Abstract
Single fruit weight is an important goal of crop production and horticultural species domestication, but its genetic mechanism is still unclear. In this study, the fruits of different peach fruit types in their first rapid development period were used as materials. First, the differentially expressed genes were analyzed by RNA-seq data. Secondly, weighted gene co-expression network analysis (WGCNA) was used to calculate the correlation between genes and modules, the genes with different expression patterns were divided into 17 modules, the modules were correlated with the phenotype of single fruit, and a highly correlated blue module was obtained. Then, the possible differentially expressed genes and signal pathways among different fruit types were compared by gene set enrichment analysis (GSEA) and 43 significant pathways were obtained. Finally, 54 genes found to be repeatedly expressed in 3 of the methods were screened, and 11 genes involved in plant hormone signal transduction were selected for subsequent analysis according to their functional annotations. Combined with the changing trend of phenotype, three genes (Prupe.7G234800, Prupe.8G079200 and Prupe.8G082100) were obtained as candidate genes for single fruit weight traits. All three genes are involved in auxin signal transduction, with auxin playing an important role in plant growth and development. This discovery provides a new perspective for revealing the genetic law of single fruit weight in peach.
1. Introduction
Peach (Prunus persica L.) belongs to Prunus of Rosaceae. Peach fruit has good color, fragrance, taste and quality, and it is deeply loved by consumers. In 2021, China’s peach production was about 16,016,533 tons (Faostat, http://www.fao.org/Faostat, accessed on 9 October 2023). The single fruit weight of peach is influenced by both genetic and environmental factors. The typical way to regulate the single fruit weight of peach is by analyzing and utilizing the regulatory factors of its genetic aspects [1,2]. Therefore, studying the genetics of the single fruit weight of peaches and identifying the key functional genes that control it, as well as elucidating the molecular mechanism behind it, will be extremely beneficial. Additionally, developing new, large fruit peach varieties through molecular marker-assisted selection will expand the fruit market and enhance the peach industry in China.
Peach fruit experiences a complicated process from pollination and fertilization to development and maturity, and its growth curve shows a typical double “S-shape”. The weight of a single fruit is mainly determined by the cell number, cell volume and the gap between cells in the mesocarp. In the early stage of fruit growth and development, the cell number is mainly increased, and in the late stage of fruit growth and development, the cell volume is mainly expanded [3]. The development of plant organs is influenced by cell division and cell expansion [4,5]. By a comparison of the relationship between the single fruit weight and the number and volume of mesocarp cells among different peach varieties [6], different plants of the same variety, and different fruits of the same plant [7], it was found that the number of mesocarp cells determined by the difference of cell division ability was crucial to the single fruit weight. This has also been demonstrated in tomato [8], melon [9], strawberry [10], apple [11] and avocado [12]. The candidate genes determining single fruit weight have also been reported by predecessors. For example, the expressions of ppa017982m and ppa010443m are positively correlated with fruit diameter, indicating that they may control single fruit weight traits by regulating cell growth [13]; Prune.6G046800 belongs to the cytochrome P450(CYP)79B subfamily, and participates in the biosynthesis of IAA. At the same time, tomato plants in which Prune.6G046800 is overexpressed have been found to be shorter than controls, and it can be further considered an effective candidate gene for controlling the single fruit weight of peach [14].
At present, some achievements have been made in plant functional gene mining, character analysis and assisted breeding based on RNA-seq technology. For example, based on transcription sequencing, it has been found that the reason for the obvious difference in branch color between the parents and backcross offspring of Salix goldenrod is the different expression levels of chlorophyll synthase-related genes [15]; The petal-shaped stamens of Paeonia lactiflora’ Liantai’ (stamen primordium, stamen primordium partially valved, stamen primordium completely valved) have been used as research materials, and the transcriptome analyses of flower buds in three development stages were carried out to reveal the changes of metabolic pathway of petal-shaped stamens [16]. The candidate resistance genes of cowpea (Vigna longifolia) have been revealed based on the transcriptome data of aphid resistance and aphid sensitivity [17]. However, most studies mainly mine key genes based on GO and KEGG function enrichment analysis of differential genes. Weighted gene co-expression network analysis (WGCNA) is an important method by which to study gene function through network, and has been applied in strawberries, tomatoes and apples [18,19,20]. Gene set enrichment analysis (GSEA) is used to find a set of genes with synergistic differences from the expression matrix of all genes, so it can take into account the genes with small differences [21]. Based on the analysis of differential genes by using transcriptome data, combined with the methods of WGCNA and GSEA, this study used the fruits of different fruit types in their first rapid expansion period as materials with which to mine the key candidate genes for controlling the single fruit weight traits of peaches (Supplementary Figure S1). This work is very helpful to the development of molecular markers related to single fruit weight in peach breeding.
2. Materials and Methods
2.1. Plant Materials
In the study, the tested materials came from 6 hybrid plants of the “Mengyin Qinglang × 07-8 East-9” hybrid population, including 3 large fruit types and 3 small fruit types. Fruit samples were collected at the end of flowering, 7, 14, 21, 28, 35 and 42 days after flowering for RNA sequencing. Each fruit type contains 3 biological repeats, and each repeat contains at least 3 fruits. The fruit was peeled and the mesocarp frozen quickly with liquid nitrogen and stored at −80 °C for later use. All of the materials were collected from the National Fruit Tree Germplasm Peach Resource Garden (Xinxiang, China) of the Zhengzhou Fruit Research Institute of China Academy of Agricultural Sciences, with good growth and consistent management measures, the samples were collected from the peripheral central area of each tree.
2.2. Phenotype Identification of Single Fruit Weight
The fruit weight of large and small fruit types were weighed with an electronic balance and the average value was taken.
2.3. RNA Extracting, Database Building and Data Filtering
Total RNA was extracted using the Trizol (Invitrogen, Carlsbad, CA, USA), RNA purity and integrity were monitored by NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and a Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). RNA contamination was assessed by 1.5% agarose gel. Oligo (dT)-attached magnetic beads were used to purify mRNA. Purified mRNA was fragmented into small pieces with fragment buffer at appropriate temperature. Then, first-strand cDNA was generated using random hexamer-primed reverse transcription, followed by a second-strand cDNA synthesis and purified using AMPure XP Beads. Afterwards, A-Tailing Mix and RNA index adapters were added by incubating to end cDNA repair. The cDNA fragments obtained from previous steps were amplified by PCR, products were purified by Ampure XP Bead to obtain the final library which was then sent to Frasergene platform for sequencing. In order to ultimately obtain clean reads, we used SOAP Nuke (V2.1.0) [22] to filter the sequencing data and remove reads containing sequencing linkers and N ratios greater than 0.5% as well as reads with Qphred ≤ 20 and a number of bases accounting for more than 50% of their total length.
2.4. Comparative Analysis of Reference Sequences and Differential Expression
HISAT2 [23] (V2.1.0) was used to compare clean reads with the reference genome, and Bowtie2 [24] (V2.3.5) was used to align the quality-controlled sequence to the reference transcription sequence. Using RSEM [25], the bowtie2 comparison results were used for statistics, and the number of reads compared with each transcript of each sample was obtained. FPKM [26] (Fragments Per Kilobase Per Million bases) conversion was then performed. DESeq2 [27] (V1.22.2) was used for differential expression significance analysis, and the screening threshold was |log2 (FoldChange)| > 1 and p < 0.05.
