
Non-Enzymatic and Enzymatic Antioxidant Responses of Hypericum perforatum L. Hairy Roots upon Photooxidative Stress


Abstract
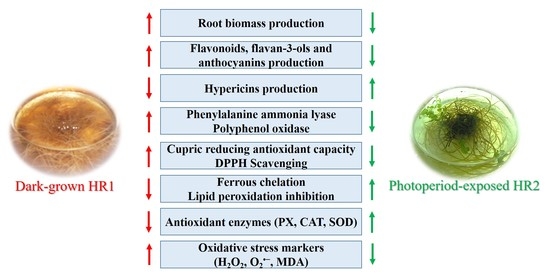
1. Introduction
- (1)
- fresh weight, dry weight, fresh weight/dry weight ratio and dry weight yield;
- (2)
- total phenolic, flavonoid, flavan-3-ol, anthocyanin and hypericin contents;
- (3)
- phenylalanine ammonia lyase and polyphenol oxidase activities;
- (4)
- cupric ion reducing antioxidant capacity, DPPH radical scavenging activity, ferrous chelating activity and lipid peroxidation inhibition;
- (5)
- guaiacol peroxidase, catalase and superoxide dismutase activities;
- (6)
- hydrogen peroxide, superoxide radical and malondialdehyde contents.
2. Materials and Methods
2.1. Establishment of Dark-Grown and Photoperiod-Exposed HR Clones
2.2. Growth Characteristics
2.3. Phenolic Compound Contents
2.4. Phenylalanine Ammonia Lyase (PAL) and Polyphenol Oxydase (PPO) Activities
2.5. Non-Enzymatic Antioxidant Capacity Assays
2.6. Antioxidant Enzyme Activities
2.7. Oxidative Stress Marker Contents
2.8. Statistical Analysis
3. Results
3.1. Growth Characteristics of Dark-Grown and Photoperiod-Exposed Hairy Roots
3.2. Phenolic Compound Production in Dark-Grown and Photoperiod-Exposed Hairy Roots
3.3. Non-Enzymatic Antioxidant Activities of Dark-Grown and Photoperiod-Exposed Hairy Roots
3.4. Antioxidant Enzyme Activities of Dark-Grown and Photoperiod-Exposed Hairy Roots
3.5. Oxidative Stress Markers of Dark-Grown and Photoperiod-Exposed Hairy Roots
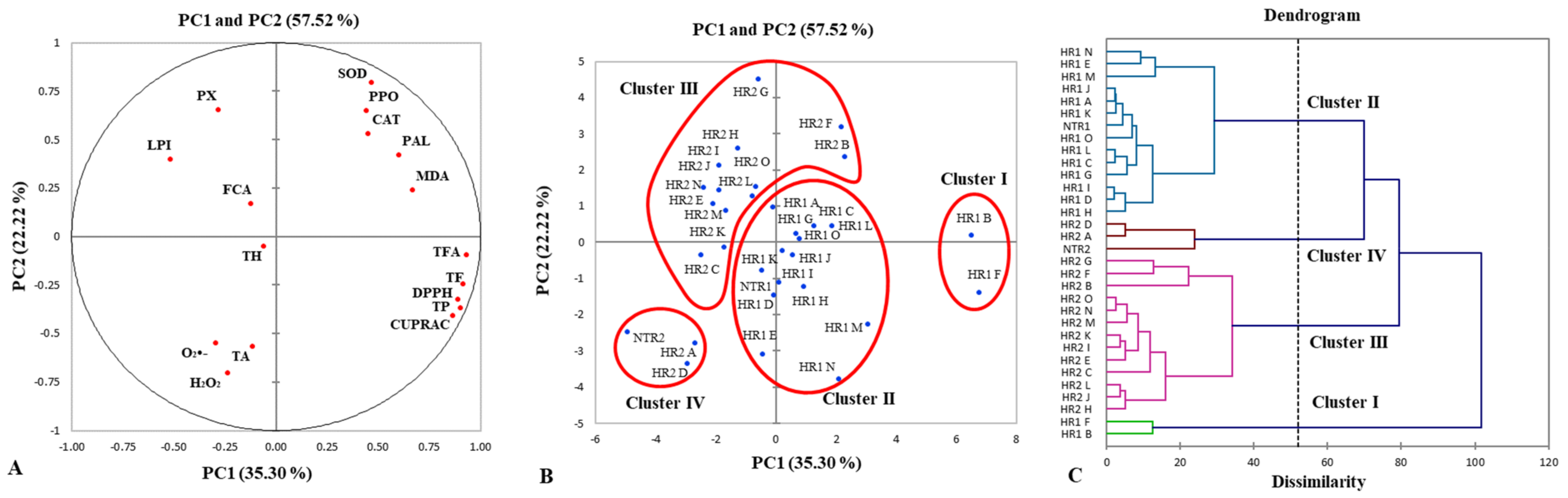
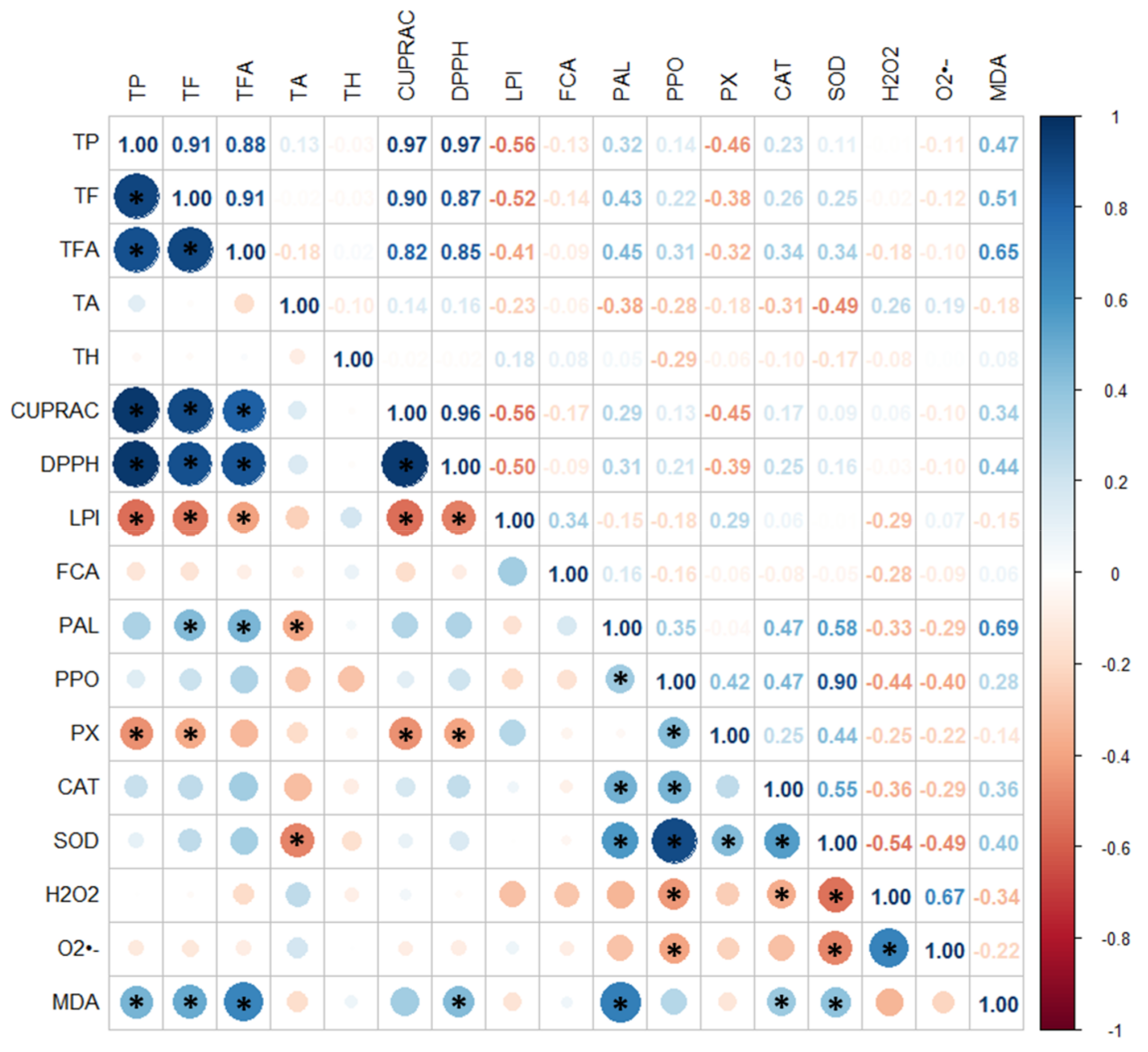
3.6. Principal Component Analysis and Hierarchical Agglomerative Clustering
4. Discussion
4.1. Growth Characteristics of Dark-Grown and Photoperiod-Exposed Hairy Roots
4.2. Phenolic Compound Composition of Dark-Grown and Photoperiod-Exposed Hairy Roots
4.3. Non-Enzymatic Antioxidant Activities in Dark-Grown and Photoperiod-Exposed Hairy Roots
4.4. Antioxidant Enzymes and Oxidative Stress Markers in Dark-Grown and Photoperiod-Exposed Hairy Roots
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Velingkar, V.S.; Gupta, G.L.; Hegde, N.B. A current update on phytochemistry, pharmacology and herb–drug interactions of Hypericum perforatum. Phytochem. Rev. 2017, 16, 725–744. [Google Scholar] [CrossRef]
- Murch, S.J.; Saxena, P.K. St. John’s wort (Hypericum perforatum L.): Challenges and strategies for production of chemically-consistent plants. Can. J. Plant Sci. 2006, 86, 765–771. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Chou, Y.-C.; Liao, K.-C.; Shieh, C.-J.; Deng, T.-S. Optimization of Light Intensity, Temperature, and Nutrients to Enhance the Bioactive Content of Hyperforin and Rutin in St. John’s Wort. Molecules 2020, 25, 4256. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.Y.; Hamid, S.; Kamili, A.N.; Hassan, Q.P. Sneak peek of Hypericum perforatum L.: Phytochemistry, phytochemical efficacy and biotechnological interventions. J. Plant Biochem. Biotechnol. 2019, 28, 357–373. [Google Scholar] [CrossRef]
- Gadzovska, S.; Maury, S.; Ounnar, S.; Righezza, M.; Kascakova, S.; Refregiers, M.; Spasenoski, M.; Joseph, C.; Hagège, D. Identification and quantification of hypericin and pseudohypericin in different Hypericum perforatum L. in vitro cultures. Plant Physiol. Biochem. 2005, 43, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Gadzovska, S.; Maury, S.; Delaunay, A.; Spasenoski, M.; Hagège, D.; Courtois, D.; Joseph, C. The influence of salicylic acid elicitation of shoots, callus, and cell suspension cultures on production of naphtodianthrones and phenylpropanoids in Hypericum perforatum L. Plant Cell Tissue Organ Cult. 2012, 113, 25–39. [Google Scholar] [CrossRef]
