
- Watts, J.F.; Wolstenholme, J. An Introduction to Surface Analysis by XPS and AES; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Bagus, P.S.; Ilton, E.S.; Nelin, C.J. The interpretation of XPS spectra: Insights into materials properties. Surf. Sci. Rep. 2013, 68, 273–304. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Shao, X.-B.; Xing, Z.-W.; Liu, S.-Y.; Miao, K.-X.; Qi, S.-C.; Peng, S.-S.; Liu, X.-Q.; Sun, L.-B. Atomically dispersed calcium as solid strong base catalyst with high activity and stability. Green Energy Environ. 2024, 9, 1619–1626. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Bancroft, G.M.; Henderson, G.S.; Ho, R.; Dalby, K.N.; Huang, Y.; Yan, Z. Bridging, non-bridging and free (O2−) oxygen in Na2O–SiO2 glasses: An X-ray photoelectron spectroscopic (XPS) and nuclear magnetic resonance (NMR) study. J. Non-Cryst. Solids 2011, 357, 170–180. [Google Scholar] [CrossRef]
- Foix, D.; Martinez, H.; Gonbeau, D.; Granier, D.; Pradel, A.; Ribes, M. Thiogermanate glasses—Influence of the modifier cation—A combined XPS and theoretical study. Phys. Chem. Chem. Phys. 2005, 7, 180–186. [Google Scholar] [CrossRef]
- Sreenivasan, H.; Kinnunen, P.; Adesanya, E.; Patanen, M.; Kantola, A.M.; Telkki, V.-V.; Huttula, M.; Cao, W.; Provis, J.L.; Illikainen, M. Field Strength of Network-Modifying Cation Dictates the Structure of (Na-Mg) Aluminosilicate Glasses. Front. Mater. 2020, 7, 267. [Google Scholar] [CrossRef]
- Lin, S.; Ionescu, C.; Pike, K.J.; Smith, M.E.; Jones, J.R. Nanostructure evolution and calcium distribution in sol–gel derived bioactive glass. J. Mater. Chem. 2009, 19, 1276–1282. [Google Scholar] [CrossRef]
- Massiot, P.; Centeno, M.A.; Gouriou, M.; Domínguez, M.I.; Odriozola, J.A. Sol–gel obtained silicophosphates as materials to retain caesium at high temperatures. J. Mater. Chem. 2003, 13, 67–74. [Google Scholar] [CrossRef]
- Massera, J.; Mishra, A.; Guastella, S.; Ferraris, S.; Verné, E. Surface functionalization of phosphate-based bioactive glasses with 3-aminopropyltriethoxysilane (APTS). Biomed. Glas. 2016, 2, 1–10. [Google Scholar] [CrossRef]
- Roy, B.; Baier, F.; Rosin, A.; Gerdes, T.; Schafföner, S. Structural characterization of the near-surface region of soda–lime–silica glass by X-ray photoelectron spectroscopy. Int. J. Appl. Glas. Sci. 2022, 14, 229–239. [Google Scholar] [CrossRef]