2.5. Analysis of WGCNA
Using gene expression information, WGCNA was constructed by R language package [28], and 17 modules were obtained. According to the gene expression and module eigenvalue, the correlation between gene and module was calculated, and the correlation result between gene and module was obtained. In order to further explain the possible functions and influences of module genes, we performed GO [29] and KEGG [30,31] on the gene of the target module.
2.6. GSEA Analysis
GSEA (http://software.broadinstitute.org/gsea/index.jsp (accessed on 9 October 2023)) was used to analyze and compare the possible differentially expressed genes and signal pathways among different fruit types based on a JAVA environment provided by the official website [21]. The parameters are as follows: set_max-500, plot_top_x-50, nperm-1000, set_min-15, with other parameters left as default.
2.7. Fluorescence Quantitative PCR Analysis
Eleven candidate genes related to the single fruit weight of peach were screened for qRT-PCR analysis to verify the reliability of transcriptome data. Using NCBI Primer-BLAST (Primer 3) software (National Center for Biotechnology Information, Bethesda, MA, USA), specific primers (Supplementary Table S5, including internal control gene primer) were designed, which were synthesized by Shenggong Bioengineering (Shanghai, China) Co., Ltd. The qRT-PCR of the target gene was carried out using the Roche SYBR Green kit (Shanghai, China) and real-time fluorescent quantitative PCR instrument with Light Cycler 480 Software. Each sample was repeated three times, and the relative expression of the gene was calculated by 2 −ΔΔCT method [32]. The 20 μL reaction system was set up as follows: SYBR Green Mix 10 μL, forward primer 0.5 μL, reverse primer 0.5 μL, cDNA 2 μL, and ddH2O 7 μL. The reaction procedure is as follows: 45 cycles of pre-denaturation at 95 °C for 5 min, denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s.
3. Results
3.1. Changes of Single Fruit Weight Traits during Peach Fruit Development
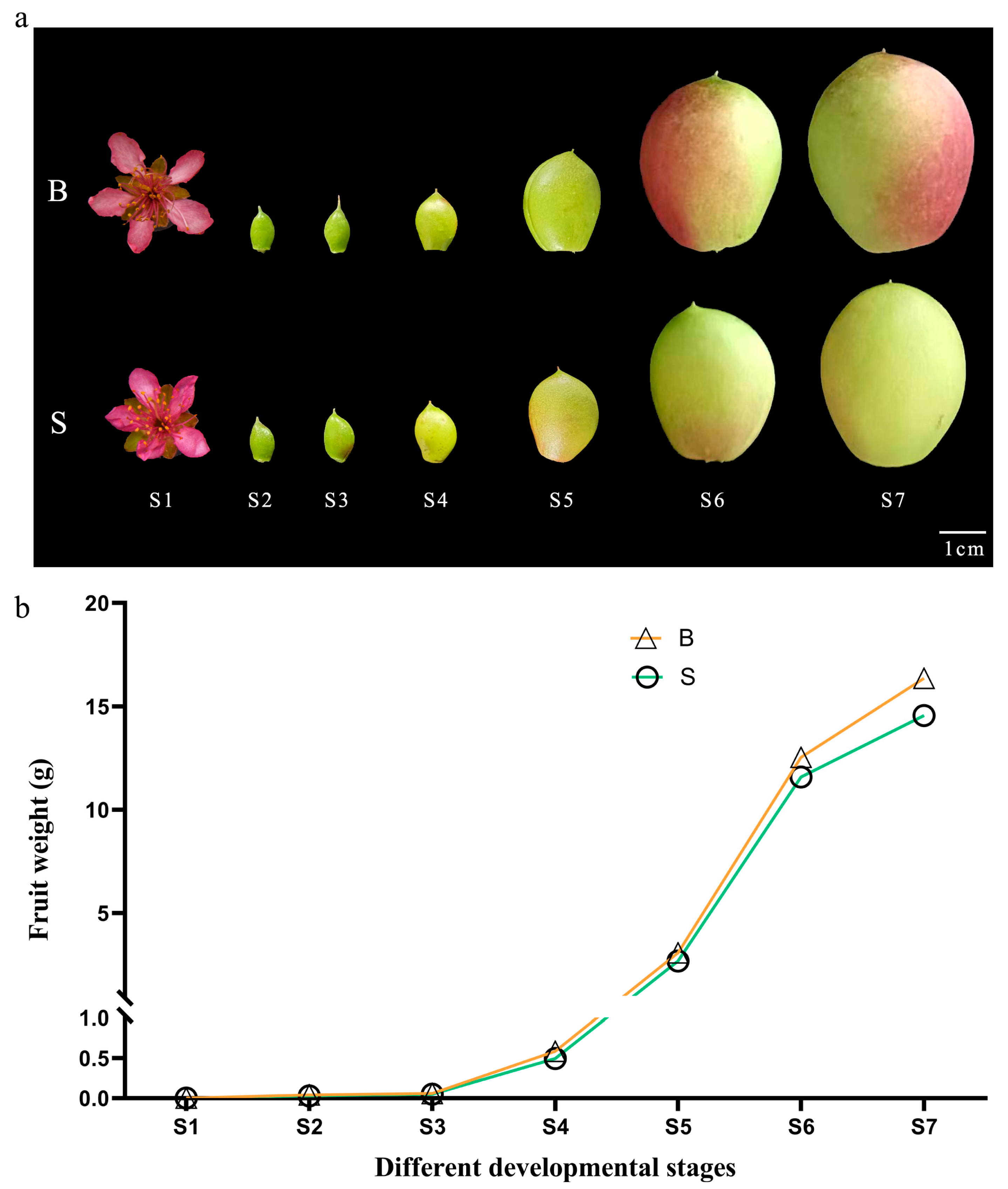
Peach fruit undergoes a complicated process from pollination and fertilization to development and maturity. The growth and development of large fruit type and small fruit type all conform to the law of ‘fast-slow-fast-slow’, that is, a double-S curve. Previous studies have shown that the first rapid growth period of peach fruit plays a decisive role in single fruit weight traits. Therefore, the changes of single fruit weight in seven stages of fruit development (late flowering stage, 7, 14, 21, 28, 35 and 42 days after flowering) were measured, and the results are shown in Figure 1. In the process of fruit development, the dynamic changes of the large fruit type and small fruit type are basically the same, and each show an upward trend. During S1–S5, there is not much difference in single fruit weight between the large fruit type and small fruit type. In S6 and S7, there are clear differences between them.
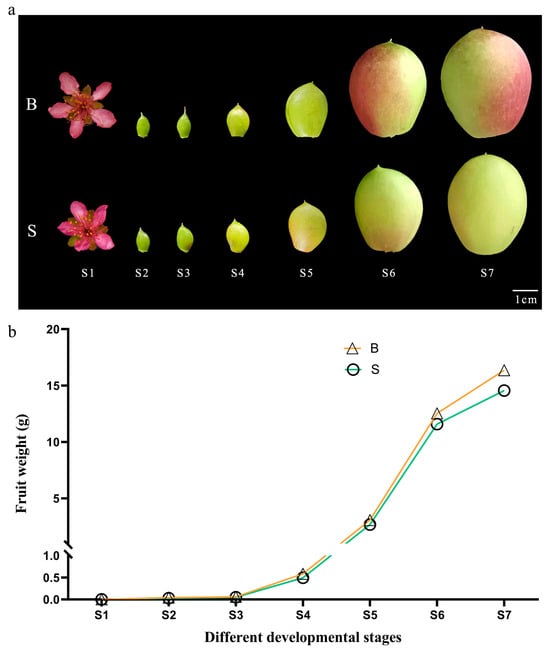
Figure 1.