- Liu, X.-N.; Zhang, X.-Q.; Sun, J.-S. Effects of cytokinins and elicitors on the production of hypericins and hyperforin metabolites in Hypericum sampsonii and Hypericum perforatum. Plant Growth Regul. 2007, 53, 207–214. [Google Scholar] [CrossRef]
- Kwiecień, I.; Smolin, J.; Beerhues, L.; Ekiert, H. The impact of media composition on production of flavonoids in agitated shoot cultures of the three Hypericum perforatum L. cultivars ‘Elixir,’ ‘Helos,’ and ‘Topas’. Vitr. Cell Dev. Biol. Plant 2018, 54, 332–340. [Google Scholar] [CrossRef]
- Tusevski, O.; Stanoeva, J.P.; Markoska, E.; Brndevska, N.; Stefova, M.; Simic, S.G. Callus cultures of Hypericum perforatum L. a novel and efficient source for xanthone production. Plant Cell Tissue Organ Cult. 2016, 125, 309–319. [Google Scholar] [CrossRef]
- Cui, X.-H.; Chakrabarty, D.; Lee, E.-J.; Paek, K.-Y. Production of adventitious roots and secondary metabolites by Hypericum perforatum L. in a bioreactor. Bioresour. Technol. 2010, 101, 4708–4716. [Google Scholar] [CrossRef]
- Tocci, N.; Simonetti, G.; D’Auria, F.D.; Panella, S.; Palamara, A.T.; Valletta, A.; Pasqua, G. Root cultures of Hypericum perforatum subsp. angustifolium elicited with chitosan and production of xanthone-rich extracts with antifungal activity. Appl. Microbiol. Biotechnol. 2011, 91, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Brasili, E.; Miccheli, A.; Marini, F.; Praticò, G.; Sciubba, F.; Di Cocco, M.E.; Cechinel, V.F.; Tocci, N.; Valletta, A.; Pasqua, G. Metabolic Profile and Root Development of Hypericum perforatum L. In vitro Roots under Stress Conditions Due to Chitosan Treatment and Culture Time. Front. Plant Sci. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, A.S.; Khanahmadi, M.; Ebrahimi, M.; Moradi, K.; Behroozi, P.; Noormohammadi, N. Effect of different quality of light on growth and production of secondary metabolites in adventitious root cultivation of Hypericum perforatum. Plant Signal. Behav. 2019, 14, 1640561. [Google Scholar] [CrossRef]
- Wu, S.-Q.; Yu, X.-K.; Lian, M.-L.; Park, S.-Y.; Piao, X.-C. Several factors affecting hypericin production of Hypericum perforatum during adventitious root culture in airlift bioreactors. Acta Physiol. Plant. 2014, 36, 975–981. [Google Scholar] [CrossRef]
- Sobhani, A.; Khanahmadi, M.; Jalali, A.; Moradi, K.; Noormohammadi, N.; Ebrahimi, M. Development of a low-cost disposable bioreactor for pilot scale production of Hypericum perforatum L. adventitious roots. Ind. Crops Prod. 2020, 160, 113096. [Google Scholar] [CrossRef]
- Gantait, S.; Mukherjee, E. Hairy root culture technology: Applications, constraints and prospect. Appl. Microbiol. Biotechnol. 2020, 105, 35–53. [Google Scholar] [CrossRef]
- Vinterhalter, B.; Ninkovic, S.; Cingel, A.; Vinterhalter, D. Shoot and root culture of Hypericum perforatum L. transformed with Agrobacterium rhizogenes A4M70GUS. Biol. Plant. 2006, 50, 767–770. [Google Scholar] [CrossRef]
- Komarovská, H.; Giovannini, A.; Košuth, J.; Čellárová, E. Agrobacterium rhizogenes-Mediated Transformation of Hypericum tomentosum L. and Hypericum tetrapterum Fries. Z. Nat. C 2009, 64, 864–868. [Google Scholar] [CrossRef]
- Tusevski, O.; Stanoeva, J.; Stefova, M.; Kungulovski, D.; Pancevska, N.; Sekulovski, N.; Panov, S.; Simic, S. Hairy roots of Hypericum perforatum L.: A promising system for xanthone production. Open Life Sci. 2013, 8, 1010–1022. [Google Scholar] [CrossRef]
- Zubrická, D.; Mišianiková, A.; Henzelyová, J.; Valletta, A.; De Angelis, G.; D’Auria, F.D.; Simonetti, G.; Pasqua, G.; Čellárová, E. Xanthones from roots, hairy roots and cell suspension cultures of selected Hypericum species and their antifungal activity against Candida albicans. Plant Cell Rep. 2015, 34, 1953–1962. [Google Scholar] [CrossRef]