- Barr, T.L. The nature of the relative bonding chemistry in zeolites: An XPS study. Zeolites 1990, 10, 760–765. [Google Scholar] [CrossRef]
- Serra, J.; González, P.; Liste, S.; Serra, C.; Chiussi, S.; León, B.; Pérez-Amor, M.; Ylänen, H.; Hupa, M. FTIR and XPS studies of bioactive silica based glasses. J. Non-Cryst. Solids 2003, 332, 20–27. [Google Scholar] [CrossRef]
- Deng, B.; Shi, Y.; Zhou, Q.; Bauchy, M. Revealing the structural role of MgO in aluminosilicate glasses. Acta Mater. 2022, 222, 117417. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Possible Benefit in Biomedical Glass | Ref. |
|---|---|---|
| Mg2+ | Stimulates bone growth, osteoblast activity | [11] |
| Zn2+ | Antibacterial and regenerative action, important in bone metabolism | [12] |
| Sr2+ | Stimulates bone formation, inhibits bone resorption | [13] |
| Cu2+ | Antibacterial action, angiogenesis (formation of blood vessels) | [14] |
| B3+ | Stimulates bone and cartilage regeneration | [15] |
| Li+ | Potential neuroprotective and bone regenerative properties | [16] |
| Fe | May influence magnetic properties and strength | [17] |
| Mo, Mn | Less common, but can be incorporated for specific applications | [18,19] |
| Al | High quantities can be toxic, must be controlled | [20] |
| Sample ID | Water Type | Si-OH (%) | Si-O-Si (BO, %) | Si-O− (NBO, %) | Notes |
|---|---|---|---|---|---|
| 7 | DI (control) | 75.0 | 25.0 | - | Hydrated, sol–gel, balanced BO |
| 4 | Água Prata | 68.4 | 15.3 | 16.3 | Ion-induced NBO formation |
| 4700 | Água Prata | 21.0 | 77.0 | 2.0 | Densified, restructured network |
| 10 | Água Azeda | 49.2 | 50.8 | - | No resolved NBOs |
| Sample | BE(eV) | Area (%) | Assignment |
|---|---|---|---|
| 7 | 102.4 | 82.4 | Q2/Q3—NBO-rich (disordered) |
| 7 | 103.3 | 17.6 | Q3/Q4—Minor ordered sites |
| 4700 | 103.1 | 70.6 | Q3—Bridging oxygen (ordered) |
| 4700 | 103.7 | 29.4 | Q4—Fully polymerized domains |
| BE (eV) | Assignment | Description |
|---|---|---|
| 132.5 | PO43− (fully oxidized) | Main phosphate environment, tetrahedral and chemically uniform |
| 133.6 | Modified PO43− (P–O–Sr/Zn) | Phosphate perturbed by interaction with network-modifying cations |