Development pattern of single fruit weight of peach with different fruit types. (a) Pictures of peach fruit at different development stages. (b) Single fruit Weight of peach fruits with different fruit types in seven different development stages. ‘S’ indicates the small fruit type, ‘B’ indicates the large fruit type. S1–S7: the end of flowering, 7, 14, 21, 28, 35 and 42 days after flowering, respectively.
3.2. The Quality Evaluation of RNA-Seq Sequencing Data
A total of 42 samples were taken from the mesocarp of hybrid plants of both large and small fruit type, at the end of the flowering period and at 7, 14, 21, 28, 35 and 42 days after flowering, with 3 replicates in 7 periods, and the transcriptome was sequenced. Finally, 436.10 Gb of raw data were obtained, and 424.74 Gb clean data remained after treatment. The clean data of each sample reached 7.41 Gb, and the GC content was greater than 44.99%. The range of clean reads compared with the peach reference genome (LoveII) was 93.96–98.16% (Supplementary Table S1). Principal component analysis revealed that the differences between groups and the samples within groups are good (Supplementary Figure S2a), and the correlation coefficient of the three repetitions of the same group of samples is greater than 0.71 (Supplementary Figure S2b). In addition, the gene expression level of each sample is basically similar (Supplementary Figure S3). To sum up, the sequencing data quality of the transcriptomics ensures the validity and reliability of the subsequent biological data analysis.
3.3. Statistics of Differentially Expressed Genes
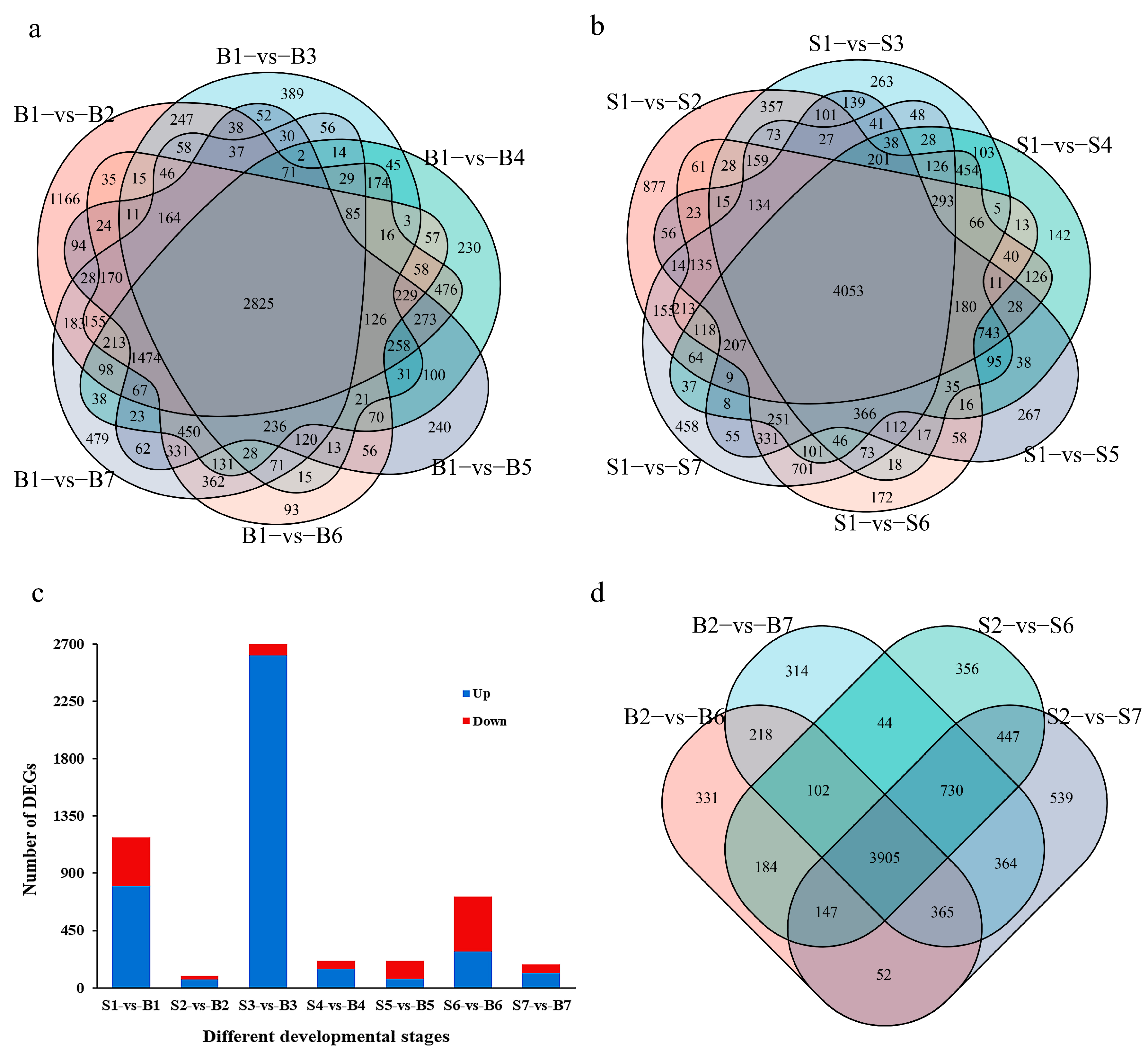
In order to find the key genes for controlling single fruit weight, differential genes were screened with |log2 (FoldChange)| > 1 and p < 0.05 as the standard. Compared with the last flowering period, the results show that 12791 differential genes were identified in the large fruit type. A total of 13,222 differential genes were identified in the small fruit type. Among these, 2825 genes were differentially expressed in the whole development period of the large fruit type, and 4053 genes were differentially expressed in the whole development period of the small fruit type (Figure 2a,b). Comparing the differences between groups during peach fruit development, it was found that there were 1182 differentially expressed genes (804 upregulated and 378 downregulated) in S1 stage. In S2 stage, the number of differentially expressed genes was the least, with 96 (63 upregulated and 33 downregulated). The number of differentially expressed genes in S3 was up to 2787 (2612 upregulated and 175 downregulated). The number of differentially expressed genes in S4 and S5 was the same, with 213 (21 days, 150 were upregulated and 63 were downregulated; 28 days, 72 up and 141 down). There were 715 differentially expressed genes (287 upregulated and 428 downregulated) and 184 differentially expressed genes (115 upregulated and 69 downregulated) in S6 and S7, respectively (Figure 2c). Among these, three genes were differentially expressed among all of the groups during the development of peach fruit (Supplementary Figure S4). To sum up, the number of different genes between large fruit type and small fruit type is large in their whole fruit development period, and the number of downregulated genes is more than that of upregulated genes. There is little difference between different fruit types in the same development period, and the number of differential genes is highest in the S3 period. It is speculated that the metabolic activity of peach fruit in this period is strong, which may be the key period for the formation of different fruit types. At the same time, combined with the phenotypic data, there were differences between the two fruit types in S6 and S7. Compared with S2 (the first fruit picking), 3905 genes were expressed in each period for subsequent analysis (Figure 2d).