- Koperdáková, J.; Komarovská, H.; Košuth, J.; Giovannini, A.; Čellárová, E. Characterization of hairy root-phenotype in transgenic Hypericum perforatum L. clones. Acta Physiol. Plant. 2008, 31, 351–358. [Google Scholar] [CrossRef]
- Komarovská, H.; Košuth, J.; Giovannini, A.; Smelcerovic, A.; Zuehlke, S.; Čellárová, E. Effect of the Number of rol Genes Integrations on Phenotypic Variation in Hairy Root-Derived Hypericum perforatum L. Plants. Z. Nat. C 2010, 65, 701–712. [Google Scholar] [CrossRef]
- Vinterhalter, B.; Zdravković-Korać, S.; Mitić, N.; Bohanec, B.; Vinterhalter, D.; Savić, J. Effect of sucrose on shoot regeneration in Agrobacterium transformed Hypericum perforatum L. roots. Acta Physiol. Plant. 2015, 37, 1–12. [Google Scholar] [CrossRef]
- Tusevski, O.; Vinterhalter, B.; Milošević, D.K.; Soković, M.; Ćirić, A.; Vinterhalter, D.; Korać, S.Z.; Stanoeva, J.P.; Stefova, M.; Simic, S.G. Production of phenolic compounds, antioxidant and antimicrobial activities in hairy root and shoot cultures of Hypericum perforatum L. Plant Cell Tissue Organ Cult. 2016, 128, 589–605. [Google Scholar] [CrossRef]
- Tusevski, O.; Stanoeva, J.P.; Stefova, M.; Spasenoski, M.; Simic, S.G. State of antioxidant systems and phenolic compounds’ production in Hypericum perforatum L. hairy roots. Acta Physiol. Plant. 2019, 41, 132. [Google Scholar] [CrossRef]
- Tusevski, O.; Todorovska, M.; Stanoeva, J.P.; Stefova, M.; Simic, S.G. In Vitro and in Silico Insights on the Biological Activities, Phenolic Compounds Composition of Hypericum perforatum L. Hairy Root Cultures. Phyton 2022, 92, 921–941. [Google Scholar] [CrossRef]
- Sharma, P.; Padh, H.; Shrivastava, N. Hairy root cultures: A suitable biological system for studying secondary metabolic pathways in plants. Eng. Life Sci. 2012, 13, 62–75. [Google Scholar] [CrossRef]
- Tusevski, O.; Stanoeva, J.P.; Stefova, M.; Simic, S.G. Phenolic Profile of Dark-Grown and Photoperiod-Exposed Hypericum perforatum L. Hairy Root Cultures. Sci. World J. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Mukherjee, C.; Sircar, D.; Chatterjee, M.; Das, S.; Mitra, A. Combating photooxidative stress in green hairy roots of Daucus carota cultivated under light irradiation. J. Plant Physiol. 2014, 171, 179–187. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Zhishen, J.; Mengcheng, T.; Jianming, W. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem. 1999, 64, 555–559. [Google Scholar] [CrossRef]
- Arnous, A.; Makris, D.P.; Kefalas, P. Correlation of Pigment and Flavanol Content with Antioxidant Properties in Selected Aged Regional Wines from Greece. J. Food Compos. Anal. 2002, 15, 655–665. [Google Scholar] [CrossRef]
- Meyers, K.J.; Watkins, C.B.; Pritts, M.P.; Liu, R.H. Antioxidant and Antiproliferative Activities of Strawberries. J. Agric. Food Chem. 2003, 51, 6887–6892. [Google Scholar] [CrossRef] [PubMed]
- Das, J.R.; Bhat, S.G.; Gowda, L.R. Purification and Characterization of a Polyphenol Oxidase from the Kew Cultivar of Indian Pineapple Fruit. J. Agric. Food Chem. 1997, 45, 2031–2035. [Google Scholar] [CrossRef]
- Apak, R.; Güçlü, K.; Özyürek, M.; Karademir, S.E. Novel Total Antioxidant Capacity Index for Dietary Polyphenols and Vitamins C and E, Using Their Cupric Ion Reducing Capability in the Presence of Neocuproine: CUPRAC Method. J. Agric. Food Chem. 2004, 52, 7970–7981. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Decker, E.A.; Welch, B. Role of ferritin as a lipid oxidation catalyst in muscle food. J. Agric. Food Chem. 1990, 38, 674–677. [Google Scholar] [CrossRef]
