
Transcriptomic Comparison of Liver Tissue across Different Largemouth Bass (Micropterus salmoides) Strains

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish and Sample Collection
2.2. Growth Performance
2.3. Extraction of RNA, Library Preparation and Sequencing
2.4. De Novo Assembly, Unigene Annotation and Functional Classification
2.5. Analysis of Differentially Expressed Unigenes (DEGs) and Functional Enrichment Analyses
2.6. Analysis of Protein–Protein Interaction (PPI) and Scoring of the Gene Network
2.7. Statistical Analysis
3. Results
3.1. Growth Performance of the Four Largemouth Bass Strains
3.2. Sequencing and Annotation of Unigenes
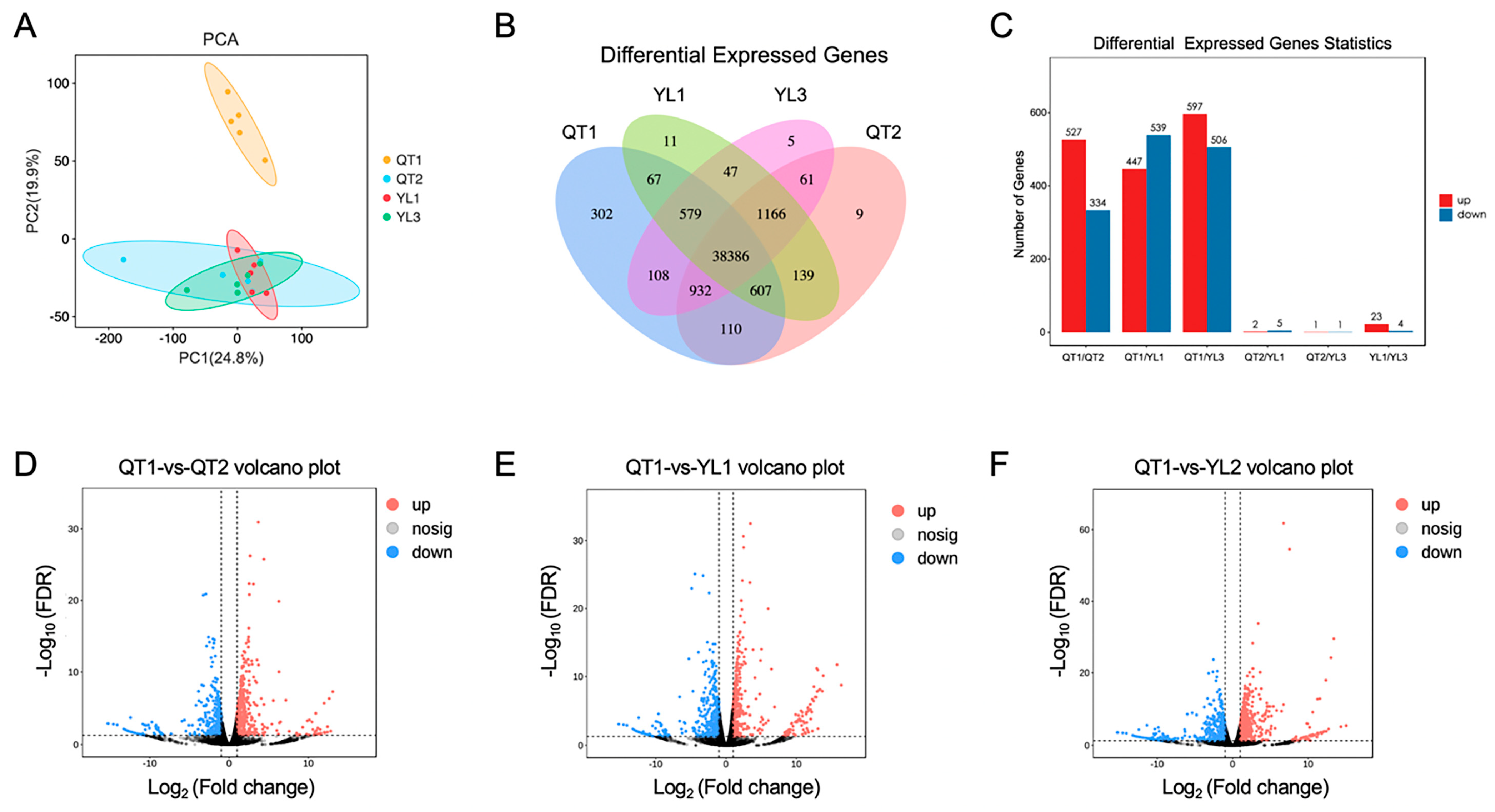
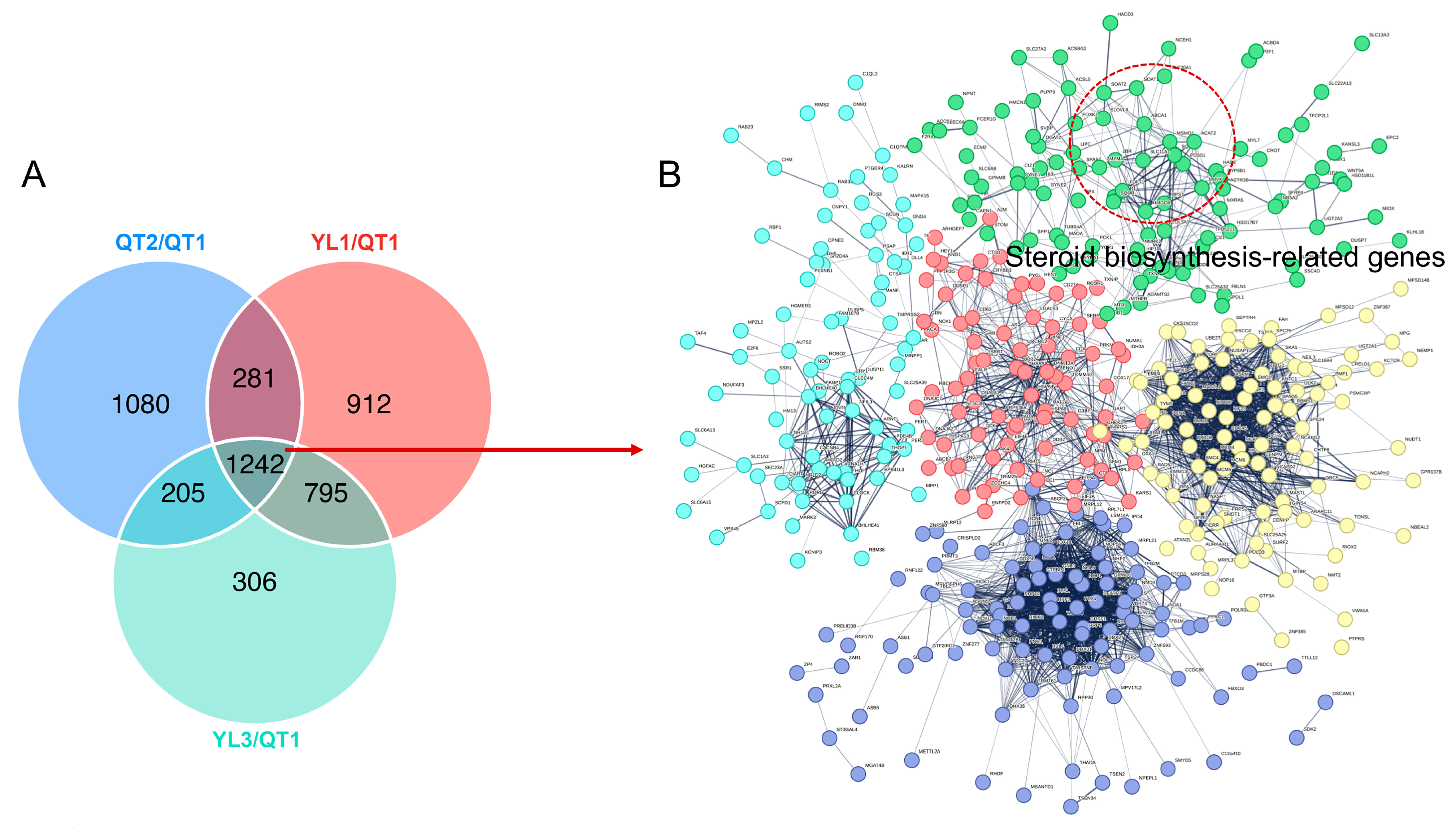
3.3. Analysis of Differentially Expressed Genes
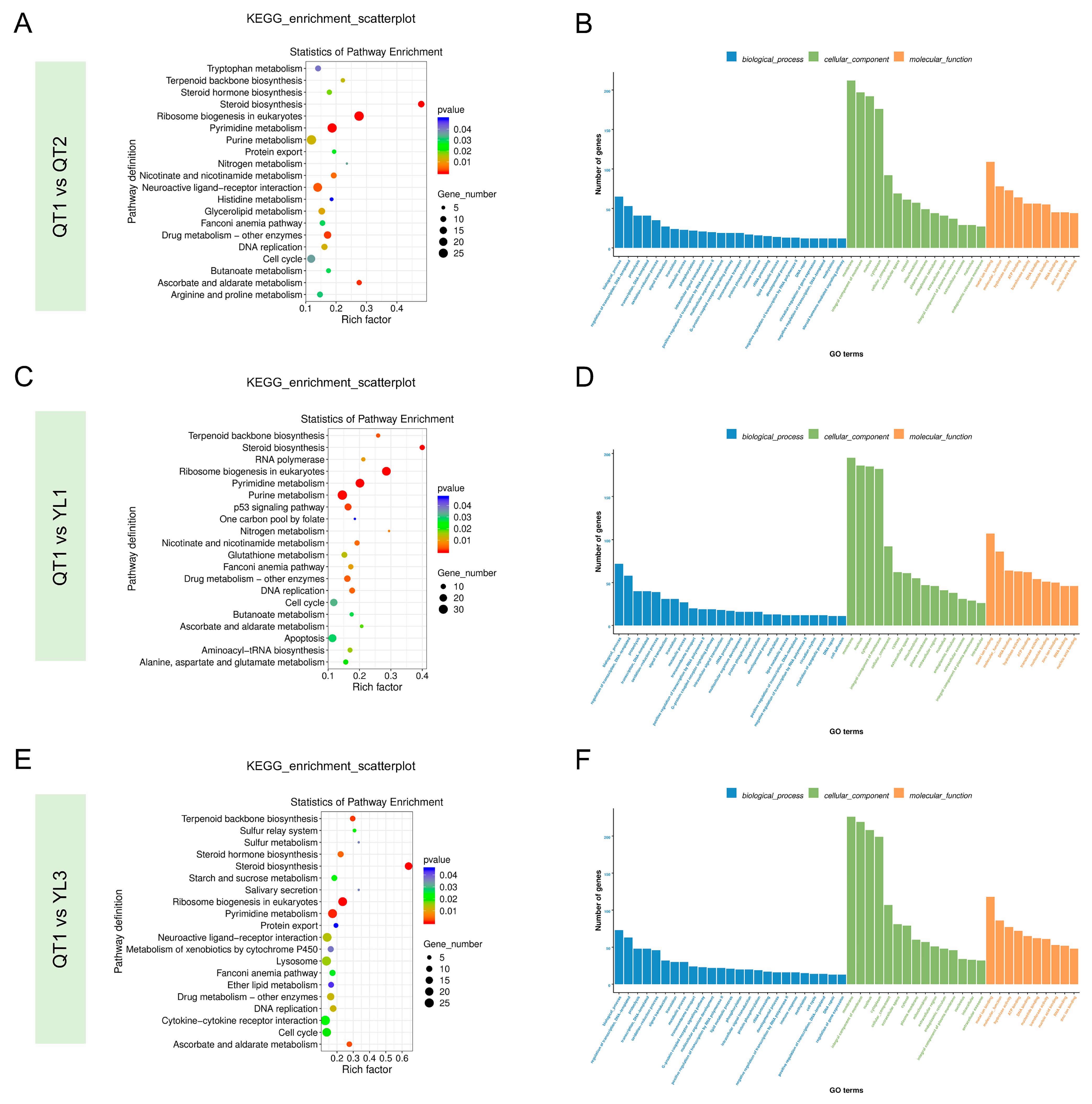
3.4. Functional Annotation of Unigenes
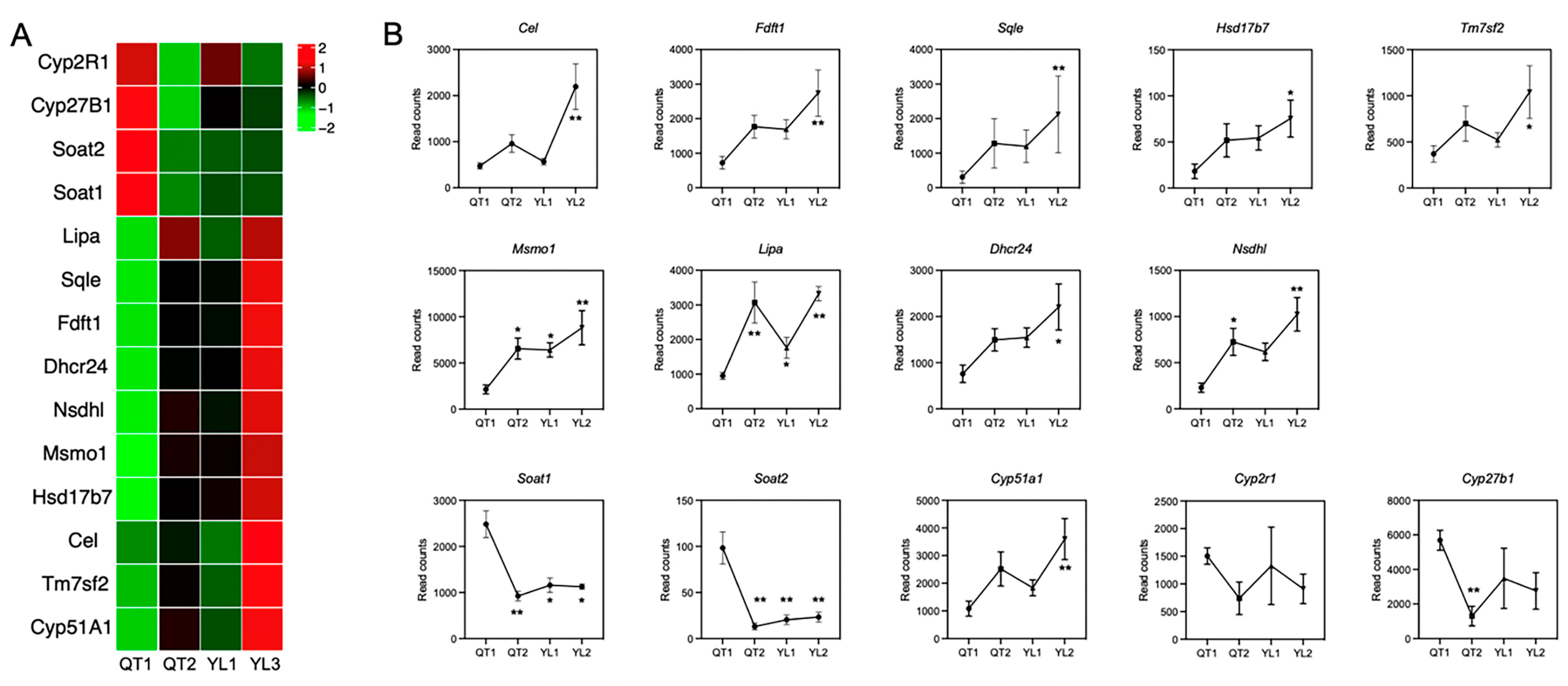
3.5. Expression Level of Genes Involved in Liver Steroid Biosynthesis
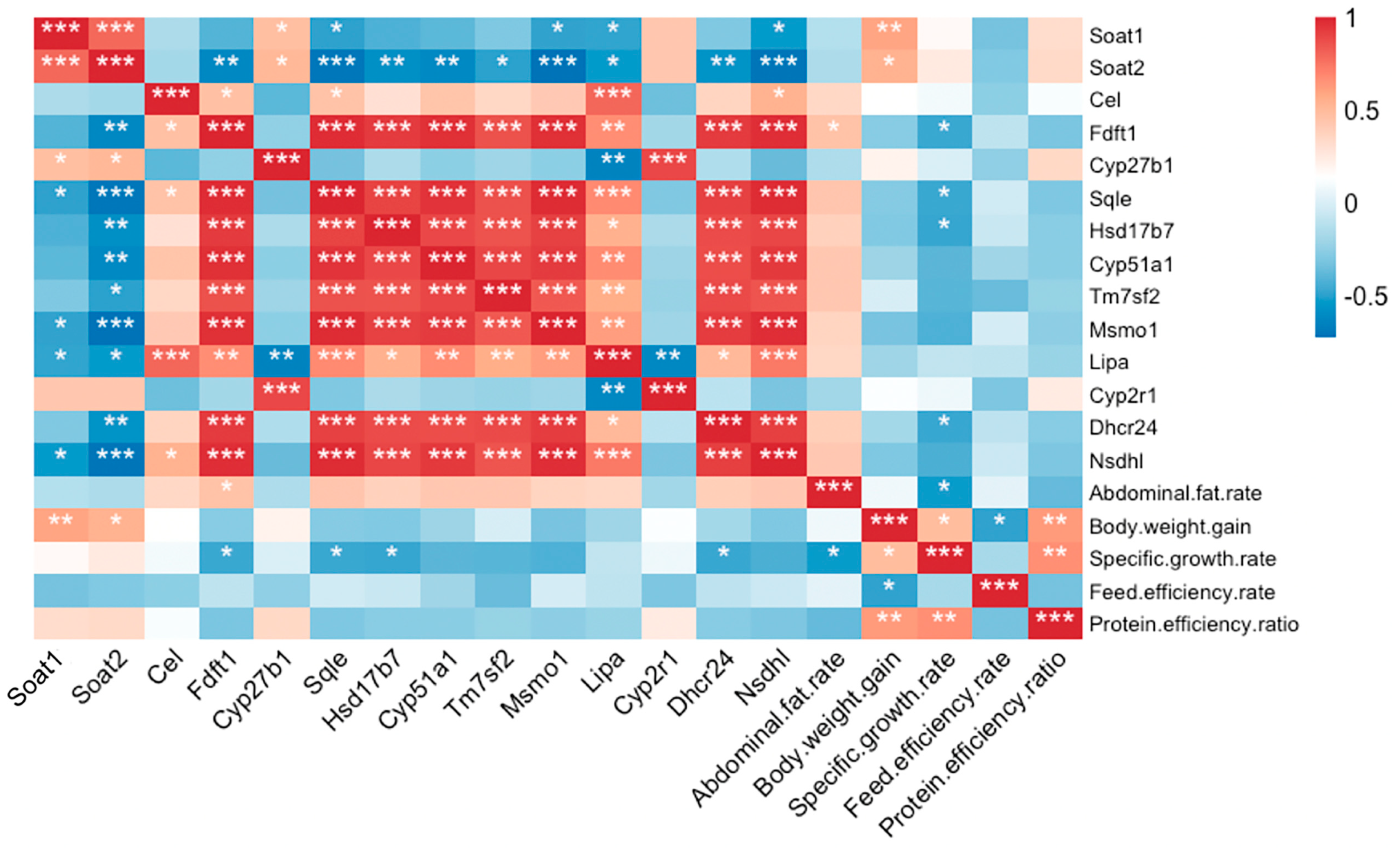
3.6. Correlation Analysis
3.7. Co-Expression Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marijani, E. Prevalence and Antimicrobial Resistance of Bacteria Isolated from Marine and Freshwater Fish in Tanzania. Int. J. Microbiol. 2022, 2022, 4652326. [Google Scholar] [CrossRef]
- Zou, J.H.; Hu, P.; Wang, M.Y.; Chen, Z.W.; Wang, H.; Guo, X.L.; Gao, J.; Wang, Q.C. Liver Injury and Metabolic Dysregulation in Largemouth Bass (Micropterus salmoides) after Ammonia Exposure. Metabolites 2023, 13, 274. [Google Scholar] [CrossRef]