| Sample | BE (eV) | Area (%) | Assignment |
|---|---|---|---|
| 4 | 346.6 | 58.8 | Ca–O–P (calcium phosphate) |
| 4 | 347.5 | 9.6 | Ca–O–Si (calcium in silicate domains) |
| 4 | 350.4 | 31.6 | Ca 2p1/2 spin–orbit pair |
| 7 | 345.5 | 25.9 | Disordered Ca2+ (e.g., Ca–OH) |
| 7 | 347.5 | 48.3 | Ca–O–P (calcium phosphate) |
| 7 | 350.7 | 25.8 | Ca 2p1/2 spin–orbit pair |
| Sample | Synthesis Water | Key XPS Observations | Key Observations |
|---|---|---|---|
| 7 | DI | Broad, symmetric O 1s; disordered Si 2p; intense P 2p; low Na 1s; Ca 2p shows Ca–O–P + disordered component | Phosphate-rich, under-condensed network with weak ionic shielding; Ca loosely coordinated; low densification |
| 4 | Prata | Asymmetric O 1s; Na 1s and P 2p broadened; Ca 2p shows Ca–O–P + Ca–O–Si; no disordered Ca | Presence of multivalent cations (e.g., Sr2+, Zn2+) induces moderate network perturbation and partial integration of Ca |
| 4700 | Prata | Sharpened O 1s, Si 2p, and Ca 2p; most intense Na 1s; P 2p narrow and symmetric | Thermal densification promotes polymerization (Q3/Q4), reduction of OH groups, and consolidation of coordination environments |
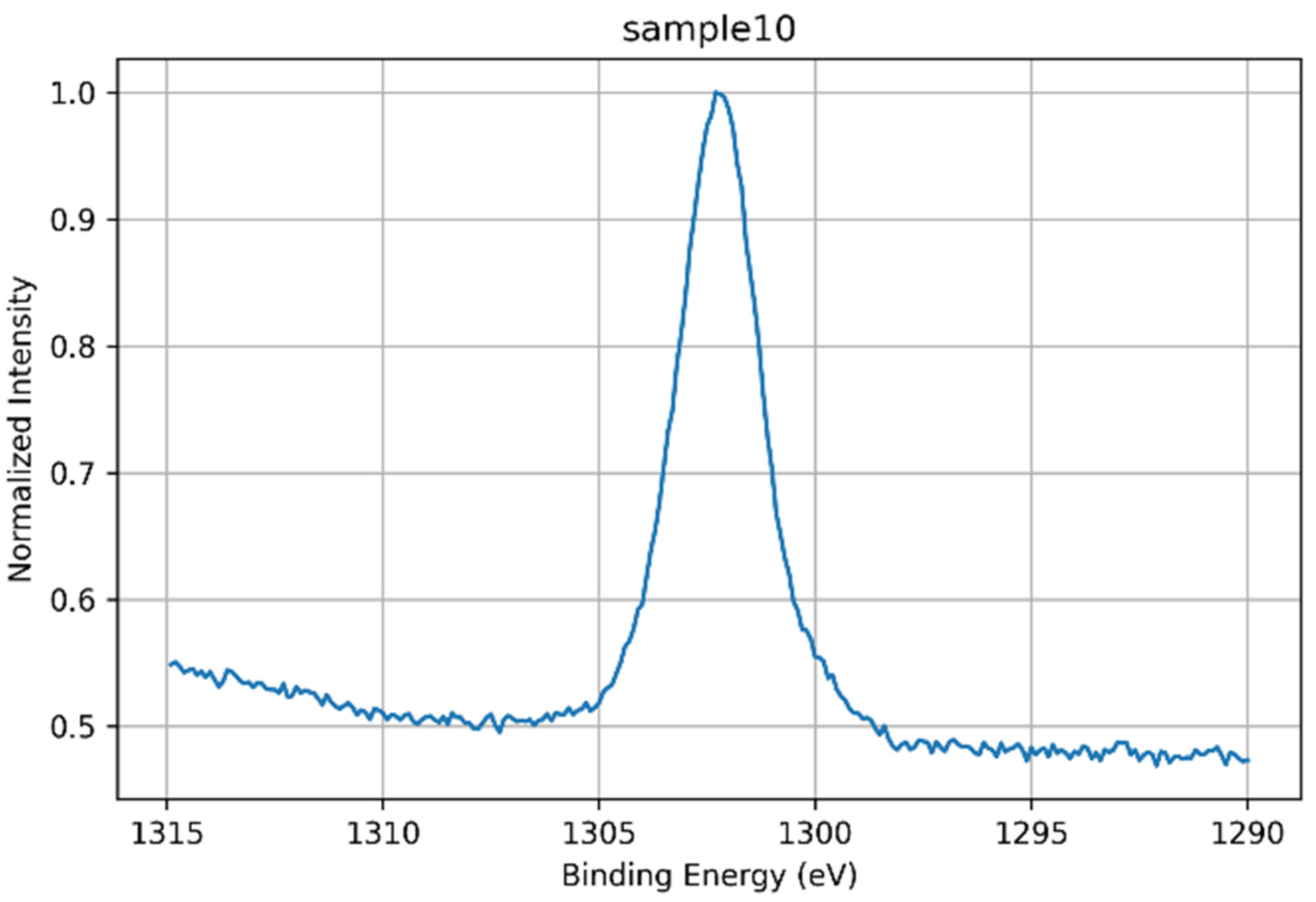
| 10 | Azeda | Broad O 1s; Na 1s depleted; Mg 1s detected; P 2p absent; Ca 2p sharp (Ca–O–Si) | Ion exchange with Mg2+ and other ions disrupts Na+ role; silicate network less hydrated, more ordered despite no heat treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasconcelos, H.C.; Meirelles, M.; Özmenteş, R. XPS Investigation of Sol–Gel Bioactive Glass Synthesized with Geothermal Water. Surfaces 2025, 8, 50. https://doi.org/10.3390/surfaces8030050
Vasconcelos HC, Meirelles M, Özmenteş R. XPS Investigation of Sol–Gel Bioactive Glass Synthesized with Geothermal Water. Surfaces. 2025; 8(3):50. https://doi.org/10.3390/surfaces8030050
Chicago/Turabian StyleVasconcelos, Helena Cristina, Maria Meirelles, and Reşit Özmenteş. 2025. "XPS Investigation of Sol–Gel Bioactive Glass Synthesized with Geothermal Water" Surfaces 8, no. 3: 50. https://doi.org/10.3390/surfaces8030050
APA StyleVasconcelos, H. C., Meirelles, M., & Özmenteş, R. (2025). XPS Investigation of Sol–Gel Bioactive Glass Synthesized with Geothermal Water. Surfaces, 8(3), 50. https://doi.org/10.3390/surfaces8030050