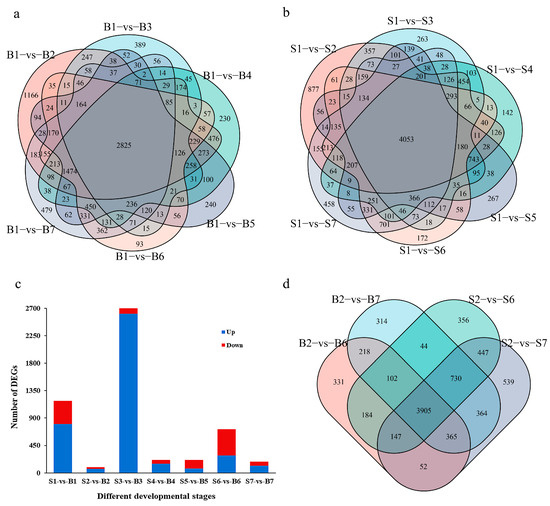
Figure 2.
Analysis of differentially expressed genes. (a) Venn diagram of differentially expressed genes in different stages of big fruit type (B1 as control). (b) Venn diagram of differentially expressed genes in different stages of small fruit type (S1 as control). (c) Number of differentially expressed genes in different fruit types at the same time. (d) Venn diagram of differentially expressed genes in different fruit types at the S6 and S7 stages (S2 stage as control). ‘B1’ indicates the end of flowering of the large fruit type. ‘B2’ indicates the 7 days after flowering of the large fruit type. ‘B3’ indicates the 14 days after flowering of the large fruit type. ‘B4’ indicates the 21 days after flowering of the large fruit type. ‘B5’ indicates the 28 days after flowering of the large fruit type. ‘B6’ indicates the 35 days after flowering of the large fruit type. ‘B7’ indicates the 42 days after flowering of the large fruit type. ‘S1’ indicates the end of flowering of the small fruit type. ‘S2’ indicates the 7 days after flowering of the small fruit type. ‘S3’ indicates the 14 days after flowering of the small fruit type. ‘S4’ indicates the 21 days after flowering of the small fruit type. ‘S5’ indicates the 28 days after flowering of the small fruit type. ‘S6’ indicates the 35 days after flowering of the small fruit type. ‘S7’ indicates the 42 days after flowering of the small fruit type.
3.4. Screening Key Candidate Genes Based on WGCNA
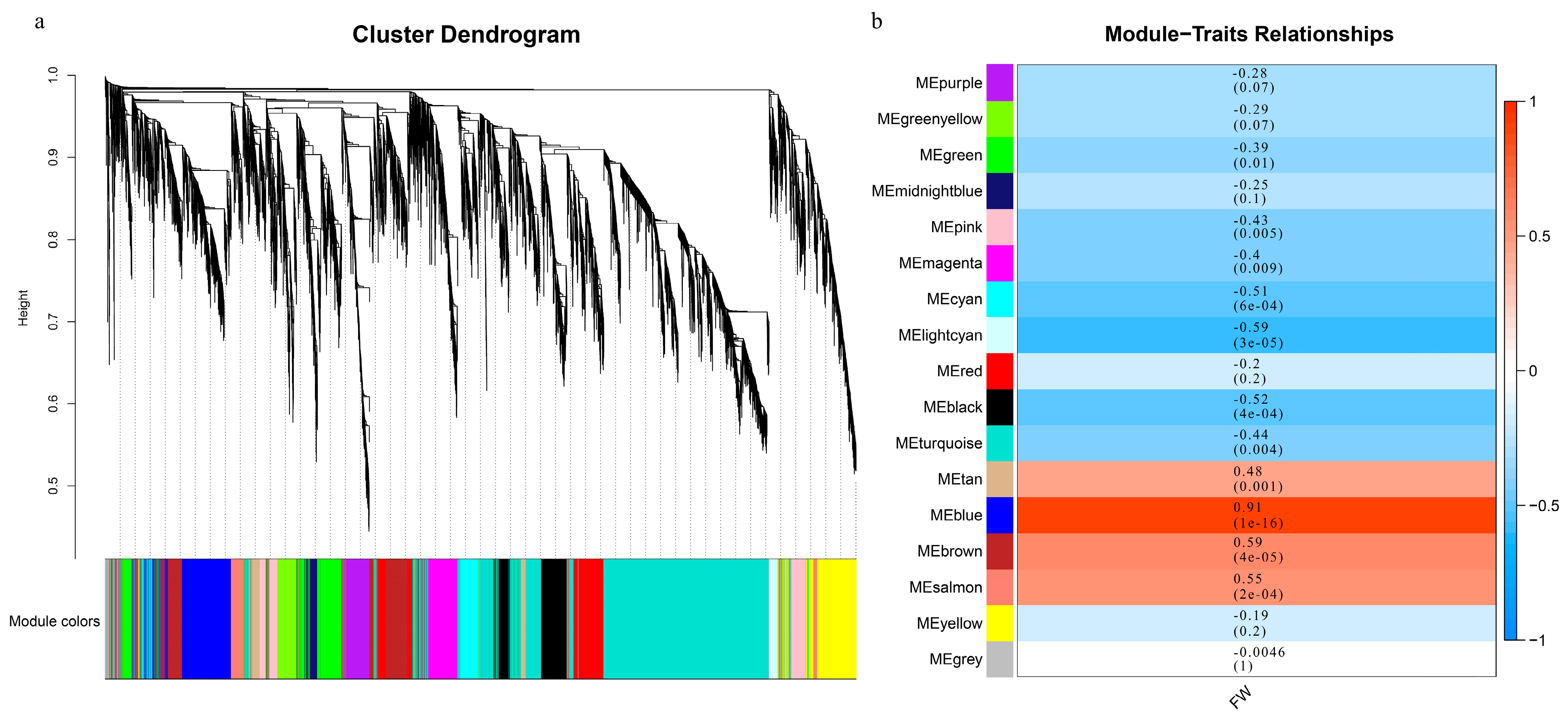
In our study, the related genes affecting single fruit weight traits of different peach fruit types were further identified by WGCNA analysis. According to the correlation coefficient between 12,076 differentially expressed genes, the co-expression modules of all samples were constructed, and 17 modules were finally obtained (Figure 3a). Among these, there are, at most, 4036 genes in the turquoise module and, at least, 115 genes in the grey module (Supplementary Table S2). The correlation analysis between the gene expression module and the single fruit weight revealed that the blue module was significantly correlated (r2 = 0.91), and that there were 958 genes in this module (Figure 3b). The expression patterns of all genes contained in the blue module are displayed by thermogram, and the changes of the module characteristic values among different samples are presented by histogram (Supplementary Figure S5). Almost all of the samples have negative module eigenvalues in the first five periods and positive module eigenvalues in the last two periods. In order to further explore whether the differentially expressed genes in the blue module are the reasons for the different traits of single fruit weight, we conducted GO analysis and KEGG function enrichment analysis. GO analysis enriched 36 terms, including 20 biological processes (BP), 2 cellular components (CC) and 14 biological functions (MF), with the largest number of cellular anatomical entity genes. KEGG metabolic pathway analysis showed that 17 signaling pathways were significantly enriched, among which the number of genes in the carbohydrate metabolism pathway was the largest (Supplementary Figure S6).