- Lutz-Carrillo, D.J.; Nice, C.C.; Bonner, T.H.; Forstner, M.R.J.; Fries, L.T. Admixture analysis of Florida largemouth bass and northern largemouth bass using microsatellite loci. Trans. Am. Fish. Soc. 2006, 135, 779–791. [Google Scholar] [CrossRef]
- Chen, Y.J.; Yuan, R.M.; Liu, Y.J.; Yang, H.J.; Liang, G.Y.; Tian, L.X. Dietary vitamin C requirement and its effects on tissue antioxidant capacity of juvenile largemouth bass, Micropterus salmoides. Aquaculture 2015, 435, 431–436. [Google Scholar] [CrossRef]
- Hou, H.C.; Ren, A.Q.; Yu, L.X.B.; Ma, Z.; Zhang, Y.; Liu, Y. An Environmental Impact Assessment of Largemouth Bass (Micropterus salmoides) Aquaculture in Hangzhou, China. Sustainability 2023, 15, 12368. [Google Scholar] [CrossRef]
- Guo, Y.H.; Bai, J.J.; Chang, O.Q.; Lao, H.H.; Ye, X.; Luo, J.R. Molecular structure of the largemouth bass (Micropterus salmoides) Myf5 gene and its effect on skeletal muscle growth. Mol. Biol. Rep. 2009, 36, 1497–1504. [Google Scholar] [CrossRef]
- Bai, J.J.; Lutz-Carrillo, D.J.; Quan, Y.C.; Liang, S.X. Taxonomic status and genetic diversity of cultured largemouth bass Micropterus salmoides in China. Aquaculture 2008, 278, 27–30. [Google Scholar] [CrossRef]
- Wang, D.; Yao, H.; Li, Y.H.; Xu, Y.J.; Ma, X.F.; Wang, H.P. Global diversity and genetic landscape of natural populations and hatchery stocks of largemouth bass across American and Asian regions. Sci. Rep. 2019, 9, 16697. [Google Scholar] [CrossRef]
- Zhu, L.Y.; Zong, X.; Xiao, X.; Cheng, Y.Z.; Fu, J.; Lu, Z.Q.; Jin, M.L.; Wang, F.Q.; Wang, Y.Z. Multi-Omics Analysis of the Gut-Liver Axis Reveals the Mechanism of Liver Injury in Colitis Mice. Front. Immunol. 2022, 12, 773070. [Google Scholar] [CrossRef]