- Sun, T. Antioxidant activities of buckwheat extracts. Food Chem. 2005, 90, 743–749. [Google Scholar] [CrossRef]
- Hemeda, H.M.; Klein, B.P. Effects of Naturally Occurring Antioxidants on Peroxidase Activity of Vegetable Extracts. J. Food Sci. 1990, 55, 184–185. [Google Scholar] [CrossRef]
- Aebi, H. [13] Catalase in vitro. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1984; Volume 105, pp. 121–126. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Sergiev, I.; Alexieva, V.; Karanov, E. Effect of spermine, atrazine and combination between them on some endogenous protective systems and stress markers in plants. Comptes Rendus Acad. Bulg. Sci. 1997, 51, 121–124. [Google Scholar] [CrossRef]
- Elstner, E.F.; Heupel, A. Inhibition of nitrite formation from hydroxylammoniumchloride: A simple assay for superoxide dismutase. Anal. Biochem. 1976, 70, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts: I. kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys. 1968, 125, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, M.S.A.; Lockwood, G.B. Volatile oils from the plant and hairy root cultures of Ageratum conyzoides L. Nat. Prod. Res. 2011, 25, 909–917. [Google Scholar] [CrossRef]
- Kino-oka, M.; Nagatome, H.; Taya, M. Characterization and application of plant hairy roots endowed with photosynthetic functions. In Plant Cells; Zhong, J.-J., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 72, pp. 183–218. [Google Scholar] [CrossRef]
- Taya, M.; Sato, H.; Kino-Oka, M.; Tone, S. Characterization of pak-bung green hairy roots cultivated under light irradiation. J. Ferment. Bioeng. 1994, 78, 42–48. [Google Scholar] [CrossRef]
- Lanoue, A.; Boitel-Conti, M.; Dechaux, C.; Laberche, J.C.; Christen, P.; Sangwan-Norreel, B. Comparison of growth proper-ties, alkaloid production and water uptake of two selected Datura hairy root lines. Acta Biol. Crac. Ser. Bot. 2004, 46, 185–192. [Google Scholar]
- Sidwa-Gorycka, M.; Krolicka, A.; Orlita, A.; Maliński, E.; Golebiowski, M.; Kumirska, J.; Chromik, A.; Biskup, E.; Stepnowski, P.; Lojkowska, E. Genetic transformation of Ruta graveolens L. by Agrobacterium rhizogenes: Hairy root cultures a promising approach for production of coumarins and furanocoumarins. Plant Cell Tissue Organ Cult. 2009, 97, 59–69. [Google Scholar] [CrossRef]
- Abbasi, B.H.; Tian, C.-L.; Murch, S.J.; Saxena, P.K.; Liu, C.-Z. Light-enhanced caffeic acid derivatives biosynthesis in hairy root cultures of Echinacea purpurea. Plant Cell Rep. 2007, 26, 1367–1372. [Google Scholar] [CrossRef]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.-J.; Reddy, M.S.S.; Wang, L. The phenylpropanoid pathway and plant defence-a genomics perspective. Mol. Plant Pathol. 2002, 3, 371–390. [Google Scholar] [CrossRef]
- Murthy, H.N.; Kim, Y.-S.; Park, S.-Y.; Paek, K.-Y. Hypericins: Biotechnological production from cell and organ cultures. Appl. Microbiol. Biotechnol. 2014, 98, 9187–9198. [Google Scholar] [CrossRef]
- Qian, J.; Wu, J.; Yao, B.; Lu, Y. Preparation of a polyclonal antibody against hypericin synthase and localization of the enzyme in red-pigmented Hypericum perforatum L. plantlets. Acta Biochim. Pol. 2012, 59, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, L.; Routaboul, J.-M.; Cheynier, V.; Lepiniec, L.; Debeaujon, I. Flavonoid oxidation in plants: From biochemical properties to physiological functions. Trends Plant Sci. 2007, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, M.; D’Aprano, G.; Lacroix, M. Effect of dose rate of gamma irradiation on biochemical quality and browning of mushrooms Agaricus bisporus. Radiat. Phys. Chem. 2002, 63, 311–315. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Nagatome, H.; Tsutsumi, M.; Kino-Oka, M.; Taya, M. Development and characterization of a photoautotrophic cell line of pak-bung hairy roots. J. Biosci. Bioeng. 2000, 89, 151–156. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FW (g) | DW (g) | FW/DW Ratio | DWY | |||||