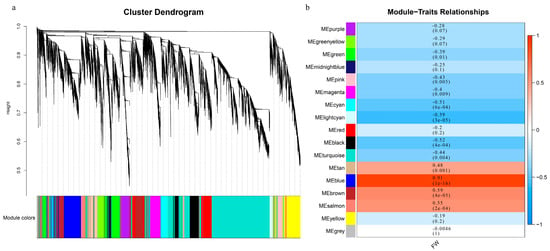
Figure 3.
WGCNA analysis of DEGs between different developmental stages and single fruit weight. (a) Cluster diagram of co-expression modules. Different colors correspond to different co-expression modules. (b) Relationships between modules and traits. Behavior clustering module, listed as a single fruit phenotype. The values in the cells in which the rows and columns cross represent the correlation coefficient between modules and phenotypes, and the numbers in brackets are p values.
3.5. Gene Set Enrichment Analysis GSEA
GSEA is aimed at gene sets rather than at single genes, and can contain genes with small differences for enrichment. On this basis, and when combined with phenotype, the result is more perfect [21]. A total of 24157 genes of two fruit types were enriched and analyzed by GSEA. Among the 107 enriched pathways (Supplementary Table S3), the genes in 39 pathways were upregulated in the large fruit type, including 21 pathways with an FDR q-val of less than 0.25, 13 pathways with nominal p-values of less than 0.05, and 10 pathways with nominal p-values less than 0.01. There are 13 paths with an FDR q-val less than 0.25 and nominal p-values less than 0.05, including MAPK signaling pathway plant, ABC transporters and plant hormone signal transduction (Supplementary Figure S7). MAPK plays a key role in two important processes of plant growth and development, cell division and cell differentiation [33]. ABA is considered a growth inhibitory hormone, usually involved in coping with many environmental stresses, such as cold, salt and drought [34], but some studies have also revealed some functions of ABA in regulating fruit development. ABA is considered to inhibit the fruit growth in the early stage of strawberry, which is accompanied by low ABA levels in the early development and a sharp increase during maturity [35]. Genes were upregulated in 68 pathways in the small fruit type, including 38 pathways with an FDR q-val less than 0.25, 30 pathways with a nominal p-value less than 0.05, 26 pathways with a nominal p-value less than 0.01, and 30 pathways with an FDR q-val less than 0.25 and a nominal p-value less than 0.05. When including carbon fixation in photo synthetic organizations, DNA replication and RNA degradation it is often found that the average level of replication in different plant organs is related to the size of organs (fruits, leaves, flowers and roots). It has been reported that tetraploid fruits caused by chromosome doubling are much larger than diploid fruits in horticultural plants [36,37,38].
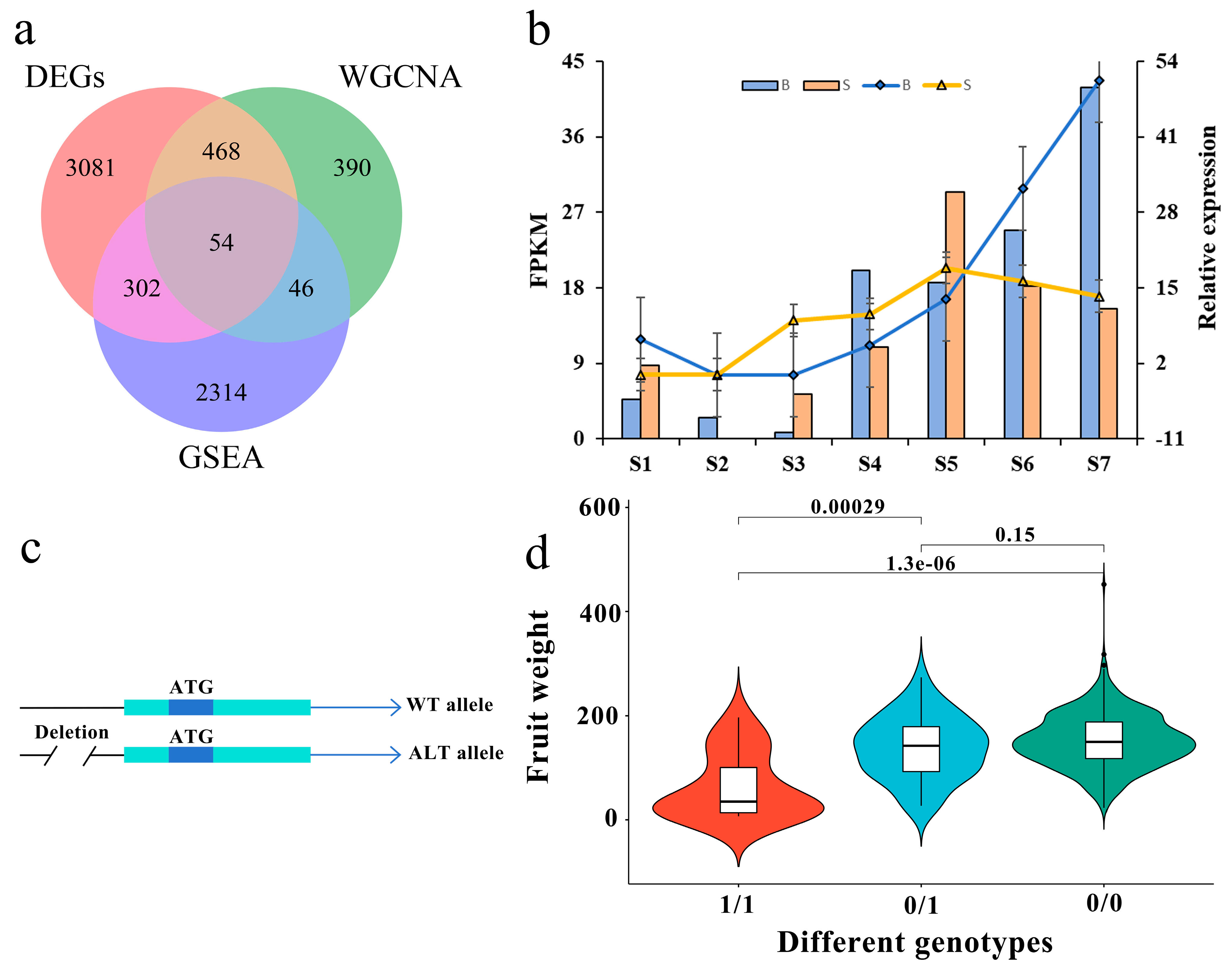
3.6. Screening of Key Genes for Single Fruit Weight by Three Methods
Genes based on DEGs (3905), WGCNA (958) and GSEA (2716) were used to screen the key genes of single fruit weight. Figure 4a shows that there are 54 genes that are duplicated in three parts. Combined with the existing gene function annotations (Supplementary Table S4), it is found that 11 genes are involved in plant hormone signal transduction. Previous studies have shown that plant hormones participate in the whole growth and development process of plants and respond to environmental and endogenous signals, and many studies have emphasized the role of plant hormones and their interactions in regulating fruit weight traits [39,40,41,42,43]. By analyzing the relative expression levels of 11 genes (Figure 4b and Supplementary Figure S8), it was found that the expression patterns of these genes were consistent with the results of RNA-seq, indicating the accuracy of RNA-seq data. The expression of three genes (Prupe.7G234800, Prupe.8G079200 and Prupe.8G082100) is located in the auxin synthesis pathway (Supplementary Figure S9) and is consistent with the development trend of fruits. Both Prupe.8G079200 and Prupe.8G082100 edit the SAUR-like auxin-responsive protein, while Prupe.7G234800 edits the auxin-responsive protein IAA17. At the same time, a deletion of 1304bp was found in the promoter region of Prupe.8G079200 (Figure 4c), and the genotype of the deletion site was highly correlated with the single fruit weight traits (Figure 4d), suggesting that it plays a key role in the development of single fruit weight traits in peach.