- Aung, T.N.; Qu, Z.P.; Kortschak, R.D.; Adelson, D.L. Understanding the Effectiveness of Natural Compound Mixtures in Cancer through Their Molecular Mode of Action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef]
- Guan, C.; Cen, H.F.; Cui, X.; Tian, D.Y.; Tadesse, D.; Zhang, Y.W. Proline improves switchgrass growth and development by reduced lignin biosynthesis. Sci. Rep. 2019, 9, 20117. [Google Scholar] [CrossRef]
- Gonçalves, M.F.M.; Nunes, R.B.; Tilleman, L.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; Alves, A. Dual RNA Sequencing of during Infection Unveils Host-Pathogen Interactions. Int. J. Mol. Sci. 2019, 20, 6083. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.S.; Ruiz Daniels, R.; Dobie, R.; Naseer, S.; Clark, T.C.; Henderson, N.C.; Boudinot, P.; Martin, S.A.M.; Macqueen, D.J. Single cell transcriptomics of Atlantic salmon (Salmo salar L.) liver reveals cellular heterogeneity and immunological responses to challenge by Aeromonas salmonicida. Front. Immunol. 2022, 13, 984799. [Google Scholar] [CrossRef] [PubMed]
- Harvey, T.N.; Sandve, S.R.; Jin, Y.; Vik, J.O.; Torgersen, J.S. Liver slice culture as a model for lipid metabolism in fish. PeerJ 2019, 7, e7732. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Marandel, L.; Skiba-Cassy, S.; Corraze, G.; Dupont-Nivet, M.; Quillet, E.; Geurden, I.; Panserat, S. Regulation by Dietary Carbohydrates of Intermediary Metabolism in Liver and Muscle of Two Isogenic Lines of Rainbow Trout. Front. Physiol. 2018, 9, 1579. [Google Scholar] [CrossRef]
- Cox, A.G.; Goessling, W. The lure of zebrafish in liver research: Regulation of hepatic growth in development and regeneration. Curr. Opin. Genet. Dev. 2015, 32, 153–161. [Google Scholar] [CrossRef]