|---|---|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 7.10 ± 0.71 a | 2.35 ± 0.35 a; * | 0.56 ± 0.00 ab | 0.21 ± 0.02 a; * | 12.72 ± 0.54 bc | 11.21 ± 0.87 ab | 8.26 ± 0.38 c–f | 8.17 ± 0.22 bh |
| A | 13.49 ± 0.02 l | 7.83 ± 0.24 f; * | 1.03 ± 0.01 e–i | 0.61 ± 0.04 c; * | 13.10 ± 1.02 c | 12.91 ± 0.72 b | 7.64 ± 0.09 ae | 8.13 ± 0.70 bf |
| B | 18.42 ± 2.09 m | 16.27 ± 0.24 j | 1.58 ± 0.12 j | 1.38 ± 0.09 h | 11.69 ± 0.67 a–c | 11.81 ± 1.01 b | 9.26 ± 0.32 ef | 8.47 ± 0.68 bk |
| C | 12.97 ± 0.80 kl | 7.54 ± 0.48 d–f; * | 1.04 ± 0.11 e–i | 0.64 ± 0.06 c; * | 12.44 ± 0.45 a–c | 11.76 ± 0.57 b | 8.02 ± 0.73 b–f | 8.52 ± 0.18 bl |
| D | 9.99 ± 0.64 d–h | 5.21 ± 0.41 b; * | 0.85 ± 0.06 cdg | 0.35 ± 0.03 b; * | 11.79 ± 0.81 a–c | 14.72 ± 0.60 c; * | 8.47 ± 0.44 d–f | 6.79 ± 0.24 a; * |
| E | 7.71 ± 0.54 ab | 6.45 ± 0.65 c | 0.60 ± 0.02 a | 0.65 ± 0.07 c | 12.92 ± 0.95 bc | 9.96 ± 0.59 a; * | 8.03 ± 0.65 ac | 9.10 ± 0.82 e–m |
| F | 25.66 ± 1.15 n | 11.09 ± 1.00 i; * | 1.55 ± 0.14 j | 0.90 ± 0.04 fg; * | 16.55 ± 0.40 e | 12.28 ± 1.10 b; * | 5.69 ± 0.33 ab | 8.16 ± 0.47 bg; * |
| G | 8.92 ± 0.49 bce | 6.85 ± 0.78 c; * | 0.57 ± 0.01 ac | 0.58 ± 0.02 c | 15.77 ± 1.10 de | 11.88 ± 0.73 b; * | 6.37 ± 0.42 ad | 9.00 ± 0.30 c–m; * |
| H | 10.68 ± 0.27 gj | 6.89 ± 0.55 cd; * | 0.86 ± 0.05 dh | 0.61 ± 0.04 c; * | 12.48 ± 1.31 a–c | 11.39 ± 0.48 b | 8.00 ± 0.30 b–f | 8.80 ± 0.35 c–m |
| I | 10.38 ± 0.32 e–gi | 6.93 ± 0.18 ce; * | 0.82 ± 0.04 cdf | 0.56 ± 0.05 c; * | 12.71 ± 1.03 bc | 12.43 ± 0.91 b | 7.87 ± 0.48 af | 8.09 ± 0.79 be |
| J | 10.94 ± 0.17 h–j | 9.05 ± 0.92 g; * | 0.91 ± 0.03 di | 0.83 ± 0.04 de | 12.00 ± 1.44 a–c | 10.86 ± 0.68 ab | 8.22 ± 0.07 c–f | 8.29 ± 0.79 bj |
| K | 9.13 ± 0.22 bcf | 6.35 ± 0.49 c; * | 0.76 ± 0.03 b–d | 0.54 ± 0.05 c; * | 12.08 ± 1.12 a–c | 11.68 ± 1.11 ab | 8.28 ± 0.35 c–f | 8.58 ± 0.71 bm |
| L | 9.33 ± 0.44 cg | 10.76 ± 0.22 i; * | 0.87 ± 0.02 c–e | 0.87 ± 0.03 eg | 10.78 ± 0.86 ab | 12.42 ± 1.20 b | 8.69 ± 0.73 d–f | 8.02 ± 018 bc |
| M | 11.80 ± 1.09 i–k | 9.71 ± 0.01 h; * | 1.10 ± 0.05 e–i | 0.78 ± 0.03 e; * | 10.71 ± 0.44 a | 12.44 ± 0.92 b; * | 8.76 ± 0.28 d–f | 8.04 ± 0.34 bd |
| N | 8.10 ± 1.27 ac | 8.55 ± 0.21 g | 0.54 ± 0.00 a | 0.66 ± 0.03 cd; * | 14.87 ± 0.39 d | 12.96 ± 1.32 bc | 6.19 ± 0.48 a | 7.71 ± 0.30 b; * |
| O | 8.61 ± 0.34 b–d | 10.05 ± 0.64 h; * | 0.77 ± 0.04 ad | 0.82 ± 0.05 ef | 11.20 ± 0.72 a–c | 12.22 ± 1.31 b | 8.94 ± 0.89 c–f | 8.19 ± 0.05 bi |
| TP (mg GA·g−1 DW) | PAL (pkat·mg−1 P) | PPO (nkat·mg−1 P) | ||||
|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 21.27 ± 0.58 b–d | 12.86 ± 0.12 a; * | 1.14 ± 0.14 a | 0.97 ± 0.12 c | 0.14 ± 0.01 df | 0.07 ± 0.01 b; * |
| A | 20.80 ± 0.09 ab | 19.76 ± 0.21 h | 1.38 ± 0.35 a | 0.55 ± 0.02 a; * | 0.17 ± 0.01 h | 0.04 ± 0.00 a; * |
| B | 33.19 ± 0.37 k | 23.70 ± 0.61 i; * | 9.08 ± 0.08 h | 9.25 ± 0.06 h | 0.15 ± 0.01 e–g | 0.14 ± 0.01 e–i |
| C | 21.68 ± 0.36 ce | 17.57 ± 0.09 e; * | 4.55 ± 0.54 f | 0.98 ± 0.46 c; * | 0.19 ± 0.02 hi | 0.06 ± 0.00 b; * |
| D | 20.42 ± 0.14 a | 19.74 ± 0.55 h | 3.88 ± 0.36 de | 0.59 ± 0.03 a; * | 0.12 ± 0.00 c | 0.03 ± 0.00 a; * |
| E | 24.06 ± 0.33 g | 14.84 ± 0.30 b; * | 1.48 ± 0.40 a | 1.73 ± 0.50 cd | 0.06 ± 0.01 a | 0.14 ± 0.00 dh; * |
| F | 34.82 ± 0.09 l | 24.95 ± 0.28 j; * | 5.49 ± 0.30 g | 5.94 ± 0.52 g | 0.14 ± 0.01 dg | 0.22 ± 0.03 l; * |