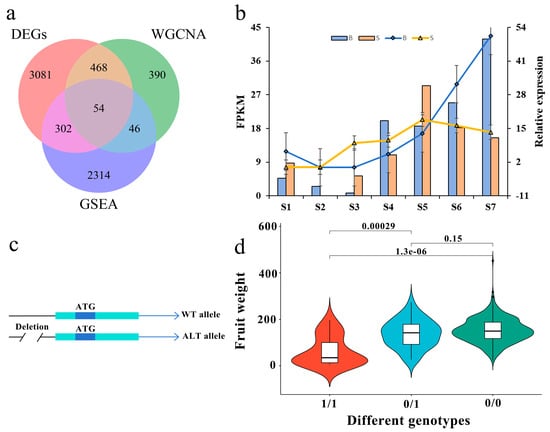
Figure 4.
On the candidate genes of single fruit weight. (a) Venn diagram of differentially expressed genes of DEGs, WGCNA and GSEA. (b) Prupe.8G079200 expression in different fruit types at different developmental stages. The column chart and left longitudinal coordinate indicate the FPKM value of RNA-seq, whereas the broken line diagram and right longitudinal coordinate show the relative expression of qRT-PCR. S1–S7: the end of flowering, 7, 14, 21, 28, 35 and 42 days after flowering, respectively. ‘S’ indicates the small fruit type, ‘B’ indicates the large fruit type. (c) Two different alleles of Prupe.8G079200. WT allele stands for the reference allele, while the ALT allele is the alternate allele that has a 1304-bp deletion (DEL) in its promoter. (d) Genotyping of 1304-bp deletion in the promoter region of Prupe.8G079200 and its relationship with single fruit weight. Mut indicates the genotype deletion, 0/1 indicates the genotype with deletion of heterozygosity, 1/1 indicates the genotype with deletion of homozygosity, and 0/0 indicates the genotype without deletion.
4. Discussion
Single fruit weight is an important goal of crop production and horticultural species domestication research [44], and is extremely important for both commercial production and consumers, but there is no relevant report on the key candidate genes of single fruit weight in peach. Therefore, it is of great significance to understand the development mechanism of single fruit weight traits of peach and to mine its key genes. RNA-seq technology is relatively mature, with low sequencing cost and simplified library preparation, and is the main direction of fruit tree sequencing research. At the same time, it has been successfully applied to jujube, grape, apple, kiwifruit and other fruit trees [45,46,47,48]. Based on RNA-seq technology, this study analyzed the differentially expressed genes in the early development stage of a large fruit type and a small fruit type and found that these genes were mostly enriched in hormone signal transduction, ubiquitin-proteasome pathway and MAPK signaling, which is consistent with previous studies on single fruit weight traits [40,48,49,50,51].
Auxin, as one of the earliest discovered plant hormones, plays an important role in the elongation and division of plant cells. The early auxin response mechanisms can be divided into three categories, namely, AUX/IAA (Auxin/Indole-3-acetic acid), GH3 (gretchen hagen3) and Saur (Small Auxin-Up RNA) [52,53], the related genes express very quickly when auxin is used [54]. SAUR encodes auxin-related protein, which is unique to plants [55] and was first discovered in the elongated hypocotyl region of soybean [56]. Since then, the SAURs gene has also been identified in mung bean, pea, Arabidopsis thaliana, tobacco, corn, rice, sorghum, tomato, potato, citrus, ramie, cotton, cucumber, watermelon, alfalfa and agastaches [57,58,59,60,61,62,63,64,65,66,67,68]. Over-expression of AtSAUR36 and 41 in Arabidopsis thaliana makes cells swell, which leads to significant elongation of hypocotyl epidermal cells [69]; AtSAUR19 and AtSAUR53 can regulate the elongation and root tip development of plant organs [70]. ZmSAUR2 gene in maize promotes the expansion and elongation of cells in coleoptile and stele sheath by regulating auxin transport in plants [71]. Previous studies have found that there are 80 SAUR genes in peach, 75 of which have no introns, suggesting that members of the gene family are highly conservative in gene structure. Real-time fluorescence quantitative PCR was used to verify the expression of Pp SAURs gene in different tissues. Taking the expression level in seeds as a reference, it was found that PpSAUR was highly expressed in leaves and roots, and that the expression level of Prupe.8G079200 and Prupe.8G082100 genes were significantly increased after IAA and GA treatment, respectively [72]. It is speculated that three candidate genes regulate the single fruit weight of peach by participating in the auxin signaling pathway, but its detailed mechanism needs further study.
5. Conclusions
In this study, peach fruits with different fruit types and in their first rapid development period were used as test materials. Based on the analysis of differential genes by transcriptome data, the genes controlling single fruit weight traits were screened by combining WGCNA and GSEA methods. Prupe.7G234800, Prupe.8G079200 and Prupe.8G082100, which participate in auxin signal transduction, were finally screened. Auxin plays an important role in the elongation and division of plant cells. Therefore, it is speculated that it is involved in the formation of single fruit weight traits of peach, but its mechanism needs further study.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/horticulturae9121335/s1, Figure S1: The correlation analysis of all samples; Figure S2: The FPKM box diagram of genes in different samples; Figure S3: Upset map of differentially expressed genes in peach fruit development with different fruit types in the same period; Figure S4: Heatmap and gene expression of blue module; Figure S5: GO and KEGG enrichment analysis of DEGs in blue module; Figure S6: Results of plant hormone signal transduction pathways analysis based on set enrichment of differential genes (GSEA) method; Figure S7: Validation of RNA-seq by qRT-PCR; Figure S8: Validation of RNA-seq by qRT-PCR; Figure S9: The auxin synthesis pathway using KEGG pathway analysis about the three candidate genes; Table S1: Transcriptome data and quality assessment; Table S2: The number of genes in each module was identified by WGCNA method; Table S3: Gene sets enriched in phenotype (the large and small fruit type) by GSEA method; Table S4: Functional annotation of 54 genes; Table S5: Primers for RT-qPCR.
Author Contributions
K.C. and L.W. conceived the project. Y.L., W.F., C.C., X.W. and J.W. collected samples. H.B. analyzed the data. H.W. conducted all experiments. H.B. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation (31872061), the National Key Research and Development Program (2019YFD1000203), the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2019-ZFRI-01), and the National Horticulture Germplasm Resources Center.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in NCBI, reference number PRJNA1049734.
Acknowledgments
We thank the anonymous reviewers and editors for their constructive suggestions on our manuscript.
Conflicts of Interest
The authors declare that they have no competing interest.