- Cao, Q.Q.; Shan, H.Y.; Zhao, J.; Deng, J.H.; Xu, M.; Kang, H.; Li, T.; Zhao, Y.; Liu, H.F.; Jiang, J. Liver fibrosis in fish research: From an immunological perspective. Fish Shellfish Immunol. 2023, 139, 108885. [Google Scholar] [CrossRef]
- Zong, X.; Cheng, Y.Z.; Xiao, X.; Fu, J.; Wang, F.Q.; Lu, Z.Q.; Wang, Y.Z.; Jin, M.L. Protective effects of sulfated polysaccharide from Enterobacter cloacae Z0206 against DSS-induced intestinal injury via DNA methylation. Int. J. Biol. Macromol. 2021, 183, 861–869. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Z.; Tao, Y.; Wu, W.; Zeng, Y.; Liao, K.; Li, X.; Chen, L. Transcriptomic and Physiological Responses of Chlorella pyrenoidosa during Exposure to 17alpha-Ethinylestradiol. Int. J. Mol. Sci. 2022, 23, 3583. [Google Scholar] [CrossRef]
- Moreno-Santillan, D.D.; Machain-Williams, C.; Hernandez-Montes, G.; Ortega, J. De Novo Transcriptome Assembly and Functional Annotation in Five Species of Bats. Sci. Rep. 2019, 9, 6222. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10. [Google Scholar] [CrossRef]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.M.; Liu, G.Y.; Wang, H.L.; Wassie, T.; Wu, X. Maternal pyrimidine nucleoside supplementation regulates fatty acid, amino acid and glucose metabolism of neonatal piglets. Anim. Nutr. 2022, 11, 309–321. [Google Scholar] [CrossRef]
- Waititu, S.M.; Yin, F.; Patterson, R.; Yitbarek, A.; Rodriguez-Lecompte, J.C.; Nyachoti, C.M. Dietary supplementation with a nucleotide-rich yeast extract modulates gut immune response and microflora in weaned pigs in response to a sanitary challenge. Animal 2017, 11, 2156–2164. [Google Scholar] [CrossRef]