| G | 20.99 ± 0.36 ac | 16.82 ± 0.24 d; * | 3.69 ± 0.24 ce | 1.83 ± 0.01 d; * | 0.17 ± 0.02 h | 0.16 ± 0.03 j |
| H | 25.38 ± 0.18 h | 16.41 ± 0.39 d; * | 3.49 ± 0.32 cd | 4.31 ± 0.50 f | 0.10 ± 0.01 b | 0.16 ± 0.02 j; * |
| I | 21.83 ± 0.03 de | 15.14 ± 0.55 bc; * | 3.30 ± 0.05 c | 4.12 ± 0.04 f; * | 0.12 ± 0.00 c | 0.13 ± 0.00 de |
| J | 22.32 ± 0.06 ef | 15.70 ± 0.95 c; * | 1.58 ± 0.15 a | 1.67 ± 0.43 cd | 0.14 ± 0.01 de | 0.14 ± 0.02 di |
| K | 23.01 ± 0.54 f | 17.59 ± 0.84 e; * | 2.36 ± 0.13 b | 1.59 ± 0.54 cd | 0.19 ± 0.03 hj | 0.09 ± 0.00 c; * |
| L | 24.88 ± 0.82 h | 18.97 ± 0.18 g; * | 3.87 ± 0.08 de | 3.12 ± 0.58 e | 0.21 ± 0.01 k | 0.13 ± 0.00 df; * |
| M | 31.10 ± 0.30 i | 18.52 ± 0.09 fg; * | 1.12 ± 0.33 a | 1.62 ± 0.41 cd | 0.13 ± 0.01 cd | 0.12 ± 0.00 d |
| N | 32.42 ± 1.29 j | 16.64 ± 0.49 d; * | 1.09 ± 0.00 a | 0.71 ± 0.04 b; * | 0.09 ± 0.00 b | 0.18 ± 0.01 k; * |
| O | 21.89 ± 0.43 de | 17.98 ± 0.30 ef; * | 2.80 ± 0.57 b | 2.89 ± 0.12 e | 0.19 ± 0.00 ij | 0.13 ± 0.01 dg; * |
| TF (mg C·g−1 DW) | TFA (mg C·g−1 DW) | TA (mg CG·g−1 DW | TH (μg H·g−1 DW) | |||||
|---|---|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 7.19 ± 0.26 bg | 3.45 ± 0.11 a; * | 2.10 ± 0.13 c | 0.72 ± 0.02 a; * | 1.17 ± 0.01 e–g | 0.83 ± 0.01 h; * | 19.54 ± 0.25 c | 62.40 ± 1.01 k; * |
| A | 7.15 ± 0.31 bf | 5.47 ± 0.10 e; * | 1.84 ± 0.01 b | 0.93 ± 0.02 b; * | 1.09 ± 0.05 de | 1.99 ± 0.00 j; * | 52.00 ± 0.51 j | 26.36 ± 0.76 d; * |
| B | 11.35 ± 0.67 k | 7.04 ± 0.46 g; * | 5.32 ± 0.00 j | 2.66 ± 0.04 k; * | 0.78 ± 0.01 b | 0.59 ± 0.07 be; * | 16.32 ± 0.25 a | 34.25 ± 0.25 f; * |
| C | 7.64 ± 0.95 c–h | 5.11 ± 0.21 e; * | 2.14 ± 0.10 c | 1.42 ± 0.03 h; * | 0.96 ± 0.09 c | 0.56 ± 0.03 bc; * | 26.54 ± 0.51 e | 58.46 ± 1.01 j; * |
| D | 7.12 ± 0.57 be | 5.22 ± 0.05 e; * | 1.63 ± 0.02 a | 1.09 ± 0.12 cd; * | 1.40 ± 0.01 h | 1.24 ± 0.04 i; * | 43.75 ± 1.52 h | 42.14 ± 0.76 h |
| E | 6.82 ± 0.15 b | 4.67 ± 0.33 d; * | 1.88 ± 0.03 b | 1.06 ± 0.05 c; * | 0.98 ± 0.17 c | 0.77 ± 0.02 g | 46.98 ± 0.51 i | 19.72 ± 0.51 b; * |
| F | 15.15 ± 0.36 l | 7.86 ± 0.47 h; * | 6.73 ± 0.26 k | 3.83 ± 0.09 l; * | 0.57 ± 0.01 a | 0.58 ± 0.02 bd | 19.54 ± 0.25 c | 44.47 ± 1.01 i; * |
| G | 7.32 ± 0.04 bh | 5.36 ± 0.15 e; * | 1.85 ± 0.04 b | 1.29 ± 0.02 g; * | 0.75 ± 0.08 b | 0.70 ± 0.06 f | 26.18 ± 0.51 e | 62.58 ± 1.78 k; * |
| H | 8.52 ± 0.29 i | 5.33 ± 0.10 e; * | 2.82 ± 0.08 h | 1.37 ± 0.02 h; * | 1.11 ± 0.07 dg | 0.55 ± 0.03 b; * | 29.41 ± 0.51 f | 24.39 ± 0.51 c; * |
| I | 6.17 ± 0.36 a | 4.11 ± 0.22 bc; * | 2.19 ± 0.08 cd | 1.16 ± 0.03 d–f; * | 1.61 ± 0.05 j | 0.55 ± 0.04 b; * | 19.37 ± 0.51 bc | 58.99 ± 1.27 j; * |
| J | 5.88 ± 0.26 a | 3.80 ± 0.10 ab; * | 2.12 ± 0.05 c | 1.13 ± 0.03 ce; * | 0.97 ± 0.15 c | 0.49 ± 0.04 a; * | 18.29 ± 0.51 b | 38.55 ± 0.76 g; * |
| K | 7.08 ± 0.31 bd | 6.02 ± 0.67 f | 2.48 ± 0.08 f | 1.35 ± 0.01 gh; * | 1.11 ± 0.01 df | 0.82 ± 0.03 gh; * | 21.52 ± 0.51 d | 73.34 ± 2.28 l; * |
| L | 7.63 ± 0.15 c–h | 5.16 ± 0.08 e; * | 2.63 ± 0.01 g | 1.94 ± 0.05 j; * | 1.05 ± 0.00 cd | 0.62 ± 0.05 c–e; * | 37.83 ± 0.76 g | 10.22 ± 0.25 a; * |
| M | 8.28 ± 0.57 i | 5.33 ± 0.10 e; * | 4.53 ± 0.16 i | 1.37 ± 0.00 h; * | 1.16 ± 0.16 e–g | 1.24 ± 0.05 i | 26.90 ± 1.01 e | 27.26 ± 0.51 d |
| N | 9.27 ± 0.10 j | 4.27 ± 0.15 c; * | 2.28 ± 0.07 de | 1.14 ± 0.00 cf; * | 1.50 ± 0.06 i | 1.23 ± 0.04 i; * | 55.77 ± 1.27 k | 31.02 ± 0.25 e; * |
| O | 7.01 ± 0.62 bc | 5.38 ± 0.08 e; * | 2.37 ± 0.07 ef | 1.54 ± 0.01 i; * | 1.17 ± 0.07 e–g | 1.27 ± 0.03 i | 37.48 ± 1.27 g | 20.08 ± 0.51 b; * |
| CUPRAC (μM T·g−1 DW) | DPPH (μM T·g−1 DW) | FCA (mg EDTA·g−1 DW) | LPI (%) | |||||