References
- Biscarini, F.; Nazzicari, N.; Bink, M.; Arús, P.; Aranzana, M.J.; Verde, I.; Micali, S.; Pascal, T.; Quilot-Turion, B.; Lambert, P.; et al. Genome-enabled predictions for fruit weight and quality from repeated records in European peach progenies. BMC Genom. 2017, 18, 432. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, J.; Xu, Y.; Ren, F.; Zhang, Z.; Yan, J.; Fu, J.; Guo, J.; Shen, Z.; Zhao, J.; et al. Population-scale peach genome analyses unravel selection patterns and biochemical basis underlying fruit flavor. Nat. Commun. 2021, 12, 3604. [Google Scholar] [CrossRef]
- Chalmers, D.J.; Ende, B.V.D. Productivity of peach trees: Factors affecting dry-weight distribution during tree growth. Ann. Bot. 1975, 39, 423–432. [Google Scholar] [CrossRef]
- Horiguchi, G.; Ferjani, A.; Fujikura, U.; Tsukaya, H. Coordination of cell proliferation and cell expansion in the control of leaf size in Arabidopsis thaliana. J. Plant Res. 2006, 119, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Masami, Y.; Takashi, H.; Masanori, M.; Hideaki, Y. Varietal differences in cell division and enlargement periods during peach (Prunus persica Batsch) fruit development. J. Jpn. Soc. Hortic. Sci. 2002, 71, 155–163. [Google Scholar]
- Bradley, M.V. Mean cell size in the mesocarp of mature peaches of different sizes. Proc. Am. Soc. Hortic. Sci. 1959, 73, 120–124. [Google Scholar]
- Bohner, J.; Bangerth, F. Cell number, cell size and hormone levels in semi-isogenic mutants of lycopersicon pimpinellifolium differing in fruit size. Physiol. Plant. 1988, 72, 316–320. [Google Scholar] [CrossRef]
- Higashi, K.; Hosoya, K.; Ezura, H. Histological analysis of fruit development between two melon (Cucumis melo L. reticulatus) genotypes setting a different size of fruit. J. Exp. Bot. 1999, 50, 1593–1597. [Google Scholar] [CrossRef]
- Cheng, G.W.; Breen, P.J. Cell Count and Size in Relation to Fruit Size Among Strawberry Cultivars. J. Am. Soc. Hortic. Sci. 1992, 117, 946–950. [Google Scholar] [CrossRef]
- Goffinet, M.C.; Robinson, T.L.; Lakso, A.N. A comparison of ‘Empire’ apple fruit size and anatomy in unthinned and hand-thinning trees. J. Pomol. Hortic. Sci. 1995, 70, 375–387. [Google Scholar] [CrossRef]
- Cowan, A.K.; Moore-Gordon, C.S.; Bertling, I.; Wolstenholme, B.N. Metabolic Control of Avocado Fruit Growth (Isoprenoid Growth Regulators and the Reaction Catalyzed by 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase). Plant Physiol. 1997, 114, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Li, Y.; Deng, C.H.; Gardiner, S.E.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, L. Comparative population genomics identified genomic regions and candidate genes associated with fruit domestication traits in peach. Plant Biotechnol. J. 2019, 17, 1954–1970. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Song, W.; Liu, X.; Liang, H. Study on branch color and transcriptome of Salix caprea var. aurea. For. Ecol. Sci. 2022, 37, 370–377. [Google Scholar]
- Fan, Y.; Zheng, Y.; Chen, L.; Xu, L.; da Silva, J.A.T.; Wu, B.; Yu, X. Transcriptomic and proteomic analyses reveal changes in the metabolic pathways of Paeonia lactiflora petaloid stamens. Sci. Hortic. 2023, 312, 111859. [Google Scholar] [CrossRef]
- MacWilliams, J.R.; DNabity, P.; Mauck, K.E.; Kaloshian, I. Transcriptome analysis of aphid-resistant and susceptible near isogenic lines reveals candidate resistance genes in cowpea (Vigna unguiculata). BMC Plant Biol. 2023, 23, 22. [Google Scholar] [CrossRef]
- Hollender, C.A.; Kang, C.; Darwish, O.; Geretz, A.; Matthews, B.F.; Slovin, J.; Alkharouf, N.; Liu, Z. Floral Transcriptomes in Woodland Strawberry Uncover Developing Receptacle and Anther Gene Networks. Plant Physiol. 2014, 165, 1062–1075. [Google Scholar] [CrossRef]
- Gao, C.; Ju, Z.; Li, S.; Zuo, J.; Fu, D.; Tian, H.; Luo, Y.; Zhu, B. Deciphering Ascorbic Acid Regulatory Pathways in Ripening Tomato Fruit Using a Weighted Gene Correlation Network Analysis Approach. J. Integr. Plant Biol. 2013, 55, 1080–1091. [Google Scholar] [CrossRef]
- Bai, Y.; Dougherty, L.; Cheng, L.; Zhong, G.-Y.; Xu, K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genom. 2015, 16, 612. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B. Aligning Short Sequencing Reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. Mitogen-activated protein kinase cascades in plant signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic Acid: Emergence of a Core Signaling Network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Li, M.; Liu, B.; Yan, M.; Yu, X.; Zi, H.; Liu, R.; Yamamuro, C. Interlinked regulatory loops of ABA catabolism and biosynthesis coordinate fruit growth and ripening in woodland strawberry. Proc. Natl. Acad. Sci. USA 2018, 115, E11542–E11550. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, J.; Zhao, T.; Du, C.; Nie, S.; Zhang, Y.; Lv, S.; Huang, S.; Wang, X. Modification of Threonine-1050 of SlBRI1 regulates BR Signalling and increases fruit yield of tomato. BMC Plant Biol. 2019, 19, 256. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Wang, H.H.; Shi, C.H.; Zhang, X.Y.; Duan, K.; Luo, J. Morphological, cytological and fertility consequences of a spontaneous tetraploid of the diploid pear (Pyrus pyrifolia Nakai) cultivar ‘Cuiguan’. Sci. Hortic. 2015, 189, 59–65. [Google Scholar] [CrossRef]
- Xue, H.; Zhang, B.; Tian, J.R.; Chen, M.M.; Zhang, Y.Y.; Zhang, Z.H.; Ma, Y. Comparison of the morphology, growth and development of diploid and autotetraploid ‘Hanfu’ apple trees. Sci. Hortic. 2017, 225, 277–285. [Google Scholar] [CrossRef]
- Pattison, R.J.; Catalá, C. Evaluating auxin distribution in tomato (Solanum lycopersicum) through an analysis of the PIN and AUX/LAX gene families. Plant J. 2012, 70, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, T.; Liu, J.; Cong, L.; Zhu, Y.; Zhai, R.; Yang, C.; Wang, Z.; Ma, F.; Xu, L. PbGA20ox2 Regulates Fruit Set and Induces Parthenocarpy by Enhancing GA4 Content. Front. Plant Sci. 2020, 11, 113. [Google Scholar] [CrossRef]
- Pang, Y.; Zang, X.Y.; Pang, F.T.; Zhou, T.H.; Tian, F.Z. Changes of CTK and few nitrogen index during development of flower and fruit in Zhanhua jujube. J. N. China Agric. 2017, 5, 101–104. [Google Scholar]
- Zhao, Z.H.; Liu, M.J.; Zhao, J.; Dai, L.; Liu, P. Study on changes of endogenous hormone content in fruit development of ‘Dongzao’ and ‘Linyilizao’. Acta Hortic. Sin. 2014, 41, 2628. [Google Scholar]