- Liu, G.; Liu, H.; Tian, W.; Liu, C.; Yang, H.; Wang, H.; Gao, L.; Huang, Y. Dietary nucleotides influences intestinal barrier function, immune responses and microbiota in 3-day-old weaned piglets. Int. Immunopharmacol. 2023, 117, 109888. [Google Scholar] [CrossRef]
- Gao, L.M.; Liu, Y.L.; Zhou, X.; Zhang, Y.; Wu, X.; Yin, Y.L. Maternal supplementation with uridine influences fatty acid and amino acid constituents of offspring in a sow-piglet model. Br. J. Nutr. 2021, 125, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, S.; Xie, C.; Wang, R.; Zhang, Y.; Zhou, X.; Wu, X. Short-Term Oral UMP/UR Administration Regulates Lipid Metabolism in Early-Weaned Piglets. Animals 2019, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Koyama, H.; Kurajoh, M.; Shoji, T.; Tsutsumi, Z.; Moriwaki, Y. Biochemistry of uridine in plasma. Clin. Chim. Acta 2011, 412, 1712–1724. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; Franks, S.; et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef]
- Sherwood, W.B.; Kothalawala, D.M.; Kadalayil, L.; Ewart, S.; Zhang, H.M.; Karmaus, W.; Arshad, S.H.; Holloway, J.W.; Rezwan, F.I. Epigenome-Wide Association Study Reveals Duration of Breastfeeding Is Associated with Epigenetic Differences in Children. Int. J. Environ. Res. Public Health 2020, 17, 3569. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, P.S.; Li, J.; Chen, J.P. Putative Genes and Pathways Involved in the Acne Treatment of Isotretinoin via Microarray Data Analyses. BioMed Res. Int. 2020, 2020, 5842795. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Xia, Y. Convergent perturbation of the human domain-resolved interactome by viruses and mutations inducing similar disease phenotypes. PLoS Comput. Biol. 2019, 15, e1006762. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Tsai, C.Y.; Arnold, S.J.; Huang, G.J. Ablation of hippocampal neurogenesis in mice impairs the response to stress during the dark cycle. Nat. Commun. 2015, 6, 8373. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Feng, X.P.; Wang, H.L.; Meng, C.H.; Zhang, J.; Qian, Y.; Zhong, J.F.; Cao, S.X. Transcriptome analysis reveals corresponding genes and key pathways involved in heat stress in Hu sheep. Cell Stress Chaperon. 2019, 24, 1045–1054. [Google Scholar] [CrossRef]
- Chen, L.H.; Han, J.P.; Deng, X.M.; Tan, S.L.; Li, L.L.; Li, L.; Zhou, J.F.; Peng, H.; Yang, G.X.; He, G.Y.; et al. Expansion and stress responses of AP2/EREBP superfamily in Brachypodium distachyon. Sci. Rep. 2016, 6, 21623. [Google Scholar] [CrossRef]
- Castillo-Castrejon, M.; Yang, I.V.; Davidson, E.J.; Borengasser, S.J.; Jambal, P.; Westcott, J.; Kemp, J.F.; Garces, A.; Ali, S.A.; Saleem, S.; et al. Preconceptional Lipid-Based Nutrient Supplementation in 2 Low-Resource Countries Results in Distinctly Different IGF-1/mTOR Placental Responses. J. Nutr. 2021, 151, 556–569. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Group | |||
|---|---|---|---|---|
| QT1 | QT2 | YL1 | YL3 | |
| Initial body weight, g | 6.05 ± 0.08 | 6.14 ± 0.07 | 6.03 ± 0.15 | 6.17 ± 0.11 |
| Survival rate 1, % | 100.00 ± 0.00 | 98.89 ± 1.92 | 98.89 ± 1.92 | 100.00 ± 0.00 |
| Final body length, cm | 13.67 ± 0.45 | 13.57 ± 0.15 | 13.27 ± 0.32 | 13.30 ± 0.41 |