|---|---|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 109.58 ± 0.36 b | 69.75 ± 1.97 a; * | 54.37 ± 0.60 b | 29.48 ± 2.33 a; * | 3.03 ± 0.21 c | 4.65 ± 0.22 f; * | 38.90 ± 3.15 egh | 84.23 ± 3.43 fh; * |
| A | 101.71 ± 2.30 a | 107.27 ± 1.94 j | 47.75 ± 2.82 a | 50.28 ± 2.79 i | 4.21 ± 0.05 e | 5.67 ± 0.07 h; * | 47.21 ± 2.28 i | 81.23 ± 5.21 e; * |
| B | 171.77 ± 8.90 j | 127.54 ± 0.36 k; * | 93.03 ± 0.50 j | 60.91 ± 3.68 j; * | 4.64 ± 0.06 j | 6.56 ± 0.27 i; * | 27.49 ± 1.70 b | 80.31 ± 2.10 e; * |
| C | 130.28 ± 0.12 df | 86.91 ± 2.90 d; * | 58.38 ± 0.10 c | 47.75 ± 0.40 g–i; * | 4.35 ± 0.00 eg | 6.61 ± 0.18 i; * | 36.37 ± 1.01 dfg | 84.20 ± 1.33 fg; * |
| D | 118.31 ± 5.20 c | 105.22 ± 0.48 j; * | 54.72 ± 0.10 b | 45.35 ± 0.60 e–g; * | 4.26 ± 0.03 ef | 4.25 ± 0.26 de | 18.96 ± 1.96 a | 79.94 ± 1.98 e; * |
| E | 152.44 ± 0.24 h | 75.02 ± 1.57 b; * | 60.07 ± 1.29 ce | 36.20 ± 1.00 b; * | 3.10 ± 0.19 c | 3.41 ± 0.11 a | 32.81 ± 3.78 cd | 82.53 ± 3.17 ef; * |
| F | 212.06 ± 2.06 l | 136.07 ± 6.25 l; * | 97.18 ± 0.40 k | 68.92 ± 2.82 k; * | 4.37 ± 0.36 eh | 5.39 ± 0.28 g; * | 44.29 ± 2.02 hi | 84.39 ± 1.92 fi; * |
| G | 126.86 ± 3.27 d | 91.62 ± 0.36 eh; * | 52.49 ± 0.57 b | 41.83 ± 0.98 c; * | 2.64 ± 0.07 b | 3.80 ± 0.11 b; * | 35.89 ± 1.86 de | 90.94 ± 0.04 km; * |
| H | 135.96 ± 7.71 fg | 97.95 ± 2.78 i; * | 61.41 ± 0.80 d–f | 45.40 ± 1.20 d–fh; * | 4.42 ± 0.09 ej | 4.11 ± 0.11 cd | 66.17 ± 0.97 k | 88.32 ± 1.14 jk; * |
| I | 126.52 ± 0.36 d | 81.27 ± 0.95 c; * | 60.05 ± 2.96 cd | 38.73 ± 0.20 b; * | 5.31 ± 0.11 l | 4.48 ± 0.17 ef; * | 29.36 ± 0.34 bc | 74.84 ± 1.52 d; * |
| J | 119.16 ± 3.51 c | 88.96 ± 1.69 de; * | 53.24 ± 0.20 b | 38.45 ± 1.79 b; * | 3.64 ± 0.09 d | 5.27 ± 0.26 g; * | 26.77 ± 2.87 b | 85.84 ± 0.95 g–j; * |
| K | 129.51 ± 0.73 de | 94.61 ± 2.66 f–i; * | 60.21 ± 1.69 cf | 42.68 ± 1.59 cd; * | 4.39 ± 0.06 ei | 3.88 ± 0.18 bc; * | 37.77 ± 1.13 ef | 61.74 ± 2.55 b; * |
| L | 134.30 ± 1.45 e–g | 97.12 ± 0.94 i; * | 69.37 ± 1.29 g | 43.66 ± 1.39 ce; * | 3.58 ± 0.10 d | 5.39 ± 0.06 g; * | 28.10 ± 0.34 b | 41.30 ± 2.24 a; * |
| M | 165.78 ± 1.69 i | 91.45 ± 4.48 eg; * | 88.80 ± 4.08 i | 50.28 ± 1.60 i; * | 2.38 ± 0.02 a | 5.68 ± 0.25 h; * | 47.86 ± 1.09 i | 71.91 ± 0.69 c; * |
| N | 201.88 ± 1.94 k | 90.85 ± 1.69 ef; * | 96.90 ± 1.99 k | 44.30 ± 0.10 cf; * | 4.46 ± 0.09 f–j | 4.37 ± 0.04 e | 40.91 ± 2.22 fh | 89.53 ± 1.31 kl; * |
| O | 131.91 ± 2.90 dg | 103.62 ± 2.14 j; * | 73.80 ± 0.40 h | 49.91 ± 1.14 i; * | 4.95 ± 0.17 k | 3.85 ± 0.06 b; * | 54.56 ± 0.71 j | 91.44 ± 0.46 lm; * |
| PX (nkat·mg−1 P) | CAT (nkat·mg−1 P) | SOD (U·mg−1) | ||||
|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 1.24 ± 0.02 m | 0.78 ± 0.20 de; * | 1.24 ± 0.21 c | 1.36 ± 0.06 bc | 13.87 ± 0.76 deg | 6.89 ± 1.08 c; * |
| A | 0.94 ± 0.08 hik | 0.35 ± 0.01 b; * | 4.65 ± 0.18 h | 0.33 ± 0.04 a; * | 18.66 ± 1.60 h | 5.27 ± 0.19 b; * |
| B | 0.22 ± 0.06 b | 0.05 ± 0.01 a; * | 11.56 ± 0.03 k | 3.35 ± 0.00 ef; * | 18.52 ± 1.60 h | 22.10 ± 0.62 j; * |
| C | 0.88 ± 0.13 gil | 0.51 ± 0.02 bc; * | 3.19 ± 0.06 i | 1.70 ± 0.10 c; * | 20.90 ± 1.94 i | 8.39 ± 0.22 d; * |
| D | 0.49 ± 0.01 ef | 0.09 ± 0.00 a; * | 1.03 ± 0.03 a | 0.18 ± 0.01 a; * | 12.37 ± 0.36 ce | 2.55 ± 0.28 a; * |
| E | 0.58 ± 0.06 f | 0.84 ± 0.06 eh; * | 2.24 ± 0.09 e | 1.69 ± 0.12 c; * | 4.58 ± 0.46 a | 16.08 ± 0.00 g–i; * |
| F | 0.07 ± 0.04 a | 1.45 ± 0.01 j; * | 2.60 ± 0.04 g | 8.54 ± 0.80 k; * | 20.20 ± 1.70 hi | 25.63 ± 0.56 l; * |
| G | 0.36 ± 0.00 c–e | 2.39 ± 0.14 k; * | 5.55 ± 0.08 j | 7.50 ± 0.29 j; * | 21.28 ± 2.35 i | 24.30 ± 2.00 k |
| H | 0.56 ± 0.09 f | 1.33 ± 0.00 j; * | 1.06 ± 0.07 ab | 3.14 ± 0.02 e; * | 10.82 ± 0.43 c | 22.95 ± 1.83 j; * |