- Feng, C.Z.; Chen, Y.; Wang, C.; Kong, Y.H.; Wu, W.H.; Chen, Y.F. Arabidopsis RAV1 transcription factor, phosphorylated by SnRK2 kinases, regulates the expression of ABI3, ABI4, and ABI5 during seed germination and early seedling development. Plant J. 2014, 80, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, J.; Xiao, K.; Yang, W.C. Origin of the domesticated horticultural species and molecular bases of fruit shape and size changes during the domestication, taking tomato as an example. Hortic. Plant J. 2017, 3, 125–132. [Google Scholar] [CrossRef]
- Liu, M.-J.; Zhao, J.; Cai, Q.-L.; Liu, G.-C.; Wang, J.-R.; Zhao, Z.-H.; Liu, P.; Dai, L.; Yan, G.; Wang, W.-J.; et al. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 2014, 5, 5315. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Sun, X.; Yan, X.; Xu, X.; Yan, Q.; Gao, M.; Fei, Z.; Wang, X. Grape Transcriptome Response to Powdery Mildew Infection: Comparative Transcriptome Profiling of Chinese Wild Grapes Provides Insights Into Powdery Mildew Resistance. Phytopathology 2021, 111, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Kamber, T.; Buchmann, J.P.; Pothier, J.F.; Smits, T.H.M.; Wicker, T.; Duffy, B. Fire blight disease reactome: RNA-seq transcriptional profile of apple host plant defense responses to Erwinia amylovora pathogen infection. Sci. Rep. 2016, 6, 21600. [Google Scholar] [CrossRef]
- Nardozza, S.; Cooney, J.; Boldingh, H.L.; Hewitt, K.G.; Trower, T.; Jones, D.; Thrimawithana, A.H.; Allan, A.C.; Richardson, A.C. Phytohormone and Transcriptomic Analysis Reveals Endogenous Cytokinins Affect Kiwifruit Growth under Restricted Carbon Supply. Metabolites 2020, 10, 23. [Google Scholar] [CrossRef]
- Walsh, C.K.; Sadanandom, A. Ubiquitin chain topology in plant cell signaling: A new facet to an evergreen story. Front. Plant Sci. 2014, 5, 122. [Google Scholar] [CrossRef]
- Stone, S.L.; Williams, L.A.; Farmer, L.M.; Vierstra, R.D.; Callis, J. Keep on going, a RING E3 ligase essential for Arabidopsis growth and development, is involved in abscisic acid signaling. Plant Cell 2007, 18, 3415–3428. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, Z.; Li, S.; Lian, Q.; Fu, P.; He, Y.; Qiao, J.; Xu, K.; Liu, L.; Wu, M.; et al. Genomic analyses of diverse wild and cultivated accessions provide insights into the evolutionary history of jujube. Plant Biotechnol. J. 2020, 19, 517–531. [Google Scholar] [CrossRef]
- Nemhauser, J.L.; Hong, F.; Chory, J. Different Plant Hormones Regulate Similar Processes through Largely Nonoverlapping Transcriptional Responses. Cell 2006, 126, 467–475. [Google Scholar] [CrossRef]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 375–385. [Google Scholar] [CrossRef]
- Ljung, K.; Bhalerao, R.P.; Sandberg, G. Sites and homeostatic control of auxin biosynthesis in Arabidopsis during vegetative growth. Plant J. 2001, 28, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Niek, S.; Marian, B. The SAUR gene family: The plant’s toolbox for adaptation of growth and development. J. Exp. Bot. 2018, 70, 17–27. [Google Scholar]
- McClure, B.A.; Guifoyle, T. Characterizaton of a class of small auxin-inducible soybean polyadenylated RNAs. Plant Mol. Biol. 1987, 9, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.T.; Mori, H.; Imaseki, H. cDNA Cloning of Indole-3-Acetic Acid-Regulated Genes: Aux22 and SAUR from Mung Bean (Vigna radiata) Hypocotyl Tissue. Plant Cell Physiol. 1992, 33, 93–97. [Google Scholar] [CrossRef]
- Guilfoyle, T.J.; Hagen, G.; Li, Y.; Ulmasov, T.; Liu, Z.B.; Strabala, T.; Gee, M. Auxin-Regulated Transcription. Funct. Plant Biol. 1993, 20, 489–502. [Google Scholar] [CrossRef]
- Gil, P.; Liu, Y.; Orbović, V.; Verkamp, E.; Poff, K.L.; Green, P.J. Characterization of the Auxin-Inducible SAUR-AC1 Gene for Use as a Molecular Genetic Tool in Arabidopsis. Plant Physiol. 1994, 104, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Bilang, J.; Theunissen, B.H.; Perrot-Rechenmann, C. Identification of new early auxin markers in tobacco by mRNA differential display. Plant Mol. Biol. 1998, 37, 385–389. [Google Scholar] [CrossRef]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family inrice (Oryza sativa). Genomics 2006, 88, 360–367. [Google Scholar] [CrossRef]
- Wang, S.; Bai, Y.; Shen, C.; Wu, Y.; Zhang, S.; Jiang, D.; Guilfoyle, T.J.; Chen, M.; Qi, Y. Auxin-related gene families in abiotic stress response in Sorghum bicolor. Funct. Integr. Genom. 2010, 10, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, S.; He, Y.; Guan, X.; Zhu, X.; Cheng, L.; Wang, J.; Lu, G. Genome-wide analysis of SAUR gene family in Solanaceae species. Gene 2012, 509, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kong, Y.; Wang, J. Research advances in auxin-responsive SAUR genes. Life Sci. 2014, 26, 407–413. [Google Scholar]
- Chen, Y.; Hao, X.; Cao, J. Small auxin upregulated RNA (SAUR) gene family in maize: Identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J. Integr. Plant Biol. 2014, 56, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shang, Q. Entification and expression analysis of cucumber SAUR gene family. Acta Hortic. Sin. 2019, 46, 1093–1111. [Google Scholar]
- Deng, G.; Huang, X.; Xie, L.; Tan, S.; Gbokie, T.; Bao, Y.; Xie, Z.; Yi, K. Identification and Expression of SAUR Genes in the CAM Plant Agave. Genes 2019, 10, 555. [Google Scholar] [CrossRef]
- Zhang, T.; Jiang, B.; Xie, R.; Xu, M.; Li, H.; Liu, K. Genome-wide identification and analysis of Medicago truncatula Small auxin upregulated RNA (SAUR) gene family uncover their roles in nodule formation. J. Plant Biochem. Biotechnol. 2020, 30, 126–137. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Praveen, K.K.; Sunethra, D.; Nihal, D. SAUR53 regulates organ elongation and apical hook development in Arabidopsis. Plant Signal. Behav. 2018, 13, e1514896. [Google Scholar]
- Knauss, S.; Rohrmeier, T.; Lehle, L. The Auxin-induced Maize Gene ZmSAUR2 Encodes a Short-lived Nuclear Protein Expressed in Elongating Tissues. J. Biol. Chem. 2003, 278, 23936–23943. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Zhai, Y.; Tian, Y.; Zhang, Y.; Yang, L.; Wen, Z.; Chen, H. Genome-wide identification of peach SAUR gene family and characterization of PpSAUR5 gene. Acta Hortic. Sin. 2023, 50, 1–14. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).