| Final body weight, g | 55.58 ± 0.85 | 54.08 ± 1.15 | 53.17 ± 3.09 | 56.01 ± 1.37 |
| Condition factor 2, g/cm 3 | 2.29 ± 0.13 | 2.32 ± 0.08 | 2.28 ± 0.06 | 2.37 ± 0.11 |
| XLiver/body ratio 3, % | 2.04 ± 0.33 | 2.05 ± 0.13 | 2.03 ± 0.27 | 2.15 ± 0.17 |
| Organ/body ratio 4, % | 9.11 ± 0.56 | 9.08 ± 0.32 | 9.42 ± 0.50 | 9.36 ± 0.38 |
| Abdominal fat rate 5, % | 2.49 ± 0.19 a | 2.54 ± 0.07 a | 2.82 ± 0.19 ab | 3.01 ± 0.21 b |
| Body weight gain 6, g | 828.9 ± 11.0 a | 792.8 ± 10.3 a | 788.9 ± 7.9 b | 819.1 ± 13.2 b |
| Specific growth rate 7, %/day | 4.05 ± 0.02 a | 4.05 ± 0.02 b | 3.97 ± 0.02 a | 4.03 ± 0.02 a |
| Average food intake, g/day | 0.90 ± 0.01 | 0.90 ± 0.02 | 0.91 ± 0.03 | 0.92 ± 0.01 |
| Feed efficiency rate 8 | 0.99 ± 0.03 a | 1.01 ± 0.02 a | 1.05 ± 0.02 b | 1.00 ± 0.01 a |
| Protein efficiency ratio 9 | 2.32 ± 0.10 a | 2.20 ± 0.03 a | 2.13 ± 0.06 b | 2.23 ± 0.03 ab |
| Raw Reads | Valid Reads | Valid% | Q20% | Q30% | GC% | |
|---|---|---|---|---|---|---|
| QT1 | 41,263,680 | 40,606,568 | 98.41 | 98.23 | 94.26 | 49.51 |
| 48,528,916 | 47,838,206 | 98.58 | 98.34 | 94.5 | 49.79 | |
| 43,071,948 | 42,371,898 | 98.37 | 98.19 | 94.17 | 49.41 | |
| 46,583,924 | 45,965,530 | 98.67 | 98.29 | 94.38 | 48.87 | |
| QT2 | 40,637,870 | 39,709,516 | 97.72 | 98.21 | 94.06 | 48.44 |
| 44,131,564 | 43,591,280 | 98.78 | 98.61 | 95.11 | 49.1 | |
| 44,273,842 | 43,705,154 | 98.72 | 98.6 | 95.13 | 49.16 | |
| 38,160,526 | 37,616,558 | 98.57 | 98.6 | 95.12 | 49.07 | |
| YL1 | 50,196,110 | 49,594,184 | 98.8 | 98.55 | 94.96 | 49.5 |
| 48,186,328 | 47,599,102 | 98.78 | 98.56 | 95.01 | 49.3 | |
| 40,125,834 | 38,890,244 | 96.92 | 98.25 | 94.19 | 49.68 | |