| I | 0.82 ± 0.08 gh | 1.06 ± 0.02 i; * | 1.21 ± 0.13 bc | 3.51 ± 0.17 f; * | 12.06 ± 0.60 cd | 15.60 ± 0.66 fi; * |
| J | 0.54 ± 0.00 f | 0.70 ± 0.07 ce; * | 2.24 ± 0.03 e | 1.27 ± 0.28 b; * | 15.66 ± 1.96 g | 15.82 ± 0.13 g–i |
| K | 0.76 ± 0.01 g | 0.59 ± 0.01 cd; * | 1.98 ± 0.20 d | 3.94 ± 0.07 g; * | 14.73 ± 1.70 fg | 10.53 ± 0.16 e; * |
| L | 0.93 ± 0.08 h–j | 1.01 ± 0.22 f–i | 2.62 ± 0.07 g | 2.43 ± 0.06 d | 23.45 ± 0.74 j | 14.80 ± 1.35 fg; * |
| M | 0.30 ± 0.11 bc | 1.28 ± 0.06 j; * | 2.51 ± 0.12 fg | 2.37 ± 0.06 d | 13.12 ± 0.25 d–f | 14.34 ± 0.26 f |
| N | 0.31 ± 0.05 bd | 0.83 ± 0.09 eg; * | 3.21 ± 0.30 i | 5.33 ± 0.63 h; * | 8.43 ± 0.04 b | 15.78 ± 1.04 g–i; * |
| O | 0.98 ± 0.11 j–l | 0.81 ± 0.13 ef | 2.38 ± 0.08 ef | 6.79 ± 0.40 i; * | 18.52 ± 0.36 h | 15.04 ± 0.69 fh; * |
| H2O2 (μM·g−1 FW) | O2•− (nM·min−1·g−1 FW) | MDA (nM·g−1 FW) | ||||
|---|---|---|---|---|---|---|
| HR1 | HR2 | HR1 | HR2 | HR1 | HR2 | |
| NTR | 0.51 ± 0.00 h | 0.79 ± 0.08 l; * | 0.24 ± 0.01 h | 0.84 ± 0.01 k; * | 0.66 ± 0.00 ce | 0.39 ± 0.05 a; * |
| A | 0.37 ± 0.05 b | 0.47 ± 0.04 j; | 0.14 ± 0.00 b | 0.26 ± 0.05 i; * | 0.63 ± 0.01 cd | 0.55 ± 0.05 be |
| B | 0.41 ± 0.02 c–g | 0.20 ± 0.00 a; * | 0.28 ± 0.01 j | 0.09 ± 0.00 a; * | 1.42 ± 0.01 k | 1.14 ± 0.06 l; * |
| C | 0.56 ± 0.03 i | 0.45 ± 0.01 ij; * | 0.21 ± 0.03 d | 0.20 ± 0.01 c–f | 0.94 ± 0.03 i | 0.58 ± 0.00 c–f; * |
| D | 0.49 ± 0.04 h | 0.54 ± 0.01 k | 0.22 ± 0.00 dg | 0.48 ± 0.05 j; * | 0.68 ± 0.02 d–f | 0.56 ± 0.04 c–e; * |
| E | 0.65 ± 0.05 j | 0.44 ± 0.03 i; * | 0.19 ± 0.00 c | 0.25 ± 0.00 g–i; * | 0.44 ± 0.01 b | 0.98 ± 0.00 k; * |
| F | 0.32 ± 0.04 a | 0.27 ± 0.01 b | 0.26 ± 0.00 i | 0.11 ± 0.01 a; * | 1.05 ± 0.05 j | 0.72 ± 0.00 i; * |
| G | 0.50 ± 0.01 h | 0.29 ± 0.03 bf; * | 0.22 ± 0.01 df | 0.11 ± 0.01 a; * | 0.43 ± 0.04 b | 0.85 ± 0.14 j; * |
| H | 0.43 ± 0.00 g | 0.31 ± 0.00 c–g; * | 0.21 ± 0.01 de | 0.23 ± 0.01 fg | 0.94 ± 0.09 i | 0.49 ± 0.01 b; * |
| I | 0.37 ± 0.01 bc | 0.30 ± 0.01 bg; * | 0.28 ± 0.00 j | 0.17 ± 0.01 bd; * | 0.81 ± 0.03 h | 0.60 ± 0.01 eh; * |
| J | 0.39 ± 0.01 bf | 0.32 ± 0.00 e–g; * | 0.12 ± 0.00 a | 0.15 ± 0.01 b | 0.72 ± 0.04 fg | 0.54 ± 0.01 bd; * |
| K | 0.38 ± 0.04 bd | 0.36 ± 0.04 h | 0.30 ± 0.01 k | 0.23 ± 0.01 fh; * | 0.61 ± 0.07 c | 0.50 ± 0.02 b |
| L | 0.43 ± 0.02 g | 0.27 ± 0.01 bc; * | 0.23 ± 0.01 e–g | 0.17 ± 0.03 bc | 0.69 ± 0.00 eg | 0.74 ± 0.03 i |
| M | 0.38 ± 0.00 be | 0.29 ± 0.01 be; * | 0.34 ± 0.01 l | 0.23 ± 0.00 fi; * | 1.03 ± 0.03 j | 0.62 ± 0.01 fh; * |
| N | 0.51 ± 0.05 h | 0.28 ± 0.04 bd; * | 0.25 ± 0.01 hi | 0.24 ± 0.02 g–i | 0.34 ± 0.02 a | 0.54 ± 0.03 bc; * |
| O | 0.44 ± 0.01 g | 0.23 ± 0.01 a; * | 0.26 ± 0.01 i | 0.17 ± 0.00 be; * | 0.73 ± 0.09 fg | 0.81 ± 0.04 j |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tusevski, O.; Gadzovska Simic, S. Non-Enzymatic and Enzymatic Antioxidant Responses of Hypericum perforatum L. Hairy Roots upon Photooxidative Stress. Horticulturae 2023, 9, 581. https://doi.org/10.3390/horticulturae9050581
Tusevski O, Gadzovska Simic S. Non-Enzymatic and Enzymatic Antioxidant Responses of Hypericum perforatum L. Hairy Roots upon Photooxidative Stress. Horticulturae. 2023; 9(5):581. https://doi.org/10.3390/horticulturae9050581
Chicago/Turabian StyleTusevski, Oliver, and Sonja Gadzovska Simic. 2023. "Non-Enzymatic and Enzymatic Antioxidant Responses of Hypericum perforatum L. Hairy Roots upon Photooxidative Stress" Horticulturae 9, no. 5: 581. https://doi.org/10.3390/horticulturae9050581
APA StyleTusevski, O., & Gadzovska Simic, S. (2023). Non-Enzymatic and Enzymatic Antioxidant Responses of Hypericum perforatum L. Hairy Roots upon Photooxidative Stress. Horticulturae, 9(5), 581. https://doi.org/10.3390/horticulturae9050581