| 48,112,390 | 47,515,170 | 98.76 | 98.6 | 95.13 | 49.68 | |
| YL3 | 38,414,580 | 37,946,352 | 98.78 | 98.62 | 95.18 | 49.69 |
| 36,765,132 | 36,245,694 | 98.59 | 98.5 | 94.82 | 49.02 | |
| 45,187,452 | 44,623,852 | 98.75 | 98.55 | 94.99 | 49.43 | |
| 49,797,460 | 49,214,324 | 98.83 | 98.6 | 95.1 | 49.75 |
| Index | All | GC% | Median Length | Total Assembled Bases | N50 |
|---|---|---|---|---|---|
| Transcript | 89,056 | 45.45 | 901 | 135,827,643 | 2854 |
| Unigene | 42,529 | 44.99 | 378 | 48,407,951 | 2675 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, F.; Zhang, X.; Yao, G.; Chen, X.; Qi, M.; Zhou, Q.; Zhu, N.; Meng, Q.; Zhang, Y.; Ding, X. Transcriptomic Comparison of Liver Tissue across Different Largemouth Bass (Micropterus salmoides) Strains. Fishes 2023, 8, 558. https://doi.org/10.3390/fishes8110558
Zhou F, Zhang X, Yao G, Chen X, Qi M, Zhou Q, Zhu N, Meng Q, Zhang Y, Ding X. Transcriptomic Comparison of Liver Tissue across Different Largemouth Bass (Micropterus salmoides) Strains. Fishes. 2023; 8(11):558. https://doi.org/10.3390/fishes8110558
Chicago/Turabian StyleZhou, Fan, Xuelin Zhang, Gaohua Yao, Xiaoming Chen, Ming Qi, Qin Zhou, Ningyu Zhu, Qinghui Meng, Yu Zhang, and Xueyan Ding. 2023. "Transcriptomic Comparison of Liver Tissue across Different Largemouth Bass (Micropterus salmoides) Strains" Fishes 8, no. 11: 558. https://doi.org/10.3390/fishes8110558
APA StyleZhou, F., Zhang, X., Yao, G., Chen, X., Qi, M., Zhou, Q., Zhu, N., Meng, Q., Zhang, Y., & Ding, X. (2023). Transcriptomic Comparison of Liver Tissue across Different Largemouth Bass (Micropterus salmoides) Strains. Fishes, 8(11), 558. https://doi.org/10.3390/fishes8110558